Introduction
Epigenetic regulation alters the gene expression
through chromatin structural modification without DNA sequence
changes (1). Post-translational
modifications of histones, including acetylation, serve an
important role in regulating gene expression (2). The pattern of histone acetylation is
determined by two groups of enzymes: Histone acetyltransferases and
histone deacetylases (HDACs). The deacetylation of lysine residues
by HDAC is associated with condensed chromatin state and
transcriptional gene silencing (3).
HDACs alter the transcription of genes that regulate essential cell
functions, including cell growth, cell cycle regulation and
apoptosis (1). HDACs have been
subdivided into a family of 18 genes that represent four classes:
Class I (HDAC1, 2, 3 and 8), Class II (HDAC4, 5, 6 and 11), Class
III (HDAC11) and Sirtuins (Sirt1-7). Upregulation of HDACs has been
determined in numerous human cancer types, including oral cancer
(1,4,5).
Therefore, HDACs have become an attractive target for cancer
therapeutics.
HDAC inhibitors have emerged as a novel class of
anticancer drugs. They are structurally classified into at least
four groups: Hydroxamic acids, short-chain fatty acids, benzamides
and cyclic peptides (6,7). These agents vary notably in their
potency and specificity toward individual HDACs (8). Apicidin is a cyclic peptide isolated as
a fungal metabolite from fermentations of Fusarium spp
(9). Apicidin has been reported to
exhibit a proliferative effect in various cancer types, including
leukemia, ovarian cancer and hepatocellular carcinoma (10–12).
Apicidin primarily induces cell cycle arrest and apoptosis through
caspase activation in cancer cells (10–12).
However, specific targets of apicidin in a variety of cancer types,
including lung and pancreatic cancer, remain unclear, and research
into the molecular mechanism of apicidin for anticancer activity
remains ongoing in pre-clinical studies (13–16).
Oral cancer is a group of neoplasms located in the
oral cavity, pharyngeal regions and salivary glands (17). Oral squamous cell carcinoma (OSCC) is
the most common oral cancer type and accounts for >90% of human
oral malignancy types (18). OSCC is
frequently treated with a combination of surgery, radiotherapy and
chemotherapy (19). Despite advanced
therapeutic approaches, the incidence and mortality rates for OSCC
have not significantly improved in the past 30 years (17); therefore, improving the treatment
outcome for OSCC requires investigation into novel therapeutic
strategies. Our previous study demonstrated that the HDAC inhibitor
apicidin exerts anti-proliferative effects on human OSCC cell lines
(20). However, the members of HDACs
that are selectively inhibited by apicidin remain unclear, and
in vivo antitumor efficacy has not been examined in OSCC.
Identification of an isoform selective HDAC inhibitor may improve
the therapeutic potential and reduce the cytotoxicity associated
with cancer treatment. Therefore, the present study aimed to
examine the selective HDAC inhibitory effect of apicidin in
vitro and in vivo, and the in vivo antitumor
effect of apicidin, in a murine OSCC model.
Materials and methods
Cell culture and chemicals
The murine OSCC AT-84 cells were provided by Dr E.
J. Shillitoe (Upstate Medical University, Syracuse, NY, USA)
(21). AT-84 cells originated from a
spontaneous murine SCC in the oral mucosa of C3H mice (22) and were isolated by Hier et al
(23). The cells were maintained in
RPMI-1640 medium containing 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) and 100 U/ml
penicillin-streptomycin (Welgene, Inc., Daegu, Korea) at 37°C in an
atmosphere containing 5% CO2. Unless stated otherwise,
all chemicals were purchased from Sigma-Aldrich (Merck KGaA,
Damstadt, Germany). Apicidin (Sigma-Aldrich; Merck KGaA) was
dissolved in sterile DMSO to generate a 5 mM stock solution, which
was stored at −80°C. The cells were treated with culture media
alone as a control, or with various concentrations (0.1, 0.5, 1, 5
or 10 µM) of apicidin (the maximum final concentration of DMSO was
<0.1%) for 24 h.
MTT assay
Cells (1×104 cells/well) were seeded in a
96-well plate and incubated overnight to allow attachment. Cells
were treated with apicidin at the aforementioned concentrations for
24 h. At the end of the treatment period, 10 µl MTT (Sigma-Aldrich;
Merck KGaA) reagent (5 mg/ml) was added to each well (final
concentration, 0.5 mg/ml). After 4 h at 37°C, the supernatant was
aspirated and formazan crystals were dissolved in 100 µl DMSO. A
microplate autoreader ELISA was used to determine the absorbance at
595 nm. All experiments were performed in triplicate.
Western blot analysis
The cells were washed with PBS and harvested in a
lysis buffer (Intron Biotechnology, Inc., Seongnam, Korea). Protein
concentrations were measured using a Bradford protein assay kit,
according to the manufacturer's protocols. Samples containing equal
amounts of protein (50 µg) were resolved on SDS-PAGE in a 10–15%
gel and transferred to a polyvinylidene difluoride membrane.
Following blocking with 5% skim milk in tris-buffered saline with
0.1% Tween-20 (TBS-T) for 1 h at room temperature, the membranes
were incubated with primary antibodies (1:1,000 dilution) against
acetylated histone H4 (cat. no. 07-108; Upstate Biotechnology,
Inc., Lake Placid, NY, USA), HDAC8 (cat. no. ab187139; Abcam,
Cambridge, MA, USA), HDAC7 (cat. no. SC-11421; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), HDAC1 (cat. no. 5356), HDAC2
(cat. no. 5113), HDAC4 (cat. no. 7628), HDAC6 (cat. no. 7612),
cleaved caspase-3 (cat. no. 9664), poly(ADP-ribose) polymerase
(PARP; cat. no. 9542), microtubule associated protein 1 light chain
3B (LC3B; cat. no. 3868), autophagy-related protein 7 (ATG7; cat.
no. 2631), p62 (cat. no. 5114; Cell Signaling Technology, Inc.,
Danvers, MA, USA) and β-actin antibody (cat. no. SC-47778; Santa
Cruz Biotechnology, Inc.) overnight at 4°C. The membranes were then
washed six times with TBS-T and incubated with horseradish
peroxidase-conjugated secondary antibodies (anti-mouse, cat. no.
SC-2354; anti-rabbit, cat. no. SC-2768; 1:5,000 dilution; Santa
Cruz Biotechnology, Inc.) for 1 h at room temperature. Finally, the
membranes were washed six times with TBS-T and visualized using the
Pierce ECL western Blotting Substrate (cat. no. 32209; Thermo
Fisher Scientific, Inc.) on an ATTO chemiluminescent imaging system
(ATTO Corporation, Tokyo, Japan).
Flow cytometry analysis for apoptosis
detection
The cells were stained using a Alexa
Fluor® 488 Annexin V/Dead cell Apoptosis kit with Alexa
Fluor® 488 Annexin V and propidium iodide (PI; cat. no.
V13241; Molecular Probes; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol for apoptosis analysis. A
Cell Lab Quanta™ SC flow cytometer (Beckman Coulter
Inc., Brea, CA, USA) with Cell Lab Quanta SC software (version 1.0;
Beckman Coulter Inc.) were used to perform data acquisition and
analysis.
Detection of acidic vesicular
organelles (AVOs)
Autophagy is characterized by the formation and
promotion of AVOs. To detect the development of AVOs, the cells
were stained with acridine orange, as described previously
(20). In acridine orange-stained
cells, the cytoplasm and nucleus fluoresce bright green, whereas
acidic compartments fluoresce bright red. The treated cells were
stained with acridine orange (1 µg/ml; Sigma-Aldrich; Merck KGaA)
for 15 min at 37°C. To quantify the development of AVOs, the
increased red stained cells were analyzed using a FACScan flow
cytometer and CellQuest Pro software (version 4.0; BD Biosciences,
San Jose, CA, USA).
Autophagy inhibition and trypan blue
exclusion assay
A specific autophagy inhibitor chloroquine (CQ; 50
µM; Sigma-Aldrich; Merck KGaA) was co-treated with apicidin
(Sigma-Aldrich; Merck KGaA) in AT-84 cells for 24 h at 37°C.
Following treatment, cell proliferation was measured using a trypan
blue exclusion assay. The trypan blue exclusion assay was based on
the capability of viable cells to exclude the dye. After 5 min of
treatment of cells with 0.4% trypan blue (Sigma-Aldrich; Merck
KGaA) at room temperature, they were loaded into a hemocytometer
and counted for the dye uptake. The number of viable cells was
calculated as the percentage of the total cell population.
In vivo study on AT-84 cells bearing
CH3 mice
AT-84 cells were used for the in vivo
experiments, previously established by Pang et al (24). Male C3H mice (6 weeks old, 20–22 g)
were purchased from Samtaco Bio Korea (Osan, Korea). All animals
were housed under controlled temperature (22±2°C) and relative
humidity (50–60%) during a 12 h light-dark cycle. The animals were
provided food and tap water ad libitum. Mice were inoculated
subcutaneously in the right flank with 1×107 AT-84
cells. When the tumor size reached 150–200 mm3, the mice
were divided randomly into two groups, with six mice/group.
Apicidin (5 mg/kg) or vehicle control (0.1% DMSO) was administered
to the mice intraperitoneally every other day for 14 days. The
animals were monitored daily and tumor volume was measured with
calipers and calculated with the formula: (ab2)/2, where
a is the longest diameter and b is the shortest diameter of the
tumor. The mice were sacrificed at the end of the treatment period.
The tumor tissue was excised, weighed and fixed in 4%
paraformaldehyde at room temperature over 24 h. All experiments
were performed under protocols approved by the Ethical Committee of
Animal Care and Use at Pusan National University Hospital (Busan,
Korea; approval no. PNUH-2016-090).
Histopathology
The animals were sacrificed on day 15, and the
tumors were carefully removed and fixed in 4% paraformaldehyde at
room temperature over 24 h. The tissues were embedded in paraffin
wax and cut into 2 µm sections. The slides were de-paraffinized
with xylene and rehydrated a graded alcohol series (100, 90, 70 and
50% ethyl alcohol) for 10 min at room temperature, and stained with
10% hematoxylin (Vector Laboratories, Inc., Burlingame, CA, USA)
for 5 min and 0.5% eosin for 1 min at room temperature for
histological examination. The result was viewed under a light
microscope at magnification, ×200.
Immunohistochemistry and terminal
deoxynucleotidyl transferase-mediated dUTP nick end labeling
(TUNEL) assay
The sections were incubated in 3%
H2O2 in 70% methanol for 10 min to remove
endogenous peroxidase. Subsequently, they were blocked with 1%
bovine serum albumin (Sigma-Aldrich; Merck KGaA) in PBS for 1 h at
room temperature. Following this, the sections were incubated with
proliferating cell nuclear antigen (PCNA; 1:100 dilution; cat. no.
M0879; Dako, Carpinteria, CA, USA), LC3B (1:100 dilution; cat. no.
3868; Cell Signaling Technology, Inc.), p62 (1:100 dilution; cat.
no. 5114; Cell Signaling Technology, Inc.) and HDAC8 (1:100
dilution; cat. no. ab217702; Abcam) antibodies overnight at 4°C.
After washing three times with PBS-T, the sections were subjected
to the avidin-biotin peroxidase complex (ABC) method using
universal an ABC kit with biotinylated horse anti-mouse IgG/rabbit
IgG secondary antibodies and ABC reagent (R.T.U.
VECTASTAIN® Universal Elite ABC kit; cat. no. PK-7200;
Vector Laboratories, Inc.), according to the manufacturer's
protocol. Additionally, peroxidase activity was evaluated with
3,3′-diaminobenzidine (DAB Peroxidase Substrate kit; cat. no.
SK-4100; Vector Laboratories, Inc.). Finally, 10% hematoxylin for 1
min at room temperature was used to counterstain the sections. The
TUNEL assay was performed using an TdT DAB In Situ Apoptosis
Detection kit (cat. no. 4810-30-k; R&D Systems, Inc.,
Minneapolis, MN, USA), according to the manufacturer's protocol.
The result was viewed under a light microscope at magnification,
×200 and ×400. The positive cells were counted from five randomly
selected areas under magnification, ×400 and represented the mean ±
standard deviation.
Statistical analysis
Data are expressed as the mean ± SD of at least
three individual experiments. Statistical analysis was performed
using statistical software SPSS (version 20; IBM Corp., Armonk, NY,
USA). Statistical differences among groups were performed with an
one-way analysis of variance followed by Scheffe's post-hoc test.
Differences between groups were determined with a Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effect of apicidin on AT-84 cell
viability and HDAC expression
In order to analyze the effects of apicidin on cell
viability, AT-84 cells were treated with increasing concentrations
of apicidin and the cell viability was assessed with an MTT assay.
Apicidin inhibited the viability of AT-84 cells in a dose-dependent
manner, whereby concentrations of ≥1 µM resulted in a significant
reduction (Fig. 1A). Subsequently,
the effect of apicidin on the levels of histone acetylation and
specific HDACs was analyzed using western blotting. The cells were
treated with the indicated concentrations of apicidin (1 and 5 µM)
for 24 h. As depicted in Fig. 1B,
apicidin treatment notably increased the level of acetylated
histone H4 and decreased the level of HDAC8 in AT-84 cells. HDAC7
expression was also marginally reduced, but there was no change in
the expression of other HDACs in apicidin-treated AT-84 cells.
These results indicated that apicidin induces cell growth
inhibition and that it primarily inhibited HDAC8 expression in
AT-84 cells.
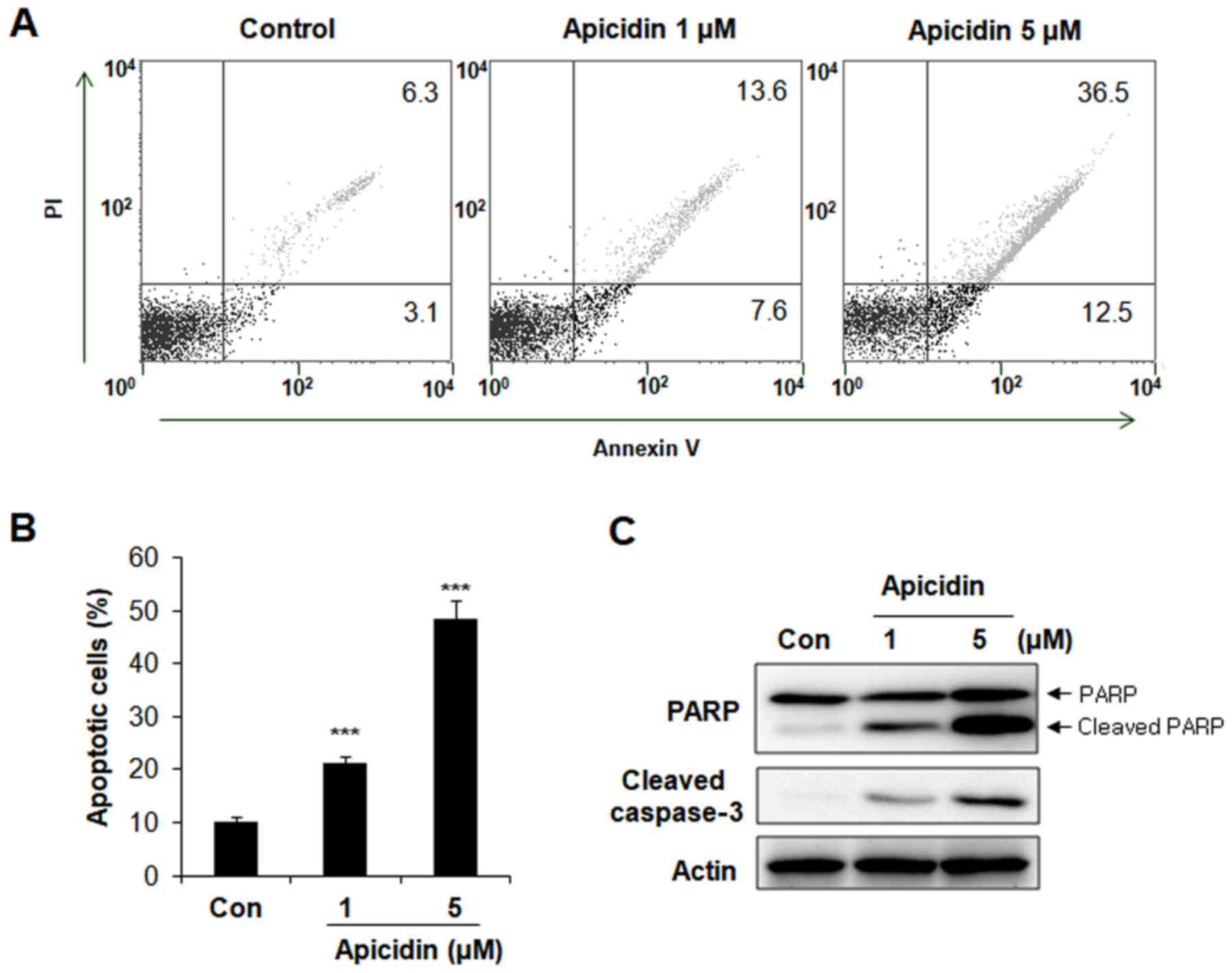
Apoptosis induction on
apicidin-treated AT-84 cells
To elucidate the mechanism underlying the reduction
in viability of AT-84 cells following treatment with apicidin,
apoptosis was measured with an Annexin V apoptosis assay. AT-84
cells were treated with 1 and 5 µM apicidin for 24 h, and
Annexin-V/PI double staining was examined with flow cytometry.
Apicidin significantly increased the percentage of Annexin
V-positive apoptotic cells in a dose-dependent manner compared with
the untreated control group (Fig. 2A and
B). The ratio of Annexin V-positive cells was 21 and 49% at
doses of 1 and 5 µM apicidin, respectively compared with 6.3% in
the control group. The expression of apoptosis-associated protein
levels was measured with western blot analysis to confirm apoptosis
involvement. As depicted in Fig. 2C,
a concentration-dependent increase in cleaved PARP and cleaved
caspase-3 expression levels were observed in apicidin-treated AT-84
cells, whereas levels of total PARP remained constant. These
results indicated that apicidin induced caspase-dependent apoptosis
in AT-84 cells.
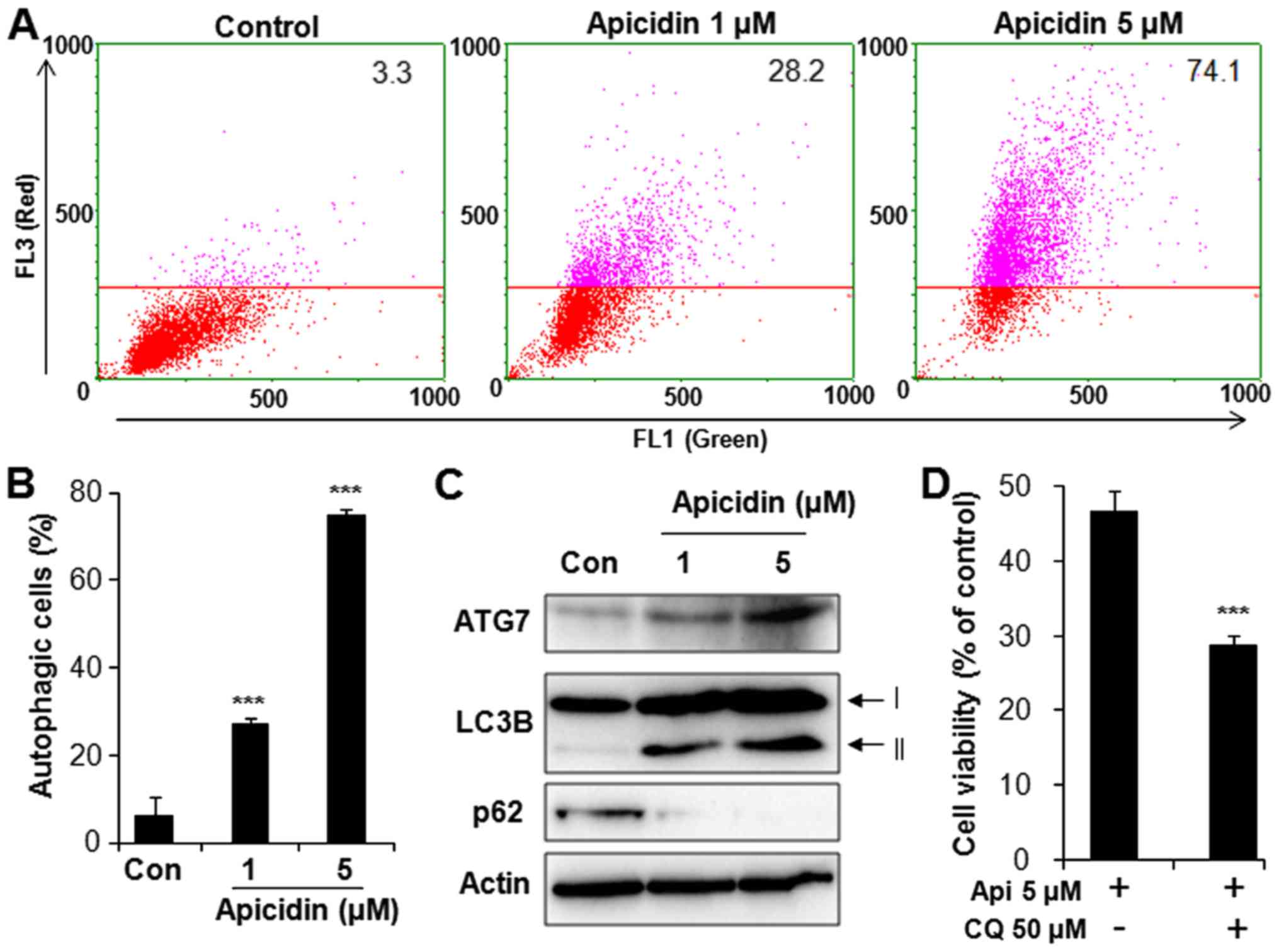
Autophagy induction on
apicidin-treated AT-84 cells
To determine whether apicidin induces autophagy in
AT-84 cells, acridine orange staining was performed and evaluated
with flow cytometry. As depicted in Fig.
3A, red fluorescence intensity in flow cytometric analysis was
dose-dependently increased with apicidin treatment in AT-84 cells,
indicating an enhancement of AVOs. The ratio of red fluorescence
intensity was 24 and 78% at doses of 1 and 5 µM apicidin,
respectively, both of which was significantly increased compared
with the untreated control group (Fig.
3B). Autophagy-associated protein expression was measured using
western blotting to confirm the induction of autophagy. Apicidin
notably induced the expression level of ATG7 and LC3B-II, a
specific marker of autophagosome promotion (Fig. 3C). The level of p62, a marker for
autophagic flux, was decreased in apicidin-treated AT-84 cells.
Additionally, to determine whether autophagy by apicidin
contributes to survival or death, AT-84 cells were co-treated with
apicidin and CQ, a specific autophagy inhibitor. Cell viability was
examined using the trypan blue exclusion assay. As depicted in
Fig. 3D, apicidin with CQ treatment
significantly reduced cell viability, compared with apicidin
without CQ. These results demonstrated that apicidin induced
autophagy in AT-84 cells and that autophagy induction by apicidin
may be associated with cell protection.
In vivo antitumor effect on apicidin
treatment
The in vivo effect of apicidin on tumor
growth was examined using mice inoculated with AT-84 cells. These
mice were administered with apicidin or vehicle every other day for
a 14-day period. As depicted in Fig.
4A, apicidin significantly decreased the tumor volume, compared
with the control group, and inhibited tumor growth up to 47%
relative to the control group at the end of the 14-day period. Mean
tumor weight was also significantly reduced in the apicidin-treated
group, compared with the vehicle-treated control group (Fig. 4B). No significant systemic toxicity,
including body weight changes or other apparent adverse effects,
was observed in the animals throughout the study period (Fig. 4C). These results indicated that
apicidin exhibited an antitumor effect against AT-84 cells in
vivo.
Histological changes and
immunohistochemistry analysis
The histological changes between apicidin-treated
and control groups were examined using hematoxylin and eosin
staining in paraffin-embedded tumor sections. Markedly increased
eosinophilic cytoplasm and nuclear changes were observed in tumor
tissues treated with apicidin, compared with the control tumor
tissues (Fig. 5). Subsequently, the
immunohistochemistry was examined with regard to the antitumor
effects of apicidin in tumor tissues. As depicted in Fig. 5A and C, tumor tissues treated with
apicidin exhibited significantly decreased PCNA and HDAC8
expression levels, compared with control tumor tissues treated with
vehicle. Furthermore, the percentage of TUNEL-positive apoptotic
cells was significantly increased, with a significant increase in
LC3B and reduction in p62 expression levels in apicidin-treated
tumor tissues, compared with vehicle-treated control tumor tissues
(Fig. 5B and C). These results
demonstrated that apicidin effectively inhibited cell proliferation
and HDAC8, and induced apoptosis and autophagy in vivo AT-84
mediated OSCC model.
 | Figure 5.Effect of apicidin on PCNA, HDAC8,
apoptosis and autophagy in AT-84 cell-derivated allografts. (A)
H&E staining of tumor sections (magnification, ×200), and
immunohistochemistry for PCNA and HDAC8 were performed on
paraffin-embedded tumor sections (magnification, ×400). (B) TUNEL
assay and immunohistochemistry for LC3B and p62 were performed on
paraffin-embedded tumor sections (magnification, ×400). (C)
Positive cells of immunostaining and TUNEL were counted, and the
results are expressed as the mean ± standard deviation. Five random
fields from each experiment were counted under a microscope at
maginification, ×400. The index was calulated assuming that the
amount of positive cells in the control was 100%. ***P<0.001,
compared with the vehicle-treated control group. H&E,
hematoxylin and eosin; LC3B, microtubule associated protein 1 light
chain 3B; PCNA, proliferating cell nuclear antigen; HDAC8, histone
deacetylase 8; TUNEL, terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling. |
Discussion
Potential preclinical efficacy of HDAC inhibitors in
oral cancer has been indicated, but there is limited knowledge
regarding the therapeutic role of the HDAC inhibitor apicidin in
OSCC (25,26). The present study investigated the
antitumor effect of the HDAC inhibitor apicidin in murine OSCC.
Inhibition of cell proliferation and the expression of HDAC were
examined in the apicidin-treated AT-84 cells. Apicidin
significantly inhibited cell viability and induced histone
acetylation in AT-84 cells, consistent with a previous study with
human OSCC cell lines (20). As HDAC
inhibitors do not inhibit all HDAC isoforms to the same extent,
they may be grouped into pan HDAC inhibitors and class or iso-form
selective HDAC inhibitors (27,28). The
development of selective HDAC inhibitors may be useful for
effective chemotherapy. Apicidin is primarily known as a class I
selective HDAC inhibitor; however, a number of studies have
provided evidence that apicidin also inhibited class II HDACs
(10,29,30), and
it is possible that specific HDAC inhibition by apicidin may have
different effects depending on the cell or tissue type. To the best
of our knowledge, isoform selective HDAC inhibition in apicidin
treatment has not been previously reported in OSCC. HDAC2 was
overexpressed in paraffin-embedded biopsies from patients with OSCC
and increased HDAC6 expression was determined in tumor tissues,
compared with normal oral squamous tissues, in previous reports
(31,32). In the results of the present study, no
notable changes were determined in the levels of HDAC2 and HDAC6
expression by apicidin treatment in AT-84 cells. A recent study
evaluated that HDAC8 overexpressed in human OSCC tissues and cell
lines, and inhibition of HDAC8, could be an effective therapeutic
tool for OSCC (5). The present
results demonstrated that the level of HDAC8 expression was
upregulated in the control, and apicidin notably inhibited HDAC8
expression in AT-84 cells. Although HDAC7 was also marginally
reduced in the apicidin-treated AT-84 cells, compared with the
control, the basal level of HDAC7 expression in the control was
reduced, compared with other HDACs in AT-84 cells. Following these
results, it was considered that HDAC8 inhibition by apicidin is
highly associated with the recovery of histone acetylation and cell
growth inhibition, rather than HDAC7 inhibition, in AT-84 cells. It
was confirmed that HDAC8 was overexpressed in human and murine OSCC
cell lines and apicidin also notably inhibited HDAC8 expression in
human OSCC cell lines (data not shown). Therefore, the fact that
apicidin inhibited HDAC8 may indicate that apicidin could be an
efficient tumor-targeted drug for OSCC.
Subsequently, cell death induction by apicidin was
examined in AT-84 cells. Apoptosis is the first identified
programmed cell death process and its contribution to cancer
research has been well documented (33). The present results demonstrated that
apicidin significantly induced caspase-dependent apoptosis in AT-84
cells. Previous studies indicated that apicidin treatment primarily
promoted apoptosis through an intrinsic caspase-dependent pathway
in a number of cancer cells, in a manner similar to the present
result (20,34). Autophagy, a key homeostatic process in
which cytosolic components are degraded and recycled through
lysosomes, has been identified as a novel programmed cell death
process. However, its role in cancer treatment remains
controversial (30,35). In the present study, formation of AVOs
increased LC3B-II and ATG7 expression, and degradation of p62
indicated that apicidin notably induced autophagy in AT-84 cells.
Although autophagy is considered a cell death mechanism, the
current consensus is that the role of autophagy in cell death is
primarily protective (36). Apicidin
induced apoptosis and pro-survival autophagy in human OSCC cell
lines (20). Additionally, the
inhibition of HDAC8 induced autophagy as a protective function in
human OSCC cells (5). In the present
study, it was demonstrated that autophagy caused by apicidin may
also have a protective effect on murine OSCC cells, although
further confirmation is required. Overall, these results indicated
that apicidin has an anti-proliferative effect and may induce
apoptosis and autophagy in human and murine OSCC cell lines.
The in vivo anti-proliferative activity of
apicidin has been reported in a number of cancer cell-derived tumor
models, but, to the best of our knowledge, the in vivo
antitumor effect of apicidin in OSCC has not been determined
(10,12,37).
Subsequently, the antitumor effect of apicidin was evaluated using
a tumor allograft mouse model, due to the in vitro effects
of apicidin in OSCC cells. AT-84 cells induce OSCC in C3H mice, and
this animal model with AT-84 cells is a commonly preferred
syngeneic mouse model, which is realistic and efficient for oral
cancer experiments (38). The present
results demonstrated that apicidin notably inhibited AT-84
cell-derived tumor growth in the animal model. Evaluation of cell
proliferation in tissues of experimental animals is essential for
carcinogenesis and the most common method to identify proliferating
cells in tissue sections is immunohistochemical detection of PCNA
(39). Elevated expression of PCNA
has been observed in the buccal tissues of a 7,12-dimethylbenz(a)
anthracene-induced oral cancer animal model (40). PCNA has been demonstrated to be
associated with the prognosis in a number of cancer types,
including oral cancer (41). This
anti-proliferative activity was confirmed by a significant decrease
in PNCA expression in the apicidin-treated group, compared with the
vehicle-treated control group. In the immunohistochemical
examinations, apicidin treatment increased the percentage of
TUNEL-positive apoptotic cells, and reduced the expression levels
of LC3B and p62, compared with the vehicle-treated control group.
These results indicated that apicidin induced apoptosis and
autophagy in tumor tissues, which is similar to the in vitro
results. HDACs are considered to localize in the nucleus, in order
to regulate gene transcription (42).
Class I HDACs are localized exclusively in the nucleus, whereas
class II HDACs are larger proteins that are shuttled between the
cytoplasm and the nucleus (43,44).
However, previous studies have indicated that the subcellular
localization of class I HDACs can be located within the nucleus as
well as within the cytoplasm, and translocalization of HDACs may be
associated with the transcriptional activity of these HDACs on
histone or non-histone proteins (5,44,45). In the present study, strong HDAC8
expression in AT-84 cell-derived tumor tissues was observed in the
nucleus and cytoplasm. In particular, apicidin treatment
significantly inhibited the level of HDAC8 expression in the
nuclear compartments of tumor tissues, compared with the vehicle
group. Consistent with this result, in a number of cancer types,
including esophageal squamous cell carcinoma, the localization of
HDAC8 in the nucleus and cytoplasm has been detected (46). However, a previous study demonstrated
that overexpression of HDAC8 was primarily observed in cytoplasmic
compartments in human OSCC tissues from patients (5). It was demonstrated that subcellular
localization of HDAC8 may be dynamically regulated depending on the
intended function or cellular condition, and further evaluation of
HDAC8 localization in OSCC is required. Overall, these results
indicated that apicidin effectively inhibited cancer cells growth
and HDAC8 expression, in vitro and in vivo, in murine
OSCC cells.
In conclusion, the present study demonstrated that
the HDAC inhibitor apicidin exhibits antitumor activity in
vitro and in vivo in murine OSCC, resulting in the
inhibition of cell proliferation and the induction of apoptosis and
autophagy. Upregulated HDAC8 expression, a novel therapeutic target
of OSCC, was selectively inhibited by apicidin treatment in murine
OSCC. Collectively, these data may contribute to understanding the
antitumor mechanism of apicidin in OSCC and indicate the potential
of apicidin as a therapeutic agent against OSCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Education, Science and Technology (grant
nos. NRF-2011-0023907 and NRF-2016R1D1A3B03931034).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MYA conducted all the experiments in the present
study.
Ethics approval and consent to
participate
All experiments were performed under protocols
approved by the Ethical Committee of Animal Care and Use at Pusan
National University Hospital (approval no. PNUH-2016-090).
Patient consent for publication
Not applicable.
Competing interests
The author declares that they have no competing
interests.
References
|
1
|
Manal M, Chandrasekar MJ, Gomathi Priya J
and Nanjan MJ: Inhibitors of histone deacetylase as antitumor
agents: A critical review. Bioorg Chem. 67:18–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dokmanovic M, Clarke C and Marks PA:
Histone deacetylase inhibitors: Overview and perspectives. Mol
Cancer Res. 5:981–989. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Iizuka M and Smith MM: Functional
consequences of histone modifications. Curr Opin Genet Dev.
13:154–160. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marks P, Rifkind RA, Richon VM, Breslow R,
Miller T and Kelly WK: Histone deacetylases and cancer: Causes and
therapies. Nat Rev Cancer. 1:194–202. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ahn MY and Yoon JH: Histone deacetylase 8
as a novel therapeutic target in oral squamous cell carcinoma.
Oncol Rep. 37:540–546. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim HJ and Bae SC: Histone deacetylase
inhibitors: Molecular mechanisms of action and clinical trials as
anti-cancer drugs. Am J Transl Res. 3:166–179. 2011.PubMed/NCBI
|
|
7
|
Batty N, Malouf GG and Issa JP: Histone
deacetylase inhibitors as anti-neoplastic agents. Cancer Lett.
280:192–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grant S and Dai Y: Histone deacetylase
inhibitors and rational combination therapies. Adv Cancer Res.
116:199–237. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bauden M, Tassidis H and Ansari D: In
vitro cytotoxicity evaluation of HDAC inhibitor Apicidin in
pancreatic carcinoma cells subsequent time and dose dependent
treatment. Toxicol Lett. 236:8–15. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ahn MY, Kang DO, Na YJ, Yoon S, Choi WS,
Kang KW, Chung HY, Jung JH, do Min S and Kim HS: Histone
deacetylase inhibitor, apicidin, inhibits human ovarian cancer cell
migration via class II histone deacetylase 4 silencing. Cancer
Lett. 325:189–199. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheong JW, Chong SY, Kim JY, Eom JI, Jeung
HK, Maeng HY, Lee ST and Min YH: Induction of apoptosis by
apicidin, a histone deacetylase inhibitor, via the activation of
mitochondria-dependent caspase cascades in human Bcr-Abl-positive
leukemia cells. Clin Cancer Res. 9:5018–5027. 2003.PubMed/NCBI
|
|
12
|
Lai JP, Sandhu DS, Moser CD, Cazanave SC,
Oseini AM, Shire AM, Shridhar V, Sanderson SO and Roberts LR:
Additive effect of apicidin and doxorubicin in sulfatase 1
expressing hepatocellular carcinoma in vitro and in vivo. J
Hepatol. 50:1112–1121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang J, Lai Z, Huang W, Ling H, Lin M,
Tang S, Liu Y and Tao Y: Apicidin inhibited proliferation and
invasion and induced apoptosis via mitochondrial pathway in
non-small cell lung cancer GLC-82 cells. Anticancer Agents Med
Chem. 17:1374–1382. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bauden M, Tassidis H and Ansari D: In
vitro cytotoxicity evaluation of HDAC inhibitor Apicidin in
pancreatic carcinoma cells subsequent time and dose dependent
treatment. Toxicol Lett. 236:8–15. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ahn MY, Ahn JW, Kim HS, Lee J and Yoon JH:
Apicidin inhibits cell growth by downregulating IGF-1R in salivary
mucoepidermoid carcinoma cells. Oncol Rep. 33:1899–1907. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Keleş E, Lianeri M and Jagodziński PP:
Apicidin suppresses transcription of 17β-hydroxysteroid
dehydrogenase type 1 in endometrial adenocarcinoma cells. Mol Biol
Rep. 38:3355–3360. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Markopoulos AK: Current aspects on oral
squamous cell carcinoma. Open Dent J. 6:126–130. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheng YS, Rees T and Wright J: A review of
research on salivary biomarkers for oral cancer detection. Clin
Transl Med. 3:32014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park JW, Kim CH, Ha YC, Kim MY and Park
SM: Count of platelet and mean platelet volume score: Serologic
prognostic factor in patients with oral squamous cell carcinoma. J
Korean Assoc Oral Maxillofac Surg. 43:305–311. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ahn MY, Ahn SG and Yoon JH: Apicidin, a
histone deaceylase inhibitor, induces both apoptosis and autophagy
in human oral squamous carcinoma cells. Oral Oncol. 47:1032–1038.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lou E, Kellman RM, Hutchison R and
Shillitoe EJ: Clinical and pathological features of the murine
AT-84 orthotopic model of oral cancer. Oral Dis. 9:305–312. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schultz-Hector S and Haghayegh S:
Beta-fibroblast growth factor expression in human and murine
squamous cell carcinomas and its relationship to regional
endothelial cell proliferation. Cancer Res. 53:1444–1449.
1993.PubMed/NCBI
|
|
23
|
Hier MP, Black MJ, Shenouda G, Sadeghi N
and Karp S: A murine model for the immunotherapy of head and neck
squamous cell carcinoma. Laryngoscope. 105:1077–1080. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pang S, Kang MK, Kung S, Yu D, Lee A, Poon
B, Chen IS, Lindemann B and Park NH: Anticancer effect of a
lentiviral vector capable of expressing HIV-1 Vpr. Clin Cancer Res.
7:3567–3573. 2001.PubMed/NCBI
|
|
25
|
Murakami J, Asaumi J, Maki Y, Tsujigiwa H,
Kuroda M, Nagai N, Yanagi Y, Inoue T, Kawasaki S, Tanaka N, et al:
Effects of demethylating agent 5-aza-2(')-deoxycytidine and histone
deacetylase inhibitor FR901228 on maspin gene expression in oral
cancer cell lines. Oral Oncol. 40:597–603. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chung YL, Lee MY and Pui NN: Epigenetic
therapy using the histone deacetylase inhibitor for increasing
therapeutic gain in oral cancer: Prevention of radiation-induced
oral mucositis and inhibition of chemical-induced oral
carcinogenesis. Carcinogenesis. 30:1387–1397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rikiishi H: Autophagic and apoptotic
effects of HDAC inhibitors on cancer cells. J Biomed Biotechnol.
2011:8302602011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Balasubramanian S, Verner E and Buggy JJ:
Isoform-specific histone deacetylase inhibitors: The next step?
Cancer Lett. 280:211–221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim SN, Choi HY and Kim YK: Regulation of
adipocyte differentiation by histone deacetylase inhibitors. Arch
Pharm Res. 32:535–541. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ahn MY and Yoon JH: Histone deacetylase 7
silencing induces apoptosis and autophagy in salivary
mucoepidermoid carcinoma cells. J Oral Pathol Med. 46:276–283.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chang HH, Chiang CP, Hung HC, Lin CY, Deng
YT and Kuo MY: Histone deacetylase 2 expression predicts poorer
prognosis in oral cancer patients. Oral Oncol. 45:610–614. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sakuma T, Uzawa K, Onda T, Shiiba M, Yokoe
H, Shibahara T and Tanzawa H: Aberrant expression of histone
deacetylase 6 in oral squamous cell carcinoma. Int J Oncol.
29:117–124. 2006.PubMed/NCBI
|
|
33
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ahn MY, Lee J, Na YJ, Choi WS, Lee BM,
Kang KW and Kim HS: Mechanism of apicidin-induced cell cycle arrest
and apoptosis in Ishikawa human endometrial cancer cells. Chem Biol
Interact. 179:169–177. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Alirezaei M, Kemball CC, Flynn CT, Wood
MR, Whitton JL and Kiosses WB: Short-term fasting induces profound
neuronal autophagy. Autophagy. 6:702–710. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yonekawa T and Thorburn A: Autophagy and
cell death. Essays Biochem. 55:105–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ahn MY, Chung HY, Choi WS, Lee BM, Yoon S
and Kim HS: Anti-tumor effect of apicidin on Ishikawa human
endometrial cancer cells both in vitro and in vivo by blocking
histone deacetylase 3 and 4. Int J Oncol. 36:125–131.
2010.PubMed/NCBI
|
|
38
|
Nair DV and Reddy AG: Mouse models of oral
cancer: Challenges and opportunities. Int J Adv Biol Res.
7:203–207. 2017.
|
|
39
|
Muskhelishvili L, Latendresse JR, Kodell
RL and Henderson EB: Evaluation of cell proliferation in rat
tissues with BrdU, PCNA, Ki-67(MIB-5) immunohistochemistry and in
situ hybridization for histone mRNA. J Histochem Cytochem.
51:1681–1688. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Velu P, Vinothkumar V, Babukumar S and
Ramachandhiran D: Chemopreventive effect of syringic acid on
7,12-dimethylbenz(a)anthracene induced hamster buccal pouch
carcinogenesis. Toxicol Mech Methods. 27:631–640. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Saraç S, Ayhan A, Hosal AS and Kaya S:
Prognostic significance of PCNA expression in laryngeal cancer.
Arch Otolaryngol Head Neck Surg. 124:1321–1324. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guo X, Ruan H, Li X, Qin L, Tao Y, Qi X,
Gao J, Gan L, Duan S and Shen W: Subcellular localization of class
I histone deacetylases in the developing xenopus tectum. Front Cell
Neurosci. 9:5102016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Y, Shin D and Kwon SH: Histone
deacetylase 6 plays a role as a distinct regulator of diverse
cellular processes. FEBS J. 280:775–793. 2013.PubMed/NCBI
|
|
44
|
Yang WM, Tsai SC, Wen YD, Fejer G and Seto
E: Functional domains of histone deacetylase-3. J Biol Chem.
277:9447–9454. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Grozinger CM and Schreiber SL: Regulation
of histone deacetylase 4 and 5 and transcriptional activity by
14-3-3-dependent cellular localization. Proc Natl Acad Sci USA.
97:7835–7840. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nakagawa M, Oda Y, Eguchi T, Aishima S,
Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M and Tsuneyoshi
M: Expression profile of class I histone deacetylases in human
cancer tissues. Oncol Rep. 18:769–774. 2007.PubMed/NCBI
|