Introduction
Acidosis is associated with various pathological
conditions, including ischemia (restricted blood supply), diabetic
ketoacidosis, defective blood flow, defective glycolytic
metabolism, and lung and renal diseases (1–3). During
acidosis, hydrogen ions accumulate in ischemic tissue, leading to
intracellular acidosis (4). Within 5
min of the initiation of renal ischemia, the tissue pH drops from
7.2–6.5 and metabolic acidosis persists even with reperfusion
(4). Acidosis causes tissue injury
and promotes the progression of existing disease, such as stroke
(5). However, the specific effects of
acidosis on renal epithelial and endothelial cells, and the
molecular pathways responsible for these effects, remain
unknown.
A previous study suggested that G protein-coupled
receptor 4 (GPR4) may be activated by a decrease in extracellular
pH, which generates a second messenger protein (6). GPR4 acts as a pH sensor in the kidney,
mediating the accumulation of intracellular cyclic adenosine
monophosphate (cAMP), which regulates the activity of acid-base
transport proteins in the kidney collecting duct (6,7). A
previous study using GPR4-knockout mice has also indicated that
GPR4 is involved in renal acid-base homeostasis (3).
CCAAT/enhancer-binding protein homologous protein
(CHOP) is a transcriptional regulator and is an essential regulator
of the endoplasmic reticulum stress (ERS)-mediated apoptosis
pathway (8). CHOP is a pro-apoptotic
protein and serves key roles in the pathology of liver fibrosis
(9). It has been reported that the
deletion of CHOP may protect mice from liver and kidney injury
(10). A previous study reported that
acidosis activates GPR4 in order to increase the expression of
ERS-associated genes, including CHOP, in endothelial cells
(11). Our previous study
demonstrated that knockdown of GPR4 inhibited the expression of
CHOP in hypoxia/reoxygenation (HR)-treated human umbilical vein
endothelial cells (HUVECs) (12).
However, to the best of our knowledge, the effect of GPR4 on the
expression of CHOP in renal cells in response to acidosis remains
uncharacterized. Therefore, the aim of the present study was to
investigate the effects of acidosis on the apoptosis of renal
epithelial cells and endothelial cells and the molecular pathways
responsible for these effects.
Materials and methods
Cell culture and treatment
The human umbilical vein endothelial cell line,
HUVEC, was obtained from All Cells LLC. (Alameda, CA, USA), and the
human proximal tubular HK-2 cell line was obtained from the
American Type Culture Collection (Manassas, VA, USA). Cells were
cultured in Dulbecco's modified Eagle medium (DMEM; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) in 5%
CO2 at 37°C.
For hypoxia/reoxygenation (HR) treatment,
5×105 cells in 2 ml media were placed into each well of
6-well flat-bottomed plates at 37°C. Cells were exposed to hypoxia
(1% O2) for 4 h, followed by a 6 h re-oxygenation (95%
O2) period. Subsequently, HUVECs and HK-2 cells were
subjected to acidosis by adjusting the media to pH 6.4 by applying
0.1 M HCl for 12 h prior to harvesting.
siRNA transfection
Cell transfection was performed as previously
described (13). In brief, cells were
seeded onto 24-well pates at 1×105 cells/well. Cells
were then transfected with 80 nM siRNA using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. The non-target scrambled
siRNA duplexes (Shanghai GenePharma Co., Ltd., Shanghai, China;
cat. no. A06001) and GPR4 siRNA (siGPR4) duplexes were purchased
from Invitrogen; Thermo Fisher Scientific, Inc. The sequences of
si-GPR4 were as follows: Forward, 3′-CCCUCUACAUCUUUGUCAUTT-5′ and
reverse, 3′-AUGACAAAGAUGUAGAGGGTT-5′ (12). The cells were harvested after 36 h of
transfection.
Western blot analysis
Total cell protein was extracted from HUVECs and
HK-2 cells using a Nuclear Extract kit (Active Motif, Carlsbad, CA,
USA), according to the manufacturer's protocols. Equal amounts of
protein (60 µg) were separated by 10% SDS-PAGE, prior to being
transferred onto Whatman nitrocellulose membranes (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany). Subsequent to blocking in 5%
skimmed milk for 1 h at 37°C, the membranes were incubated with
primary antibodies against the following overnight at 4°C: GPR4
(dilution, 1:1,000; R&D Systems, Inc., Minneapolis, MN, USA;
cat. no. RDC0299), CHOP (dilution, 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA; cat. no. 2895), cleaved
caspase-3 (dilution, 1:1,000; Cell Signaling Technology, Inc.; cat.
no. 9661) and GAPDH (dilution, 1:2,000; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA; cat. no. sc-32233). Following 3 washes in
Tris-buffered saline with 0.1% Tween-20 (TBST), the membranes were
incubated with a horseradish peroxidase-conjugated mouse
anti-rabbit secondary antibody (dilution, 1:10,000; Cell Signaling
Technology, Inc.; cat. no. 5127) for 1 h at 37°C. The blot was
visualized using an enhanced chemiluminescence kit (GE Healthcare,
Chicago, IL, USA). Densitometry was performed using ImageJ software
(version 1.43a; National Institutes of Health, Bethesda, MD, USA)
and GAPDH was used as a loading control.
Measurement of cAMP levels
Cells were treated with 50 mM
3-isobutyl-1-methylxanthine (Sigma-Aldrich; Merck KGaA) for 30 min,
followed by HR treatment, performed as aforementioned.
Subsequently, 200 µl 0.1 M HCl was added to lyse the cells for 12 h
at 37°C. The lysate was boiled at 90°C for 5 min and centrifuged at
10,000 × g at 4°C for 5 min prior to the supernatant being
collected. The cAMP levels were measured using a cAMP ELISA kit
(cat no. KGE002; R&D Systems Inc.), according to the
manufacturer's protocol.
TUNEL assay
A TUNEL assay was performed using the In Situ Cell
Death Detection kit (Roche Diagnostics, Basel, Switzerland). In
brief, cells were fixed with 4% paraformaldehyde for 1 h at room
temperature and were permeabilized with 0.1% Triton X-100 in PBS
for 2 min on ice. Subsequent to rinsing with PBS, the cells were
incubated with the TUNEL reaction mixture for 60 min at 37°C. Cell
nuclei were counterstained with 10 mg/ml
4′,6-diamidino-2-phenylindole for 1 h at 37°C. Finally, the slides
were mounted with synthetic resin and observed under a fluorescence
microscope (original magnification, ×400; BX-53; Olympus
Corporation, Tokyo, Japan). The total number of TUNEL positive
cells was calculated in ≥3 fields of view.
Measurement of lactate dehydrogenase
(LDH) release
At 36 h after cell transfection, the supernatant was
collected from both HK-2 and HUVEC cells after centrifugation at
3,000 × g for 5 min at room temperature and was measured using the
CytoTox 96 Non-Radioactive Cytotoxicity assay (Promega Corporation,
Madison, WI, USA), according to the manufacturer's protocol. The
results are expressed as the percentage of maximal LDH released
into the culture medium.
Statistical analysis
All results are expressed as the mean ± standard
deviation. The statistical analyses were performed using GraphPad
Prism 6.0 software (GraphPad Software, Inc., La Jolla, CA, USA).
Comparisons between 2 groups were performed using unpaired
Student's t-tests, while comparisons between ≥3 groups were
performed using one-way analysis of variance, following by
Bonferroni's post-hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Acidosis enhances HR-induced GPR4 and
CHOP expression, as well as apoptosis
Renal epithelial and endothelial cells are
susceptible to ischemic injury, which may lead to renal injury
(14). In order to investigate the
effect of HR on the expression of GPR4 and CHOP in the two cell
types, HK-2 cells were confronted with HR injury and were
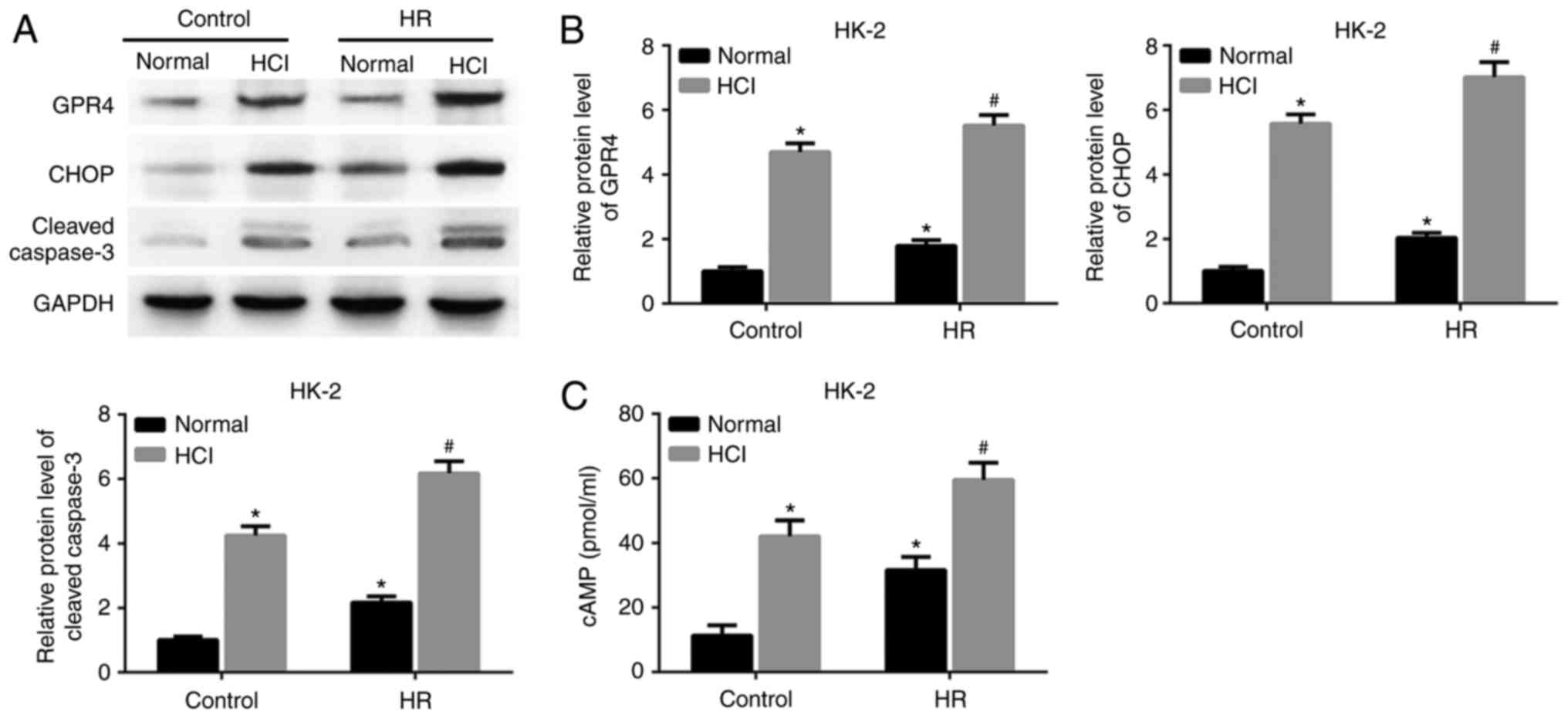
experimentally assessed. As demonstrated in Fig. 1A and B, the expression levels of GPR4
and CHOP proteins were significantly increased in both HK-2 cells
and HUVECs following HR treatment or HCl treatment alone, compared
with the control group with a normal pH. The expression levels of
GPR4 and CHOP were upregulated in the HR group treated with HCl
compared with the cells treated with HR alone. Furthermore, the
expression of cleaved caspase-3 was also increased by HCl treatment
in the HR group compared with the HR group with a normal pH.
Intracellular cAMP is a downstream effector of GPR4
(15). The expression level of cAMP
was increased in the cells exposed to HR and subsequent HCl
treatment, compared with the cells treated with HR alone (Fig. 1C). The same changes of gene
expressions (Fig. 2A and B) and cAMP
levels (Fig. 2C) existed in
HR-treated HUVECs. These results suggested that the HR-induced
increase in GPR4 and CHOP expression and the apoptosis of HK-2
cells and HUVECs may result from acidosis.
GPR4-knockdown attenuates the GPR4 and
CHOP expression, and the cell apoptosis induced by acidosis
To determine whether GPR4 function is associated
with acidosis-induced GPR4/CHOP expression or cell apoptosis, GPR4
expression was knocked down by si-GPR4 in HK-2 cells and HUVECs.
Acidosis resulted in an increase in the protein expression levels
of GPR4, CHOP and cleaved caspase-3 in the HK-2 cells and HUVECs
transfected with scrambled siRNA. The expression of these proteins
and the acidosis-induced increase in intracellular cAMP expression
levels in HK-2 cells and HUVECs were inhibited by knockdown of GPR4
(Figs. 3A-C; 4A-C).
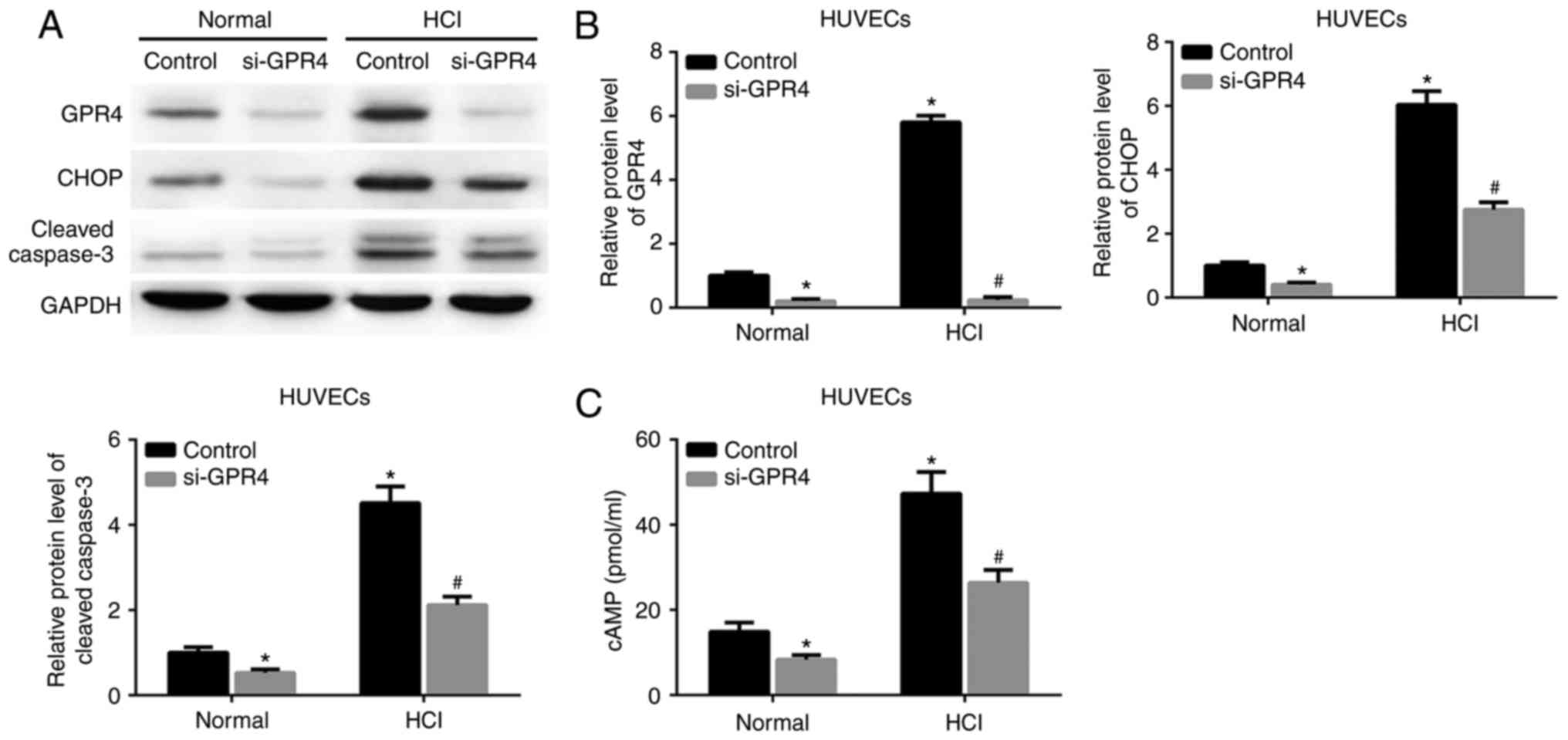 | Figure 4.Knockdown of GPR4 reduces
acidosis-induced GPR4 and CHOP expression, and suppresses cell
apoptosis and cAMP levels in HUVECs. Cells were transfected with
control siRNA or siGPR4 for 36 h, prior to being cultured in normal
culture medium (pH 7.4) or acidic medium (pH 6.4) for 12 h. (A)
Protein expression levels of GPR4, CHOP and cleaved caspase-3 were
determined by western blot assay. (B) Quantitative analyses of the
relative expression levels of GPR4, CHOP and cleaved caspase-3. (C)
Intracellular cAMP levels. *P<0.05 vs. control group under
normal conditions. #P<0.05 vs. control group under
acidic conditions. GPR4, G protein-coupled receptor 4; CHOP,
CCAAT/enhancer-binding protein homologous protein; cAMP, cyclic
adenosine monophosphate; HUVECs, human umbilical vein endothelial
cells; HCl, hydrochloric acid; siRNA/si, small interfering RNA. |
GPR4-knockdown leads to a decrease in
acidosis-induced apoptosis
To confirm the role of GPR4 in acidosis-induced cell
apoptosis, a TUNEL assay was performed. TUNEL staining indicated an
increased number of TUNEL positive HK-2/vector cells in response to
acidic pH compared with the control group under normal pH
conditions (Fig. 5A), whilst the
increase in TUNEL positive cells induced by acidosis was attenuated
by si-GPR4 treatment. These results were confirmed by measuring the
release of LDH, which is a marker of cellular injury. Under acidic
conditions, LDH release was significantly reduced in cells
transfected with si-GPR4 compared with cells transfected with
scrambled siRNA (Fig. 5B). Similarly,
GPR4 interference led to a decrease in the number of TUNEL positive
HUVEC/vector cells (Fig. 5C) and LDH
release from HUVECs (Fig. 5D). The
results indicated that GPR4 expression was required for
acidosis-induced apoptosis of the HK-2 cells and HUVECs.
Discussion
Acidosis is a pathological condition whereby acidity
over-accumulates in the body fluid (16). The condition aggravates
ischemia-associated tissue injury (17), and metabolic acidosis has been
demonstrated to exacerbate the renal injury induced by
ischemia/reperfusion (IR) (18). In
addition, acidosis has been demonstrated to induce cell apoptosis
and inhibit cell proliferation (19).
It has been demonstrated that during stroke, the pH of brain tissue
may drop to 6.0–6.5 and that this acidotic stress increases neural
cell apoptosis (20). Our previous
study indicated that HR-induced apoptosis was alleviated by
neutralization of acidity (12). The
results of the present study demonstrated that acidosis promoted
HR-induced apoptosis. Taken together, these results suggest that
HR-induced cell apoptosis in HK-2 cells and HUVECs may result from
acidosis.
It has been reported that GPR4 is involved in the
regulation of acid-base balance and kidney acid secretion (6). Acidosis may activate the expression of
GPR4 in order to regulate the expression of the genes involved in
different biological pathways, including the ERS response and cell
metabolism (3,21). A previous study demonstrated that
acidosis/GPR4 regulated endothelial cell adhesion through cAMP
production and served a role in the inflammatory response of
vascular endothelial cells (3). In
the present study, it was demonstrated that acidosis enhanced
HR-induced GPR4 protein expression in HK-2 cells and HUVECs. CHOP
is the key inducer of apoptosis in the ERS response (22), and is induced in hypoxia-treated renal
tubular epithelial cells and promotes apoptosis (23). Our previous study indicated that
hypoxia-induced CHOP expression is mediated by GPR4 (12). This result is supported by those of
the present study, in which acidosis was demonstrated to induce the
expression of CHOP. However, this effect was diminished by GPR4
knockdown in HK-2 cells and HUVECs. Therefore, acidosis-induced
CHOP expression was regulated by GPR4 in HK-2 cells and HUVECs.
An association was identified between HR and cell
apoptosis in vitro. Furthermore, the observation that
inhibition of GRP4 expression reduces the cell apoptosis induced by
acidosis contributes to the characterization of the role of the
GPR4/CHOP pathway in the pathogenesis of IR injury, and suggests a
potential molecular candidate for IR injury therapeutics. cAMP is a
ubiquitous second messenger protein that regulates numerous
cellular processes (24). Previous
studies have demonstrated that an acidic pH activates GPR4 in order
to induce the production of cAMP (15). Chen et al (3) reported that activation of GPR4 by
acidosis increased endothelial cell adhesion via the cAMP pathway.
Our previous study demonstrated that a cAMP analog, 8-bromo-cAMP,
induced CHOP expression in HUVECs (18). The present study demonstrated that
GPR4 expression reduced acidosis-induced cAMP and CHOP expression
levels in HK-2 cells and HUVECs. Taken together, these results
suggested that acidosis-induced CHOP expression may result from
GPR4 activation and cAMP accumulation.
In conclusion, the present study demonstrated that
HR-induced cell apoptosis in HK-2 cells and HUVECs may result from
acidosis. The CHOP pathway serves a key role in acidosis-induced
cell apoptosis. These results indicated that GPR4 inhibitors may be
promising and effective therapeutic tools for the treatment of
ischemic renal injury.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data that were generated or analyzed in this
study are included in this manuscript.
Authors' contributions
BD designed the present study. XLZ and YF conducted
the experiments. SC performed the statistical analysis. XPZ
interpreted the statistical analysis, and reviewed and approved the
final version to be published. All authors read and approved the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Koul PB: Diabetic ketoacidosis: A current
appraisal of pathophysiology and management. Clin Pediatr (Phila).
48:135–144. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Poschet J, Perkett E and Deretic V:
Hyperacidification in cystic fibrosis: Links with lung disease and
new prospects for treatment. Trends Mol Med. 8:512–519. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen A, Dong L, Leffler NR, Asch AS, Witte
ON and Yang LV: Activation of GPR4 by acidosis increases
endothelial cell adhesion through the cAMP/Epac pathway. PLoS One.
6:e275862011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bonventre JV: Mediators of ischemic renal
injury. Annu Rev Med. 39:531–544. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J,
Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ and Simon RP:
Neuroprotection in Ischemia: Blocking calcium-permeable
acid-sensing ion channels. Cell. 118:687–698. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sun X, Yang LV, Tiegs BC, Arend LJ, McGraw
DW, Penn RB and Petrovic S: Deletion of the pH sensor GPR4
decreases renal acid excretion. J Am Soc Nephrol. 21:1745–1755.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brown D, Breton S, Ausiello DA and
Marshansky V: Sensing, signaling and sorting events in kidney
epithelial cell physiology. Traffic. 10:275–284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nishitoh H: CHOP is a multifunctional
transcription factor in the ER stress response. J Biochem.
151:217–219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tamaki N, Hatano E, Taura K, Tada M,
Kodama Y, Nitta T, Iwaisako K, Seo S, Nakajima A, Ikai I and Uemoto
S: CHOP deficiency attenuates cholestasis-induced liver fibrosis by
reduction of hepatocyte injury. Am J Physiol Gastrointest Liver
Physiol. 294:G498–G505. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Prachasilchai W, Sonoda H, Yokota-Ikeda N,
Ito K, Kudo T, Imaizumi K and Ikeda M: The protective effect of a
newly developed molecular chaperone-inducer against mouse ischemic
acute kidney injury. J Pharmacol Sci. 109:311–314. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dong L, Li Z, Leffler NR, Asch AS, Chi JT
and Yang LV: Acidosis activation of the proton-sensing GPR4
receptor stimulates vascular endothelial cell inflammatory
responses revealed by transcriptome analysis. PLoS One.
8:e619912013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dong B, Zhou H, Han C, Yao J, Xu L, Zhang
M, Fu Y and Xia Q: Ischemia/reperfusion-induced CHOP expression
promotes apoptosis and impairs renal function recovery: The role of
acidosis and GPR4. PLoS One. 9:e1109442014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He K, Chen X, Han C, Xu L, Zhang J, Zhang
M and Xia Q: Lipopolysaccharide-induced cross-tolerance against
renal ischemia-reperfusion injury is mediated by hypoxia-inducible
factor-2α-regulated nitric oxide production. Kidney Int.
85:276–288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miao J, Lesher AM, Miwa T, Sato S,
Gullipalli D and Song WC: Tissue-specific deletion of Crry from
mouse proximal tubular epithelial cells increases susceptibility to
renal ischemia-reperfusion injury. Kidney Int. 86:726–737. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang LV, Radu CG, Roy M, Lee S, McLaughlin
J, Teitell MA, Iruela-Arispe ML and Witte ON: Vascular
abnormalities in mice deficient for the G protein-coupled receptor
GPR4 that functions as a pH sensor. Mol Cell Biol. 27:1334–1347.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang YC, Li WZ, Wu Y, Yin YY, Dong LY,
Chen ZW and Wu WN: Acid-sensing ion channel 1a contributes to the
effect of extracellular acidosis on NLRP1 inflammasome activation
in cortical neurons. J Neuroinflammation. 12:2462015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cronberg T, Rytter A, Asztély F, Söder A
and Wieloch T: Glucose but not lactate in combination with acidosis
aggravates ischemic neuronal death in vitro. Stroke. 35:753–757.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Magalhães PA, Magalhães PA, de Brito TS,
Freire RS, da Silva MT, dos Santos AA, Vale ML, de Menezes DB,
Martins AM and Libório AB: Metabolic acidosis aggravates
experimental acute kidney injury. Life Sci. 146:58–65. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sanderlin E, Justus C, Krewson E and Yang
L: Emerging roles for the pH-sensing G protein-coupled receptors in
response to acidotic stress. Cell Health Cytoskelet. 2015:99–109.
2015.
|
|
20
|
Huang Y and McNamara JO: Ischemic stroke:
‘Acidotoxicity’ is a perpetrator. Cell. 118:665–666. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dong L, Krewson EA and Yang LV: Acidosis
activates endoplasmic reticulum stress pathways through GPR4 in
human vascular endothelial cells. Int J Mol Sci. 18:pii: E2782017.
View Article : Google Scholar
|
|
22
|
Ding X, Ma M, Teng J, Shao F, Wu E and
Wang X: Numb protects human renal tubular epithelial cells from
bovine serum albumin-induced apoptosis through antagonizing
CHOP/PERK pathway. J Cell Biochem. 117:163–171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang JR, Yao FH, Zhang JG, Ji ZY, Li KL,
Zhan J, Tong YN, Lin LR and He YN: Ischemia-reperfusion induces
renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am J
Physiol Renal Physiol. 306:F75–F84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nicol X, Hong KP and Spitzer NC: Spatial
and temporal second messenger codes for growth cone turning. Proc
Natl Acad Sci USA. 108:13776–13781. 2011. View Article : Google Scholar : PubMed/NCBI
|