Introduction
Non-small cell lung cancer (NSCLC) is one of the
leading causes of cancer-associated mortality globally (1). The acquired resistance of anticancer
drugs remains a key obstacle for improving the prognosis of
patients with NSCLC (2). Epidermal
growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) have
been selected clinically as the first-line treatment for patients
with NSCLC by activating EGFR mutations (3–5).
However, the disease stage of the majority of patients inevitably
progresses despite an initial substantial and rapid response to
EGFR-TKIs (6). Previous studies
indicated that human EGFR-2 amplification, original or induced
T790M mutation, activated secondary signaling, including
phosphatidylinositol 3-kinase mutation or hepatocyte growth factor
receptor (MET) proto-oncogene, and receptor tyrosine kinase
amplification may result in acquired EGFR-TKIs resistance (6–8).
However, the initial mechanism for the acquired resistance of
EGFR-TKIs remains unclear.
Hypoxia is a notable feature of solid tumor types,
including lung cancer (9). Compared
with tumors under oxygen-rich conditions, hypoxic tumors are more
resistant to radiation and chemotherapy, more invasive, genetically
unstable, resist apoptosis and have increased metastatic potential
(10). Hypoxia activates the
hypoxia-inducible factor-1 (HIF-1) signaling pathway, which
mediates the primary biological effects of hypoxia (9). HIF-1 consists of an α and β subunit,
and HIF-1α is the functional part (11). Previous research indicates that
hypoxia increases the population of lung cancer stem cells
resistant to gefitinib in EGFR mutation-positive NSCLC, and the
HIF-1 signaling pathway is activated in EGFR-TKI-resistant lung
cancer cells (12,13). Thus, the HIF-1 signaling pathway was
targeted as a potential factor to influence the sensitivity of lung
cancer cells to EGFR-TKIs.
In the present study, the activity of the HIF-1
signaling pathway was regulated to observe if it was able to alter
change the sensitivity of lung cancer cells to EGFR-TKIs. The
present study selected
3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole (YC-1) and
dimethyloxalylglycine (DMOG) as a HIF-1 signaling pathway inhibitor
and activator, respectively. YC-1 is a chemically synthetic benzyl
indazole (14). It had been revealed
to be able to downregulate HIF-1α expression and was indicated as a
novel HIF-1α inhibitor (15). The
prolyl hydroxylase inhibitor DMOG has been used as an activator of
the HIF-1 signaling pathway (16).
It physiologically simulates a low oxygen environment by blocking
the degradation of HIF, and inducing chemical hypoxia (16,17).
Gefitinib was selected as the representative
EGFR-TKI. HCC827, the gefitinib hypersensitive EGFR exon 19 mutant
NSCLC cell line (8), was selected
for the present study. Previous research demonstrated that MET
amplification is the mechanism of acquired resistance against
gefitinib in HCC827-GR, the gefitinib resistant cell line generated
by exposing HCC827 cells to increasing concentrations of gefitinib
(8,18). Additionally, previous research
indicated that HIF-1α is involved in the regulation of MET levels
by EGFR, and EGFR regulation of MET levels in EGFR-TKIs sensitive
cell lines occurs through the HIF-1 signaling pathway in a
hypoxia-independent manner (19).
Nevertheless, in an EGFR-TKI-resistant cell line with MET
amplification, this regulation was lost (8). Subsequently, for the present study it
was speculated that the HIF-1 signaling pathway, but not the EGFR
pathway, may regulate MET levels in EGFR-TKI-resistant cell lines
caused by MET amplification. HCC827 cells can become an
EGFR-TKI-resistant cell line following MET amplification (18); therefore, the HCC827 cell line was
selected for the present study. In addition to examining the
influence of the HIF-1 signaling pathway on the sensitivity of
HCC827 cells to gefitinib, the present study also aimed to
investigate the associated mechanisms of this resistance and to
detect the levels of MET, in order to clarify whether MET levels
are associated with the sensitivity change of HCC827 cells to
gefitinib caused by the HIF-1 signaling pathway.
Materials and methods
Reagents
Reagents and suppliers were as follows: Gefitinib
(AstraZeneca UK Limited, Macclesfield, UK); YC-1; DMOG; MTT cell
viability kit (all from Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany); penicillin-streptomycin solution (Beyotime Institute of
Biotechnology, Shanghai, China); antibodies p-Met and HIF-1α (cat.
no. ab5662 and ab82832, respectively; Abcam, Cambridge, MA, USA);
and RPMI-1640 supplemented with 10% heat-inactivated fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA).
Cell line and cell culture
The present study was approved by the Ethics
Committee of Tongde Hospital of Zhejiang Province (Hangzhou,
China). Human commercially available HCC827 cell line was purchased
from the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). All cells were cultured under standard
conditions (37°C and 5% CO2) and in RPMI-1640 medium
supplemented with 10% heat-inactivated FBS and 1%
penicillin-streptomycin solution (standard medium). HCC827 cells
were observed regularly under a light microscope (×40; BX-42;
Olympus Corporation, Tokyo, Japan).
Cells were separated into four groups to research
the effect of HIF-1 signaling pathway upregulation on the
sensitivity of HCC827 cells to gefitinib: A blank control group; a
DMOG group; a gefitinib group; and a DMOG and gefitinib combined
group. Every group contained two subgroups that were treated for
different durations. For the blank control group, HCC827 cells were
cultured for 36 and 48 h (37°C and 5% CO2) in standard
medium, as aforementioned. For the DMOG group, HCC827 cells were
cultured for 36 and 48 h (37°C and 5% CO2) in standard
medium with 2 mM DMOG. For the gefitinib group, after culturing
HCC827 cells in standard medium for 24 h, gefitinib was added to
the medium at a final concentration of 20 nM and treated for 36 and
48 h. For the DMOG and gefitinib combined group, following the
culture of HCC827 cells in standard medium with 2 mM DMOG for 24 h,
gefitinib was added to the existing medium at a final concentration
of 20 nM and treated for 36 and 48 h.
Similarly, cells were also separated into four
groups to examine the effect of HIF-1 signaling pathway
downregulation on the sensitivity of HCC827 cells to gefitinib: A
blank control group, a YC-1 group, a gefitinib group, and a YC-1
and gefitinib group. For the blank control group, the HCC827 cells
were cultured for 16 and 28 h (37°C and 5% CO2) in
standard medium. For the YC-1 group, HCC827 cells were cultured for
16 and 28 h (37°C and 5% CO2) in standard medium with 40
µM YC-1. For the gefitinib group, following the culturing of HCC827
cells in standard medium for 4 h, gefitinib was added to the medium
at a final concentration of 20 nM and treated for 16 and 28 h. For
the YC-1 and gefitinib combined group, following the culture of
HCC827 cells in standard medium with 40 µM YC-1 for 4 h, gefitinib
was added to the existing medium at a final concentration of 20 nM
and treated for 16 and 28 h.
Western blot assay
Cells treated with the aforementioned different
treatments were washed with PBS (0.01 M, pH 7.2–7.3 at 4°C) three
times, and then treated with a lysis buffer containing 20 mmol/l
Tris (pH 7.5), 150 mmol/l NaCl, 1% Triton X-100 and inhibitors of
protease and phosphates (Beyotime Institute of Biotechnology) on
ice for 30 min. The cell lysis products were centrifuged for 15 min
at 12,000 × g in a 4°C refrigerated centrifuge and the supernatants
were collected. The final protein concentration was measured using
a Bicinchoninic Acid protein kit, according to the manufacturer's
protocol (Thermo Fisher Scientific, Inc.), and supernatants were
boiled for 5 min at 100°C. Subsequently, 100 µg protein lysates
were separated using 12% SDS-PAGE. The proteins were transferred to
polyvinylidene fluoride membranes and the membranes were blocked
with 5% skimmed milk with TBS and 20% Tween-20 (TBST) at room
temperature for 2 h. A total of 137 mM NaCl, 20 mM Tris and 0.05%
Tween-20 were contained in TBST of which the pH was adjusted with
HCl to pH 7.5. The blotted membrane was incubated with primary
antibodies against p-Met (1:500), HIF-1α (1:500) and GAPDH
(1:1,000; ab9484; Abcam) at room temperature for 2 h. The membrane
was washed with TBST three times and then incubated with
horseradish peroxidase-conjugated goat anti-rabbit IgG secondary
antibody (cat. no. RABHRP1-10UL; 1:1,000; Sigma-Aldrich; Merck
KGaA) at room temperature for 1.5 h. The immunoreactive bands were
washed with TBST four times and observed using enhanced
chemiluminescence plus detection reagent (Pierce; Thermo Fisher
Scientific, Inc.). GAPDH was used as an internal control. The
densitometry of the bands was quantified with an UVP Gel Imaging
System Labworks 4.6 software (UVP, LLC, Phoenix, AZ, USA).
MTT assay
HCC827 cells were seeded at a density of
2×104 cells/well in 96-well plates and maintained in
RPMI-1640 medium supplemented with 10% FBS and 1%
penicillin-streptomycin. Following overnight incubation (37°C and
5% CO2), cells were exposed to different treatments
(blank control, DMOG, gefitinib, and DMOG and gefitinib combined
for 36 and 48 h; blank control, YC-1, gefitinib, and YC-1 and
gefitinib combined for 16 and 28 h). Following treatments, an MTT
reagent (Sigma-Aldrich; Merck KGaA) was added and cells were
incubated at 37°C for 4 h. Subsequently, the medium was removed and
150 µl dimethyl sulphoxide was added to dissolve the purple
formazan salt crystals. Following this, the optical density was
measured on a microplate reader at a wavelength of 490 nm.
Colony formation assay
Cells with different treatments as aforementioned
were seeded onto culture plates. Cells were seeded at low density
(300 cells/plate) and cultured in RPMI-1640 medium supplemented
with 10% FBS and 1% penicillin-streptomycin for 2–3 weeks, in a
humidified atmosphere with 5% CO2 at 37°C. Subsequently,
the colonies were stained with 0.05% crystal violet solution for 20
min at room temperature. Finally, the number of colonies with
>10 cells were counted under an inverted light microscope (×10;
CKX41; Olympus Corporation).
Cell migration assay
Subsequent to the aforementioned treatments, HCC827
cells were plated into 6-well plates and cultured under serum
starvation conditions, in RPMI-1640 medium without FBS, to a
maximum of 60% confluence. A scratch was produced 16 h after serum
starvation of the cells. Each well was wounded by scratching with a
10 µl pipette tip, which was followed by PBS washes three times to
remove cell debris. The gap distance of the wound was measured at
three different sites using ImageJ Software (v. 1.48q; National
Institutes of Health, Bethesda, MD, USA) in pixels. Wound closure
was observed at 0, 24 and 48 h after wound simulation. Graphs were
plotted against the percentage of the migration distance that the
cells moved.
Statistical analysis
Statistical analyses were performed using SPSS
software (version 16.0.0; SPSS, Inc., Chicago, IL, USA). Data were
expressed as the mean ± standard deviation, which was based on a
minimum of three independent experiments. Differences between
groups were compared using a two-way analysis of variance followed
by a post hoc Tukey's test. Correlation between HIF-1α levels and
p-Met levels of HCC827 cells with different treatments was
evaluated by Pearson's correlation. P<0.05 was considered to
indicate a statistically significant difference.
Results
HIF-1 signaling pathway upregulation
reduces the sensitivity of HCC827 cells to gefitinib
A western blot assay was performed to detect the
expression of HIF-1α in HCC827 cells with different treatments
(blank control, DMOG, gefitinib, and DMOG and gefitinib combined
for 36 and 48 h). As depicted in Fig.
1, the protein expression levels of HIF-1α were significantly
elevated in cells treated with DMOG, compared with the blank
control for 36 and 48 h (P<0.05 and P<0.01, respectively).
Similarly, levels of HIF-1α were significantly elevated in cells
treated with DMOG and gefitinib combined, compared with the
gefitinib treatment, at 36 and 48 h (P<0.05 and P<0.01,
respectively). This indicates the activation of the HIF-1 signaling
pathway. The effect of HIF-1 signaling pathway upregulation on
HCC827 cell proliferation was assessed by performing an MTT assay.
When HCC827 cells were treated with DMOG and gefitinib combined, a
significant increase in cell proliferation was observed, compared
with gefitinib treated HCC827 cells (P<0.01; Fig. 2). This indicates that HCC827 cells
were less sensitive to gefitinib once DMOG was added, compared with
gefitinib alone. Consistent with the results from the MTT assay,
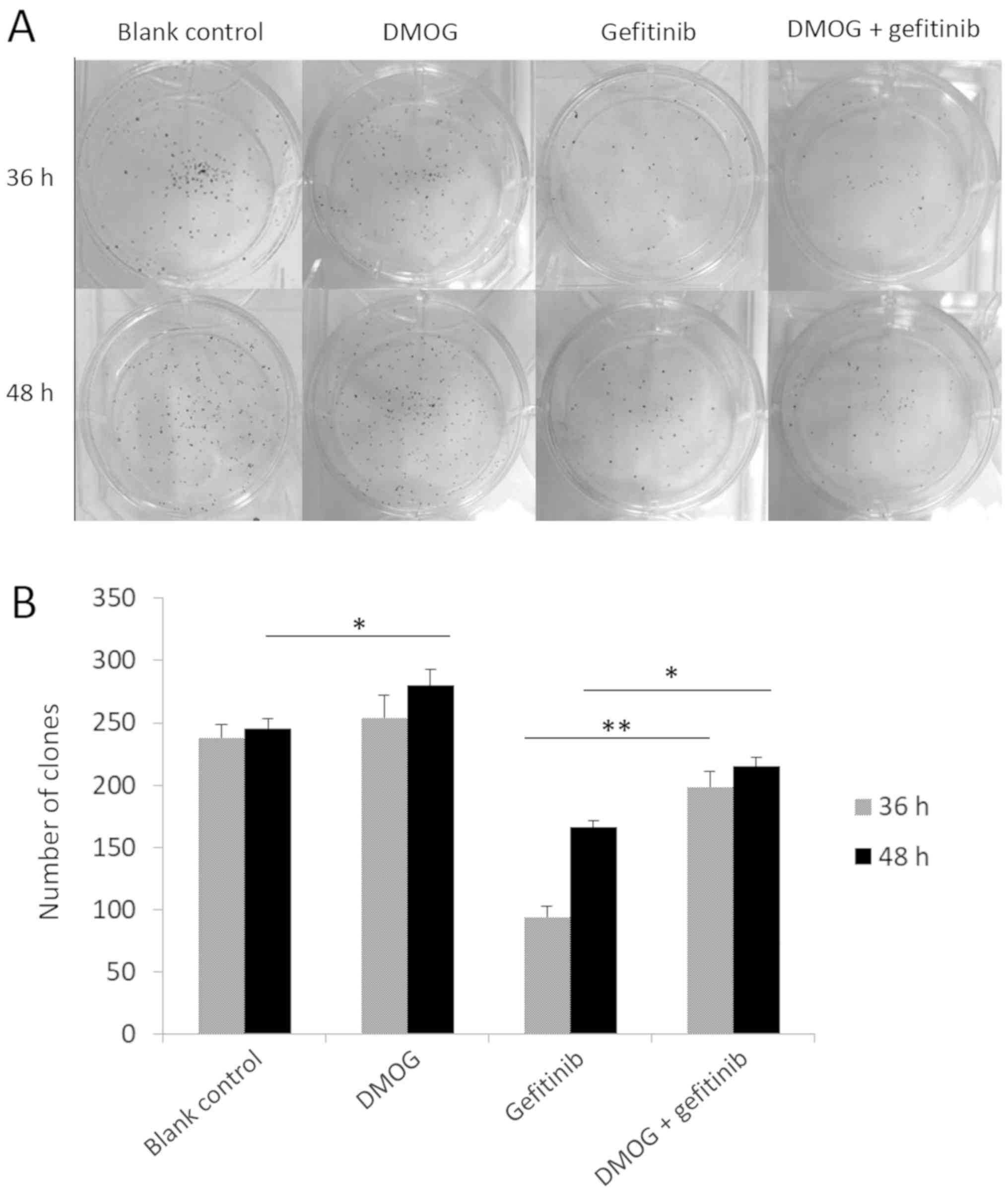
the gefitinib and DMOG combined treatment significantly improved
the colony-forming ability of HCC827 cells, compared with gefitinib
treatment alone, at 36 and 48 h (P<0.01 and P<0.05,
respectively; Fig. 3). Additionally,
it was revealed that DMOG treatment alone for 48 h significantly
improved the colony-forming ability of HCC827 cells, compared with
the blank control (P<0.05; Fig.
3). In addition to the growth ability of HCC827 cells, the
influence of HIF-1 signaling pathway upregulation on cell migration
was examined. In a wound healing assay, treatment of gefitinib and
DMOG combined for 36 and 48 h significantly enhanced cell
migration, compared with gefitinib treatment alone (P<0.01;
Fig. 4), while treatment of DMOG
alone for 24 h significantly enhanced the cell migration ability of
HCC827 cells, compared with the blank control (P<0.05; Fig 4).
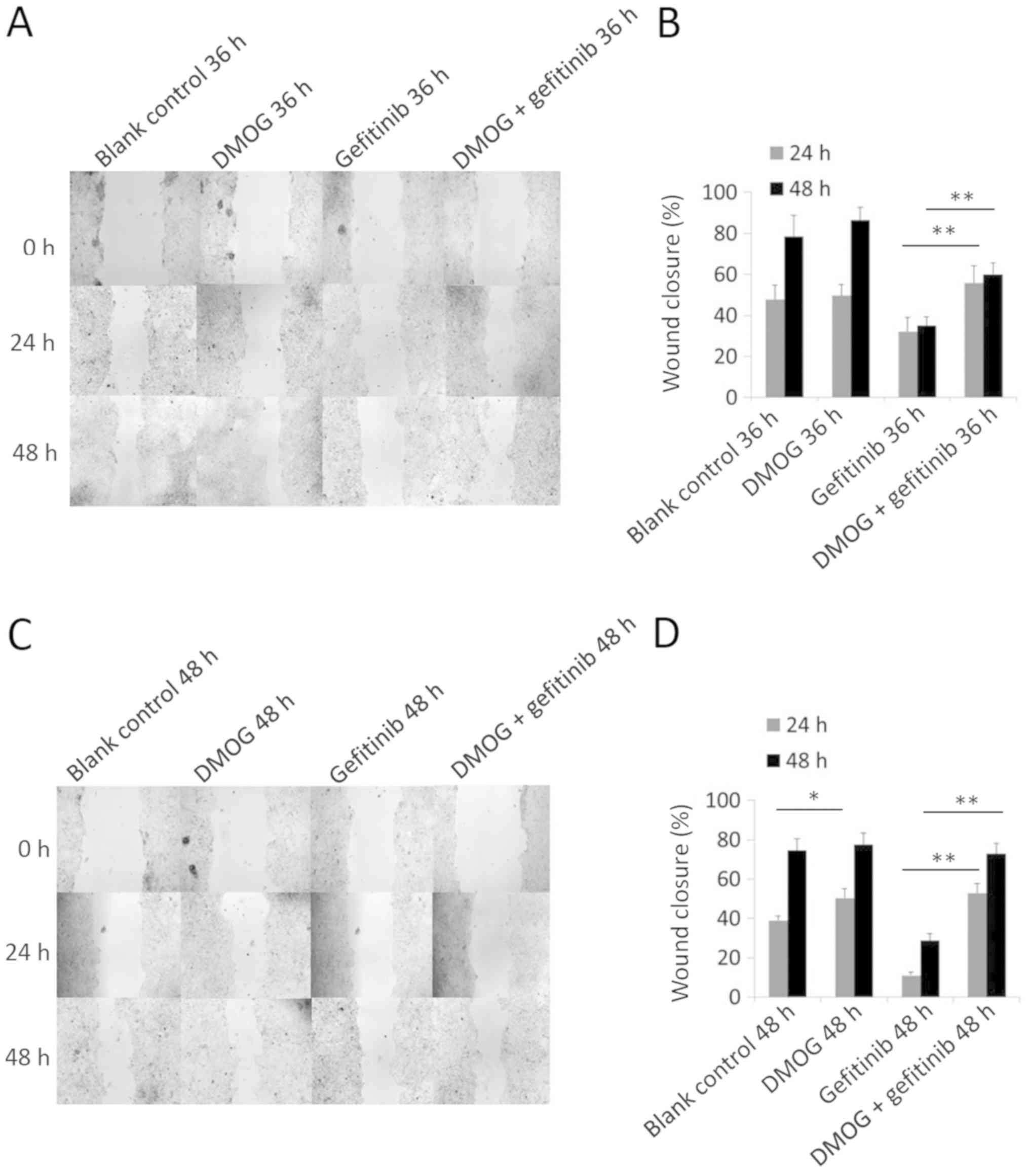 | Figure 4.Wound healing assay of HCC827 cells
with different treatments of DMOG and gefitinib. (A) A wound
healing assay was performed to determine the cell migration ability
of HCC827 cells with different treatments (blank control, DMOG,
gefitinib, and DMOG and gefitinib combined for 36 h).
Representative images captured 24 and 48 h after wounding
(magnification, ×100) depicting the cell migration. Concentrations
of DMOG and gefitinib used were 2 mM and 20 nM, respectively. (B)
Quantified wound-healing percentage of HCC827 cells 24 and 48 h
after being wounded. Error bars represent the mean ± standard
deviation from three independent experiments. (C) A wound healing
assay was performed to evaluate the cell migration ability of
HCC827 cells with different treatments (blank control, DMOG,
gefitinib, and DMOG and gefitinib combined for 48 h).
Representative images photographed 24 and 48 h after wounding
(magnification, ×100) depicting the cell migration. Concentrations
of DMOG and gefitinib used were 2 mM and 20 nM, respectively. (D)
Quantified wound-healing percentage of HCC827 cells 24 and 48 h
after being wounded. Error bars represent the mean ± standard
deviation from three independent experiments. *P<0.05 and
**P<0.01. DMOG, dimethyloxalylglycine. |
HIF-1 signaling pathway downregulation
enhances the sensitivity of HCC827 cells to gefitinib
To demonstrate the effect of the downregulation of
the HIF-1 signaling pathway, western blot analysis was performed to
detect the expression of HIF-1α in HCC827 cells with different
treatments (blank control, YC-1, gefitinib, and YC-1 and gefitinib
combined for 16 and 28 h). As depicted in Fig. 5, the protein expression levels of
HIF-1α were significantly reduced in cells treated with YC-1,
compared with the blank control for 36 and 48 h (P<0.01).
Similarly, the levels of HIF-1α were significantly reduced in cells
treated with YC-1 and gefitinib combined, compared with the
gefitinib treatment alone, for both 36 and 48 h (P<0.01). To
determine the sensitivity change of HCC827 cells to gefitinib
subsequent to the downregulation of the HIF-1 signaling pathway, an
MTT assay, a colony formation analysis and a wound-healing assay
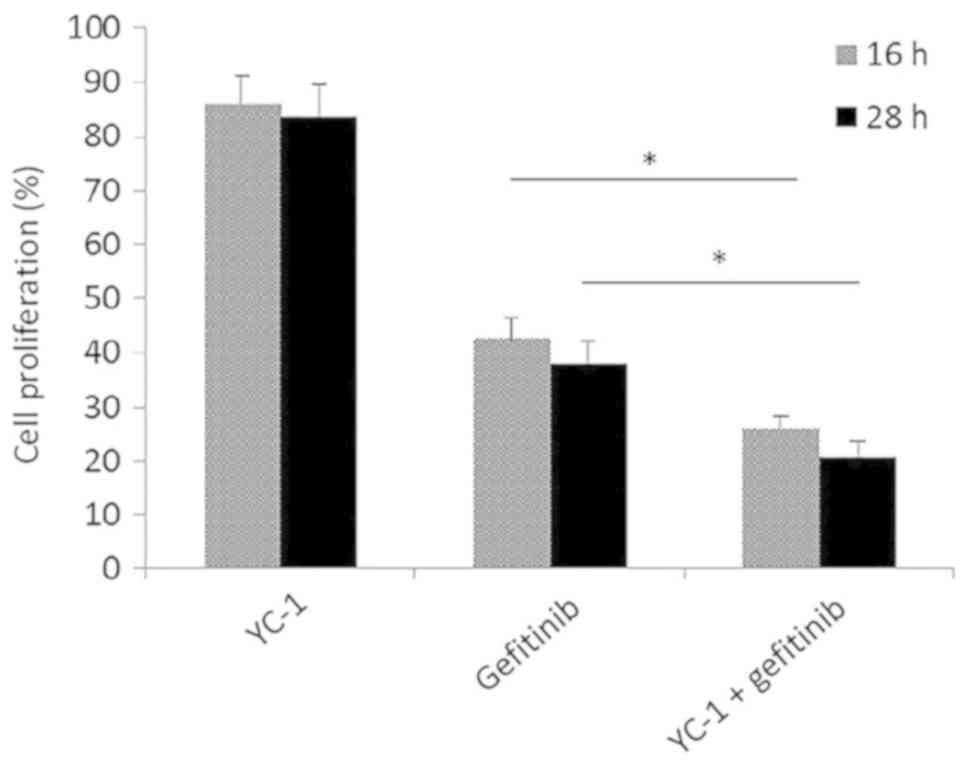
were performed. In the MTT assay, when HCC827 cells were treated
with YC-1 and gefitinib combined, a significant reduction in cell
proliferation was observed, compared with gefitinib alone treated
HCC827 cells (P<0.05; Fig. 6).
This indicated that the HCC827 cells became more sensitive to
gefitinib following YC-1 treatment, whilst the HIF-1 signaling
pathway was downregulated. YC-1 alone had a significant inhibitory
effect on the colony-forming ability of HCC827 cells, compared with
the blank control (P<0.05; Fig.
7). Gefitinib and YC-1 combined treatment resulted in
significant inhibitory effect on the colony-forming ability of
HCC827 cells, compared with gefitinib treatment alone (P<0.01;
Fig. 7). In the wound healing assay,
treatment with gefitinib and YC-1 combined significantly inhibited
cell migration, compared with gefitinib treatment alone (P<0.01;
Fig. 8).
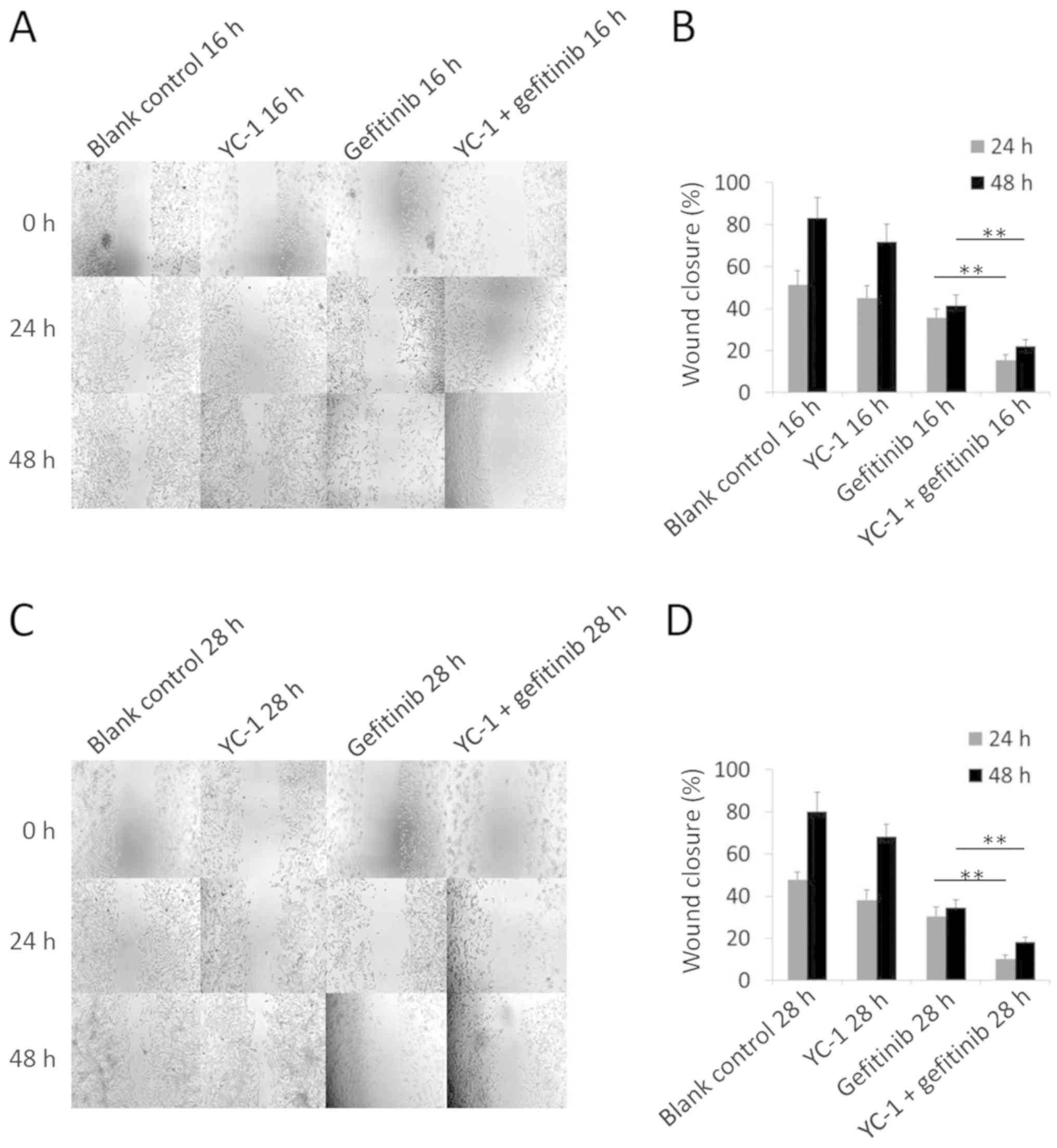 | Figure 8.Wound healing assay of HCC827 cells
with different treatments of YC-1 and gefitinib. (A) A wound
healing assay was performed to determine the cell migration ability
of HCC827 cells with different treatments (blank control, YC-1,
gefitinib, and YC-1 and gefitinib combined for 16 h).
Representative images captured 24 and 48 h after wounding
(magnification, ×100) are depicted. Concentrations of YC-1 and
gefitinib used were 40 µM and 20 nM, respectively. (B) Quantified
wound-healing percentage of HCC827 cells 24 and 48 h after being
wounded. Error bars represent the mean ± standard deviation from
three independent experiments. (C) A wound healing assay was
performed to evaluate the cell migration ability of HCC827 cells
with different treatments (blank control, YC-1, gefitinib, and YC-1
and gefitinib combined for 28 h). Representative images were
captured 24 and 48 h after wounding (magnification, ×100) are
depicted. Concentrations of YC-1 and gefitinib used were 40 µM and
20 nM, respectively. (D) Quantified wound-healing percentage of
HCC827 cells 24 and 48 h after being wounded. Error bars represent
the mean ± standard deviation from three independent experiments.
*P<0.05 and **P<0.01. YC-1,
3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole. |
Association between p-Met level of
HCC827 cell and the activity of HIF-1 signal pathway
Using western blot analysis, the protein expression
levels of HIF-1α and p-Met were examined. The protein expression
levels of HIF-1α in HCC827 cells with different treatments are
depicted and described in Figs. 1
and 5. When HCC827 cells were
treated with DMOG, the protein expression levels of HIF-1α and
p-Met were elevated, compared with the control cells (P<0.05 and
P<0.01 for 36 and 48 h, respectively; Fig. 9). When HCC827 cells were treated with
YC-1, the protein expression levels of HIF-1α and p-Met were
significantly reduced, compared with the blank control (P<0.01
for 16 and 28 h; Fig. 9). It was
revealed that the p-Met protein expression levels in HCC827 cell
were associated with the protein expression levels of HIF-1α. Based
on these results, the correlation between p-Met and HIF-1α protein
levels in HCC827 cells was investigated by Pearson's correlation
analysis (Fig. 10). The expression
level of p-Met was significantly positively correlated with HIF-1α
levels (r2=0.978, P<0.01).
 | Figure 9.Expression of HIF-1α and p-Met in
HCC827 cells with different treatments. (A) Western blot analysis
was performed to determine the HIF-1α and p-Met levels in HCC827
cells with different treatments (blank control, DMOG, gefitinib,
and DMOG and gefitinib combined for 36 and 48 h). Concentrations of
DMOG and gefitinib used were 2 mM and 20 nM, respectively. (B)
Western blot analysis was performed to evaluate the HIF-1α and
p-Met levels in HCC827 cells with different treatments (blank
control, YC-1, gefitinib, and YC-1 and gefitinib combined for 16
and 28 h). Concentrations of YC-1 and gefitinib used were 40 µM and
20 nM, respectively. (C) Quantified densitometric scanning of the
blots. Error bars represent the mean ± standard deviation from
three independent experiments. *P<0.05 and **P<0.01. HIF-1α,
hypoxia-inducible factor-1; p-Met, phosphorylated hepatocyte growth
factor receptor; DMOG, dimethyloxalylglycine; YC-1,
3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole. |
Discussion
The present study revealed that the HIF-1 signaling
pathway influenced the sensitivity of HCC827 cells to gefitinib,
thus providing insights into the effect of the HIF-1 signaling
pathway on acquired gefitinib resistance. Furthermore, p-Met levels
exerted a strong positive correlation with HIF-1α level, which may
be the molecular mechanism underlying the influence of HIF-1
signaling pathway on the sensitivity of HCC827 cells to
gefitinib.
HIF-1 mediates the primary biological effects of
hypoxia in tumorigenesis (20).
Activation of HIF-1 transcription results in the upregulation of a
number of genes, including vascular endothelial growth factor,
insulin-like growth factor 2, telomerase reverse transcriptase,
stroma-derived factor 1 and multidrug resistance 1, which encode
proteins participating in tumor angiogenesis, cell proliferation,
survival, invasion and therapy resistance (21,22).
Accumulation of HIF-1 limits the effectiveness of radiotherapy and
numerous cytotoxic drugs (23). For
HIF-1 signaling in EGFR-TKI therapy resistance, previous research
indicated that they may be associated. In an acquired EGFR-TKI
resistant cell line, the upregulation of HIF-1α was observed,
compared with an EGFR-TKI sensitive cell line (13). Furthermore, hypoxia increased the
population of lung cancer stem cells resistant to gefitinib in EGFR
mutation-positive NSCLC (PC9 and HCC827 cells) by activating
insulin-like growth factor 1 receptor (12). In the present study, it was observed
that HIF-1 signaling pathway upregulation caused by DMOG is able to
reduce the sensitivity of HCC827 cells to gefitinib (Figs. 2–4).
This is in accordant with previous studies (24–26).
This indicates that the HIF-1 signaling pathway may participate in
the forming of acquired EGFR-TKIs resistance.
When the HIF-1 pathway is upregulated, vascular
endothelial growth factor, epithelial-mesenchymal transition and
membrane-type 4 matrix metalloproteinase are activated (27–29). In
the present study, DMOG treatment alone for 48 h was able to
promote the colony-forming and migrating ability of HCC827 cells
(Figs. 3 and 4). DMOG achieves its effects on HCC827
cells through the upregulation of the HIF-1 pathway (30,31).
When DMOG and gefitinib treatments were combined, the effect of
gefitinib was substantially inhibited (Figs. 2–4).
This may verify the importance of the HIF-1 pathway inhibition in
NSCLC treatment.
YC-1, as an HIF-1 inhibitor, possesses antitumor
effects itself (32). YC-1 can
inhibit proliferation of breast cancer cells (33,34),
however its effect on lung cancer is confined to its influence on
enhancing radiotherapy sensitivity (35,36). The
present study observed that the downregulation of the HIF-1
signaling pathway caused by YC-1 is able to enhance the sensitivity
of HCC827 cells to gefitinib. When YC-1 was combined with
gefitinib, the inhibiting effect on the colony-forming, cell
migration and cell proliferation abilities of HCC827 cells was
substantially enhanced, compared with gefitinib alone (Figs. 6–8).
Due to the synergistic effect of YC-1 and gefitinib on HCC827
cells, it was speculated that YC-1 may enhance the sensitivity of
acquired EGFR-TKI-resistant lung cancer. In future studies, a
gefitinib-resistant HCC827 cell line (HCC827-GR) should be
established to examine the effect of YC-1 on enhancing the
sensitivity of HCC827-GR to gefitinib.
In order to investigate the mechanism of the
influence of the HIF-1 signaling pathway on the sensitivity of
HCC827 cells to EGFR-TKIs, the correlation of p-Met levels in
HCC827 cells with the activity of the HIF-1 signaling pathway was
analyzed. A previous study indicated that HIF-1α is involved in the
regulation of Met levels through EGFR. Additionally, EGFR
regulation of Met levels in EGFR-TKI-sensitive cell lines occurs
through the HIF-1 pathway in a hypoxia-independent manner (19,37).
Nevertheless, in an EGFR-TKI-resistant cell line with a Met gene
amplification, this regulation was lost, and if the overexpression
of a constitutively active form of HIF-1α occurred, this regulation
was also lost (12). All these
previous results indicate that the HIF-1 pathway, but not the EGFR
pathway, may regulate the Met levels in EGFR-TKI-resistant cell
lines caused by a Met gene amplification. In the present study, the
p-Met levels in HCC827 cells were significantly positively
correlated with the activity of the HIF-1 signaling pathway
(Fig. 10; P<0.01;
R2=0.978). This result is consistent with the results of
previous research. Furthermore, previous research demonstrated that
Met amplification is the primary mechanism used for forming
HCC827-GR cells through chronic exposure to gefitinib (38–40). In
future studies, the regulation of the HIF-1 pathway on Met levels
in HCC827-GR cells should be observed, and the synergistic effect
of YC-1 and gefitinib in HCC827-GR cells should be
demonstrated.
Despite the requirement for further studies to
investigate the synergistic effect of YC-1 and gefitinib in
HCC827-GR cells, the present study reveals that the HIF-1 signaling
pathway is able to influence the sensitivity of HCC827 cells to
gefitinib, and that the associated mechanism may be the regulation
of the HIF-1 pathway on p-Met. In conclusion, the HIF-1 pathway may
be an attractive target for enhancing the sensitivity of NSCLC to
EGFR-TKIs. This conclusion indicates the requirement for further
research on the HIF-1 pathway as a target for overcoming or
delaying acquired gefitinib resistance in NSCLC therapy.
Acknowledgements
The authors would like to thank Professor Kequn
Chai, Miss Guizhi Zhao and the Scientific Research and Education
Department staff of Tongde Hospital of Zhejiang Province (Hangzhou,
China) for providing advice and technical assistance for the
present study.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QJ analyzed and interpreted the data of the cell
experiments, and was a major contributor in writing the manuscript.
JZ performed the western blot assay and participated in the design
of this research. XX performed the MTT assay. FH performed the
colony formation assay. WX performed the cell migration assay. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Tongde Hospital of Zhejiang Province (Hangzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
EGFR-TKIs
|
epidermal growth factor
receptor-tyrosine kinase inhibitors
|
|
YC-1
|
3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole
|
|
HIF-1
|
hypoxia-inducible factor-1
|
|
DMOG
|
dimethyloxalylglycine
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang K and Yuan Q: Current mechanism of
acquired resistance to epidermal growth factor receptor-tyrosine
kinase inhibitors and updated therapy strategies in human nonsmall
cell lung cancer. J Cancer Res Ther. 12:C131–C137. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shepherd FA, Rodrigues Pereira J, Ciuleanu
T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S,
Smylie M, Martins R, et al: Erlotinib in previously treated
non-small-cell lung cancer. N Engl J Med. 353:123–132. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kris MG, Natale RB, Herbst RS, Lynch TJ
Jr, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H,
Sandler A, et al: Efficacy of gefitinib, an inhibitor of the
epidermal growth factor receptor tyrosine kinase, in symptomatic
patients with non-small cell lung cancer: A randomized trial. JAMA.
290:2149–2158. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thatcher N, Chang A, Parikh P, Rodrigues
Pereira J, Ciuleanu T, von Pawel J, Thongprasert S, Tan EH,
Pemberton K, Archer V and Carroll K: Gefitinib plus best supportive
care in previously treated patients with refractory advanced
non-small-cell lung cancer: Results from a randomised,
placebo-controlled, multicentre study (Iressa Survival Evaluation
in Lung Cancer). Lancet. 366:1527–1537. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu HA, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumor specimens at the time of acquired resistance to
EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Burroughs SK, Kaluz S, Wang D, Wang K, Van
Meir EG and Wang B: Hypoxia inducible factor pathway inhibitors as
anticancer therapeutics. Future Med Chem. 5:553–572. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wilson W and Hay M: Targeting hypoxia in
cancer therapy. Nat Rev Cancer. 11:393–410. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci
USA. 92:5510–5514. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Murakami A, Takahashi F, Nurwidya F,
Kobayashi I, Minakata K, Hashimoto M, Nara T, Kato M, Tajima K,
Shimada N, et al: Hypoxia increases gefitinib-resistant lung cancer
stem cells through the activation of insulin-like growth factor 1
receptor. PLoS One. 9:864592014. View Article : Google Scholar
|
|
13
|
Morgillo F, Cascone, D'Aiuto E, Martinelli
E, Troiani T, Saintigny P, De Palma R, Heymach JV, Berrino L,
Tuccillo C and Ciardiello F: Antitumour efficacy of MEK inhibitors
in human lung cancer cells and their derivatives with acquired
resistance to different tyrosine kinase inhibitors. Br J Cancer.
105:382–392. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ko FN, Wu CC, Kuo SC, Lee FY and Teng CM:
YC-1, a novel activator of platelet guanylate cyclase. Blood.
84:4226–4233. 1994.PubMed/NCBI
|
|
15
|
Chun YS, Yeo EJ, Choi E, Teng CM, Bae JM,
Kim MS and Park JW: Inhibitory effect of YC-1 on the hypoxic
induction of erythropoietin and vascular endothelial growth factor
in Hep3B cells. Biochem Pharmacol. 61:947–954. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang L, Jiang G, Zhao X and Gong Y:
Dimethyloxalylglycine promotes bone marrow mesenchymal stem cell
osteogenesis via Rho/ROCK signaling. Cell Physiol Biochem.
39:1391–1403. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li F, Li Z, Jiang Z, Tian Y, Wang Z, Yi W
and Zhang C: Enhancement of early cardiac differentiation of
dedifferentiated fat cells by dimethyloxalylglycine via notch
signaling pathway. Am J Transl Res. 8:4791–4801. 2016.PubMed/NCBI
|
|
18
|
Rho JK, Choi YJ, Kim SY, Kim TW, Choi EK,
Yoon SJ, Park BM, Park E, Bae JH, Choi CM and Lee JC: MET and AXL
inhibitor NPS-1034 exerts efficacy against lung cancer cells
resistant to EGFR kinase inhibitors because of MET or AXL
activation. Cancer Res. 74:253–262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu L, Nilsson MB, Saintigny P, Cascone T,
Herynk MH, Du Z, Nikolinakos PG, Yang Y, Prudkin L, Liu D, et al:
Epidermal growth factor receptor regulates MET levels and
invasiveness through hypoxia-inducible factor-1α in non-small cell
lung cancer cells. Oncogene. 29:2616–2627. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rankin EB and Giaccia AJ: The role of
hypoxia-inducible factors in tumorigenesis. Cell Death Differ.
15:678–685. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wigerup C, Påhlman S and Bexell D:
Therapeutic targeting of hypoxia and hypoxia-inducible factors in
cancer. Pharmacol Ther. 164:152–169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Warfel NA and El-Deiry WS: HIF-1 signaling
in drug resistance to chemotherapy. Curr Med Chem. 21:3021–3028.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Harris AL: HIF-1 signaling in drug
resistance to chemotherapy. Nat Rev Cancer. 2:38–47. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Minakata K, Takahashi F, Nara T, Hashimoto
M, Tajima K, Murakami A, Nurwidya F, Yae S, Koizumi F, Moriyama H,
et al: Hypoxia induces gefitinib resistance in non-small-cell lung
cancer with both mutant and wild-type epidermal growth factor
receptors. Cancer Sci. 103:1946–1954. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang WW, Wang YB, Wang DQ, Lin Z and Sun
RJ: Integrin beta-8 (ITGB8) silencing reverses gefitinib resistance
of human hepatic cancer HepG2/G cell line. Int J Clin Exp Med.
8:3063–3071. 2015.PubMed/NCBI
|
|
26
|
El Guerrab A, Zegrour R, Nemlin CC, Vigier
F, Cayre A, Penault-Llorca F, Rossignol F and Bignon YJ:
Differential impact of EGFR-targeted therapies on hypoxia
responses: Implications for treatment sensitivity in
triple-negative metastatic breast cancer. PLoS One. 6:e250802011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Soni S and Padwad YS: HIF-1 in cancer
therapy: Two decade long story of a transcription factor. Acta
Oncol. 56:503–515. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bertout JA, Patel SA and Simon MC: The
impact of O2 availability on human cancer. Nat Rev Cancer.
8:967–975. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu F, Hu L, Ma Y, Huang B, Xiu Z, Zhang
P, Zhou K and Tang X: Increased expression of monoamine oxidase A
is associated with epithelial to mesenchymal transition and
clinicopathological features in non-small cell lung cancer. Oncol
Lett. 15:3245–3251. 2018.PubMed/NCBI
|
|
30
|
Wu D, Chen B, Cui F, He X, Wang W and Wang
M: Hypoxia-induced microRNA-301b regulates apoptosis by targeting
Bim in lung cancer. Cell Prolif. 49:476–483. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Khong TL, Thairu N, Larsen H, Dawson PM,
Kiriakidis S and Paleolog EM: Identification of the angiogenic gene
signature induced by EGF and hypoxia in colorectal cancer. BMC
Cancer. 13:5182013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yeo EJ, Chun YS, Cho YS, Kim J, Lee JC,
Kim MS and Park JW: YC-1: A potential anticancer drug targeting
hypoxia-inducible factor 1. J Natl Cancer Inst. 95:516–525. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheng Y, Li W, Liu Y, Cheng HC, Ma J and
Qiu L: YC-1 exerts inhibitory effects on MDA-MB-468 breast cancer
cells by targeting EGFR in vitro and in vivo under normoxic
condition. Chin J Cancer. 31:248–256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chang LC, Lin HY, Tsai MT, Chou RH, Lee
FY, Teng CM, Hsieh MT, Hung HY, Huang LJ, Yu YL and Kuo SC: YC-1
inhibits proliferation of breast cancer cells by down-regulating
EZH2 expression via activation of c-Cbl and ERK. Br J Pharmacol.
171:4010–4025. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ikezawa Y, Sakakibara-Konishi J, Mizugaki
H, Oizumi S and Nishimura M: Inhibition of Notch and HIF enhances
the antitumor effect of radiation in Notch expressing lung cancer.
Int J Clin Oncol. 22:59–69. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moeller BJ and Dewhirst MW: HIF-1 and
tumour radiosensitivity. Br J Cancer. 95:1–5. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhen Q, Liu JF, Liu JB, Wang RF, Chu WW,
Zhang YX, Tan GL, Zhao XJ and Lv BL: Endothelial PAS
domain-containing protein 1 confers TKI-resistance by mediating
EGFR and MET pathways in non-small cell lung cancer cells. Cancer
Biol Ther. 16:549–557. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jang WJ, Jung SK, Kang JS, Jeong JW, Bae
MK, Joo SH, Park GH, Kundu JK, Hong YS and Jeong CH: Anti-tumor
activity of WK88-1, a novel geldanamycin derivative, in
gefitinib-resistant non-small cell lung cancers with Met
amplification. Cancer Sci. 105:1245–1253. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Y, Zhang W, Wen L, Yang H, Wen M, Yun
Y, Zhao L, Zhu X, Tian L, Luo E, et al: FOXM1 confers resistance to
gefitinib in lung adenocarcinoma via a MET/AKT-dependent positive
feedback loop. Oncotarget. 7:59245–59259. 2016.PubMed/NCBI
|
|
40
|
Zhai Y, Zhang Y, Nan K and Liang X:
Reduced expression levels of PTEN are associated with decreased
sensitivity of HCC827 cells to icotinib. Oncol Lett. 13:3233–3238.
2017. View Article : Google Scholar : PubMed/NCBI
|