Introduction
Arsenic is a widespread environmental metalloid
present in nature as oxide and sulfide compounds. Inorganic arsenic
compounds comprise numerous valence states, including arsenic
trioxide (As2O3), realgar
(As2S2), As (III), and As (V). As (III) has
been used treat tumors with a positive effect; although, due to
its' higher solubility and bioavailability, it may be more toxic
than As (V) (1). Organoarsenic
compounds comprise liver detoxification metabolites, including
dimethylarsenic acid [DMA;
(CH3)2AsO2H] and
4-[N-(S-glutathionylacetyl) amino] phenylarsonic acid (GSAO)
(2). It has been demonstrated that
As2O3 treats acute promyelocytic leukemia due
to its antitumorigenic properties (3,4).
Furthermore, in vitro studies have revealed that
As2O3 induces apoptosis in various types of
cell line, including the DU145 and PC-3 (prostate cancer) (5), MDAH 2774 (ovarian cancer) (5) and TM4 (sertoli tumor) cell lines
(6), and
CD133+/CD13+ liver cancer stem cells
(7). In addition, GSAO, an
organoarsenic compound, has been reported to inhibit proliferation
in endothelial and tumor cells, such as fibrosarcoma cells, lung
cancer, pancreatic cancer and prostate cancer cells in
vitro, and reduce angiogenesis and tumor growth in a xenograft
mouse model (8,9). Furthermore, DMA exerts an
antiproliferative and cytotoxic effect on human leukemia and
multiple myeloma cells (10).
Inorganic and organic arsenic compounds therefore represent novel
potential therapeutic agents against solid tumors and various types
of malignancy (3–10).
Previous studies have reported that tumor formation
arises from an imbalance between cell proliferation and apoptosis.
Subsequently, apoptosis induction is a potential target for cancer
therapies (11,12). Apoptosis is induced by various types
of caspase, which are aspartatespecific cysteine proteases, during
the extrinsic and intrinsic apoptosis pathways that involve death
receptors and mitochondria, respectively. The extrinsic pathway is
initiated from death receptors, e.g., tumor necrosis factor (TNF)
receptor and Fas, which bind with their associated ligands, i.e.,
TNF and Fasligand (FasL), prior to subsequent caspase-8 activation
(13). The intrinsic pathway is
caused by mitochondrial dysfunction, which leads to cytochrome
c release in the cytosol and subsequent formation of the
apoptosome when combined with cleaved caspase-9 (14). These two caspase cascades eventually
trigger caspase-3 activation and subsequent cellular morphological
alterations, including membrane blebbing, phosphatidylserine
externalization, cell detachment and chromosomal DNA fragmentation
(15). In addition, proteins from
the B-cell lymphoma-2 (Bcl-2) family are key regulators of the
apoptotic response. They serve different physiological roles in
mitochondrial integrity, including multidomain antiapoptotic (e.g.
Bcl-2 and Bcl-extra-large), multidomain proapoptotic (e.g. Bcl-2
associated X, apoptosis regulator and Bcl-2 antagonist/killer), and
Bcl-2 homology region 3 (BH3)-only proapoptotic (e.g. BH3
interacting domain death agonist and Bcl-2 modifying factor) roles
(16). These proteins can positively
and negatively regulate mitochondrial permeability and apoptotic
protein efflux (17–19). A previous study demonstrated that
As2O3 upregulates BH3-only proapoptotic, and
downregulates antiapoptotic, protein levels in myeloma (20). In addition, the extrinsic apoptotic
pathway, which involves Fas/FasL, also participates in
arsenic-induced keratinocyte apoptosis (21). The mechanisms underlying
arsenic-induced apoptosis in various types of tumor cell are
therefore complex, and have yet to be fully elucidated.
Leydig cell tumors are one type of sex cord-stromal
malignancy observed in testicular cancer, accounting for 1–3% of
all testicular neoplasms and 4–9% of tumors of the testis in
prepubertal boys. Epidemiological studies have reported that the
incidence of testicular cancer has been increasing worldwide over
the past 30 years (22). Clinically,
the major therapeutic strategy for Leydig cell tumor is radical
orchiectomy. Testis sparing surgery is preferred in order to
maintain fertility. In addition, ~10% of Leydig cell tumors respond
unfavorably to chemotherapy and irradiation (23). The present study aimed therefore to
explore alternative therapeutic strategies to treat Leydig cell
tumors. Particularly, this study aimed to determine the mechanisms
underlying the arsenicinduced cell apoptosis in Leydig cell tumors.
To do so, the effect of arsenic compounds, including sodium
arsenite and DMA, which are the most representative inorganic and
organic arsenite compounds, respectively (8), were investigated in MA-10 mouse Leydig
tumor cells, which may aid the development of potentially more
effective chemotherapy strategies.
Materials and methods
Chemicals
Sodium arsenite was purchased from Fluka (St.
Gallen, Switzerland). DMA, RNase A, Waymouth's MB 752/1 medium,
propidium iodide (PI), Folin & Ciocalteu's phenol reagent,
EDTA, EGTA, 30% acrylamide/Bisacrylamide solution, MTT and
anti-β-actin monoclonal antibody were purchased from Sigma-Aldrich;
Merck KGaA (Darmstadt, Germany). Fetal bovine serum (FBS) and
trypsin-EDTA were purchased from Gibco; Thermo Fisher Scientific,
Inc. (Waltham, MA, USA). Gentamycin sulfate was purchased from AG
Scientific Inc. (San Diego, CA, USA). Sodium chloride (NaCl),
HEPES, potassium chloride and Tris base were purchased from J.T.
Baker (Phillipsburg, NJ, USA). Disodium hydrogen phosphate,
potassium dihydrogen phosphate, and tissue culture grade sodium
bicarbonate were purchased from Riedel-de Haën (Seelze, Germany).
Hydrochloric acid, sodium dodecyl sulfate (SDS), Tween-20 and
dimethyl sulfoxide (DMSO) were purchased from Merck KGaA. Sucrose
was purchased from Panreac (Barcelona, Spain). The general caspase
inhibitor Z-VAD-fmk was purchased from R&D Systems, Inc.
(Minneapolis, MN, USA). Goat horseradish peroxidase
(HRP)-conjugated anti-rabbit immunoglobulin (Ig)G (cat. no.
NEF812001EA; 1:2,000) and HRP-conjugated antimouse IgG secondary
antibodies (cat. no. NEF822001EA; 1:2,000) were purchased from
PerkinElmer, Inc. (Waltham, MA, USA). The Annexin V-fluorescein
isothiocyanate (FITC) apoptosis detection kit was purchased from
Strong Biotech (Taipei, Taiwan). Polyclonal antibodies against
cleaved caspases-3 (cat. no. 9661; 1:2,000), cleaved caspase-8
(cat. no. 9429; 1:2,000) and cleaved caspase-9 (cat. no. 9509;
1:2,000) and poly(ADP-ribose) polymerase (PARP; cat. no. 9542;
1:2,000) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). The enhanced chemiluminescence (ECL) detection
kit was purchased from EMD Millipore (Billerica, MA, USA).
Cell culture and treatments
The MA-10 cell line was kindly donated by Dr Mario
Ascoli (University of Iowa, Iowa City, IA, USA), and maintained
using standard techniques as previously described (24,25).
Briefly, cells were cultured in Waymouth's MB 752/1 medium
supplemented with 10% FBS and placed at 37°C in a humidified
incubator containing 5% CO2. A total of 6×106
MA-10 cells were plated in a 60-mm dish. After 24 h, MA-10 cells
were treated with 0.01–100 µM sodium arsenite, 0.1 µM-10 mM
dimethylarsenic acid or PBS (Control) in the medium containing 1%
FBS for 3, 6, 12 and 24 h.
Morphological observation
The morphology of arsenic compound-treated MA-10
cells and control cells at 3, 6, 12 and 24 h after treatment was
examined using an Olympus CK40 light microscope (Olympus
Corporation, Tokyo, Japan) and images were captured using an
Olympus DP20 digital camera (Olympus Corporation; magnification,
×200). Apoptosis was characterized by assessing plasma membrane
blebbing and detached cells, as described previously (26).
Cell viability assay
Cell viability was assessed with MTT assay as
described previously (27),
following treatment with arsenic compounds. In brief, MA-10 cells
were seeded in 96-well plates at a density of 8×103
cells per well and 50 µl MTT (0.5 mg/ml) was added to each well
after 24, 48 and 72 h after treatment with arsenic compounds, and
incubated at 37°C for 4 h. The supernatant was discarded and the
MTT-formazan crystals were dissolved with 50 µl 0.5% DMSO in each
well for 1 h. The optical density (OD) values were read using a
VersaMax ELISA microplate reader (Molecular Devices, LLC,
Sunnyvale, CA, USA) at a wavelength of 570 nm.
Cell cycle analysis
Cell cycle distribution was determined by flow
cytometry following PI staining (28). After 18 h of serum starvation, MA-10
cells were treated with arsenic compounds for 3, 6, 12 and 24 h,
and then cells were detached using 1% trypsin and fixed with 70%
ethanol for 2 h at −20°C. Fixed cells were simultaneously treated
with RNase (100 µg/ml) and PI (40 µg/ml) for 30 min at room
temperature and analyzed using flow cytometry on a FACScan™
(Becton-Dickinson and Company, Franklin Lakes, NJ, USA) with an
excitation wavelength of 488 nm and a band pass filter >600 nm.
Cells in the sub-G1 phase, also termed hypodiploid,
contain less DNA, which is due to apoptosisassociated DNA
fragmentation (29).
Annexin V/PI double staining
assay
A total of 6×106 MA-10 cells were seeded
in a 60-mm dish and treated 1 day after seeding with 10 or 100 µM
sodium arsenite, 1 or 10 mM dimethylarsenic acid or PBS in the
medium containing 1% FBS for 24 h. Treated-MA-10 cells were
collected using trypsin and centrifuged at 120 × g for 10 min at
4°C, and then incubated with 100 µl Annexin V-FITC staining
solution (apoptosis detection kit; Strong Biotech Corporation,
Taipei, Taiwan) for 15 min at room temperature according to the
manufacturer's protocol. Samples were analyzed on a FACSCalibur
flow cytometer (Becton-Dickinson and Company) with an excitation
wavelength of 488 nm and band pass filters of 515 and 600 nm for
FITC and PI detection, respectively. Data were represented using
histogram plots gated into four quadrants containing negative
(Annexin V/PI), PIpositive (Annexin V/PI+), Annexin
Vpositive (Annexin V+/PI) and Annexin V/PIdoublepositive
(Annexin V+/PI+) stained cells, which
corresponded to viable, dead, early apoptotic and late apoptotic
cells, respectively (30).
Protein extraction and western
blotting
Treated cells were lysed with 100 µl ice-cold lysis
buffer for 30 min at room temperature, which contained 20 mM Tris
at pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5
mM sodium pyrophosphate and 1 mM sodium orthovanadate. Lysates were
centrifuged at 12,000 × g for 12 min at 4°C, and supernatants were
collected and stored at 20°C until future analysis. Protein
concentration was determined using the Lowry protein assay as
described previously (31). Proteins
(30 µg) were separated by 12% SDS-PAGE and transferred onto
polyvinylidene fluoride membranes. Membranes were blocked with 5%
milk dissolved in TBST for 1 h at room temperature, and incubated
with primary antibodies overnight at 4°C. Membranes were then
washed three times with TBST and incubated for 1 h with the
appropriate HRP conjugated secondary antibodies. Bands were
detected using ECL substrate and the UVP EC3 BioImaging system
(UVP, LLC, Phoenix, AZ, USA) (32–34).
Quantification of the western blotting data was performed using
ImageJ version 1.50 software (National Institutes of Health,
Bethesda, MD, USA).
Statistical analysis
All data were expressed as the means ± standard
error of the mean from three separate experiments. The statistical
significance of the differences between the control and treatment
groups at various time-points was determined using a two-way
analysis of variance followed by the Least Significance Difference
test. Statistical analysis was performed using GraphPad Prism
version 6 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
Effects of arsenic compounds on MA-10
cell morphology and viability
To determine the cytotoxicity of arsenic compounds,
MA-10 cells were treated with or without sodium arsenite (0.01–100
µM) and DMA (0.1–10 mM) for 3, 6, 12 and 24 h. At each time point,
untreated MA-10 cells were firmly attached and exhibited the
commonly anticipated polygonal-shaped morphology (Fig. 1A). Conversely, cells treated with 100
µM sodium arsenite or 10 mM DMA became gradually rounded and more
detached over time (Fig. 1A and B,
respectively). However, 12 and 24 h of treatment with 10 µM sodium
arsenite, and 24 h of treatment with 1 mM DMA, induced plasma
membrane blebbing and an enlarged and flattened appearance, which
suggested that cell apoptosis was occurring (Fig. 1A and B). Sodium arsenite appeared to
be more toxic than DMA to MA-10 cancer cells. However, low
concentrations of sodium arsenite (0.01, 0.1 and 1 µM) or DMA (0.1,
10 µM and 10 mM) did not effect cell morphology.
The effects of arsenic compounds on MA10 cell
morphology were further investigated with the MTT viability assay.
MA10 cell viability significantly decreased to 88±2.9, 84±7.4,
74±3.8 and 45±6.4% following 3, 6, 12 or and 24 h treatment with 10
µM sodium arsenite, respectively, compared with the control group
(Fig. 1C). MA-10 cell treatment with
10 mM DMA only significantly reduced cell viability to 99±6.9,
79±6.6, 84±0.8 and 49±6.0% at the same time points (Fig. 1D). These results indicated that
sodium arsenite and DMA both induced time and dosedependent cell
toxicity, and that sodium arsenite was more potent (given the
1,000-fold difference in the concentration of the two compounds
that elicited these effects).
Arsenic compounds induce MA10 cell
apoptosis
To determine whether MA10 cells were undergoing
apoptosis following treatment with arsenic compounds, cell DNA
content was quantified using PI staining. A significant increase in
the number of cells in the sub-G1 phase, which is an
apoptotic marker, was observed following 12 and 24 h treatment with
10 µM sodium arsenite or 10 mM DMA, respectively (P<0.05;
Fig. 2A and C). In addition, the
cell population in the G2/M phase was significantly
increased following 24 h treatment with 10 and 100 µM sodium
arsenite, and 12 h treatment with 1 mM DMA (12 and 24 h), which
indicated that both arsenic compounds caused G2/M cell
cycle arrest of MA10 cells (Fig. 2B and
D) (P<0.05). These results suggested that cell cycle
regulation and redistribution may be involved in sodium arsenite-
and DMA-induced MA-10 cell apoptosis.
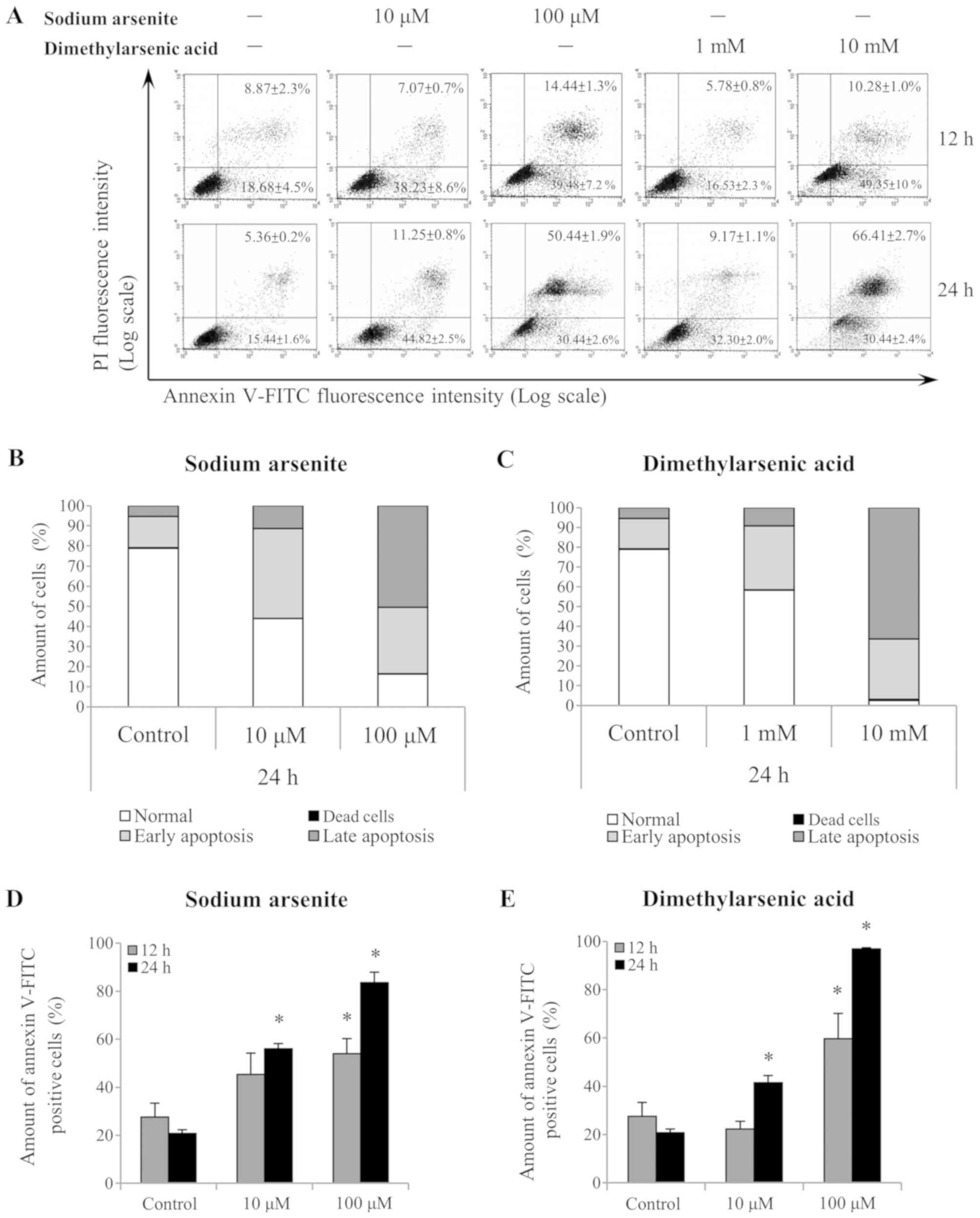
Annexin V/PI double-staining was used to refine the
apoptosis stages observed in MA-10 cells (Fig. 3A). A shift in alive (Annexin
V−/PI−), dead (Annexin
V−/PI+), early apoptotic (Annexin
V+/PI−) and late apoptotic (Annexin
V+/PI+) cell populations was observed
following 24 h treatment with 10 and 100 µM sodium arsenite, and
with 1 and 10 mM DMA (Fig. 3B and C,
respectively). Notably, the differences in the Annexin
V+-stained cells (early and late apoptotic status) were
further analyzed following sodium arsenite (Fig. 3D) and DMA (Fig. 3E) treatments. The results
demonstrated that both arsenic compounds significantly induced
MA-10 cell apoptosis following 24 h, as indicated by the increased
ratio of Annexin V+ cells (P<0.05). These results
further suggested that sodium arsenite and DMA may be cytotoxic to
Leydig cancer cells, particularly DMA.
Arsenic-induced MA-10 cell apoptosis
is mediated by caspase cascade
The caspase cascades are essential regulators of the
apoptotic signaling transduction pathway (12). Activation of these cascades triggers
PARP cleavage of its 85 kDa C-terminal fragment. PARP cleavage,
which is a common apoptosis marker, follows caspase-3 activation
and cleavage (35). To investigate
whether arsenic compounds affect the extrinsic and/or intrinsic
apoptotic pathways, the protein levels of cleaved (activated)
caspases-3, −8 and −9 were assessed by western blotting. The
results demonstrated that 12 and 24 h treatment with 10 µM sodium
arsenite significantly increased cleaved caspase-8 and −3 protein
levels (P<0.05; Fig. 4A, B and
D). In addition, 24 h treatment with 10 µM sodium arsenite
significantly increased the cleaved caspase-9 protein level
(P<0.05; Fig. 4A and C).
Furthermore, 12 h treatment with 10 µM sodium arsenite
significantly increased the cleaved PARP protein level (P<0.05;
Fig. 4A and E). Treatment with 1 mM
DMA for 24 h significantly increased the cleaved caspase-8 protein
level (P<0.05; Fig. 5A and B). In
addition, 24 h treatment with 1 and 10 mM DMA significantly
elevated the cleaved caspase-9 protein level (P<0.05; Fig. 5A and C). Furthermore, 12 and 24 h
treatment with 10 mM DMA significantly increased the cleaved
caspase-3 protein level (P<0.05; Fig.
5A and D). Finally, treatment with 10 mM DMA for 12 h
significantly increased the cleaved PARP protein level (P<0.05;
Fig. 5A and E). These results
demonstrated that arsenic compounds are involved in the intrinsic
and extrinsic apoptotic pathways in MA-10 cancer cells. In
addition, these results further indicated that sodium arsenite was
more potent than DMA in terms of activating caspase pathways in
MA10 cells.
Caspase inhibitor reverses
arsenic-induced MA-10 cell apoptosis
The membrane-permeable and irreversible pancaspase
inhibitor Z-VAD-fmk was used to determine whether the cleavage of
caspases −3, −8 and −9 could be abolished in arsenicinduced MA10
cell apoptosis. To do so, cells were pretreated with increasing
concentrations of Z-VAD-fmk (0.1, 1, 10 and 100 µM) for 2 h,
followed by 10 µM sodium arsenite or 1 mM DMA treatment for 24 h.
The results from western blotting demonstrated that 10 and 100 µM
Z-VAD-fmk treatment significantly reduced caspase-8 cleavage
induced by 10 µM sodium arsenite treatment (P<0.05; Fig. 6A and B), compared with 10 µM sodium
arsenite treatment alone. In addition, treatment with 0.1–100 µM
Z-VAD-fmk significantly reduced caspase-9 protein cleavage
(P<0.05; Fig. 6A and C), and
treatment with 100 µM Z-VAD-fmk significantly decreased caspase-3
protein activation (P<0.05; Fig. 6A
and D), compared with 10 µM sodium arsenite treatment alone.
Furthermore, 0.1, 10, and 100 µM Z-VAD-fmk treatment significantly
reduced caspase-8 activation (P<0.05; Fig. 7A and B) induced by 1 mM DMA, compared
with DMA alone. When used at a concentration range of 0.1–100 µM,
Z-VAD-fmk significantly decreased caspase-9 cleavage (P<0.05;
Fig. 7A and C) induced by 1 mM DMA,
compared with DMA alone. In addition, treatment with 100 µM
Z-VAD-fmk significantly decreased caspase-3 cleavage (P<0.05;
Fig. 7A and D) induced by 1 mM DMA,
compared with DMA alone. These results suggested that the caspase
cascades may be crucial in arsenicinduced MA-10 cell apoptosis.
Discussion
The results from the present study demonstrated that
sodium arsenite and DMA induced MA-10 cell apoptosis through
caspase pathway activation. The morphological analysis revealed
that arsenictreated MA-10 cells became more closely associated with
each other and exhibited membrane blebbing, a more rounded
appearance and detachment, findings which were consistent with a
previous study (15). It has been
reported that arsenite blocks the guanosine-5′-triphosphate binding
site of tubulin, which leads to microtubule polymerization
disruption during mitosis (36).
This may explain how arsenic compounds could target the
cytoskeleton and cause an enlarged and flattened plasma membrane
appearance, which was observed in MA-10 cells from the present
study. In addition, the toxicity of arsenic compounds varies,
according to the cellular function under consideration. For
example, AsO2- can alter the activity of cysteine-rich
enzymes by interacting with sulfhydryl groups (37). However, methylated arsenics are
metabolites of liver detoxification, which reduces acute arsenic
toxicity (38). In the present
study, cell viability was significantly decreased to 45% following
12 h treatment with 10 µM sodium arsenite, and to 48% following 12
h treatment with 10 mM DMA. These results demonstrated that sodium
arsenite was 1,000-fold more potent than DMA in inducing MA-10 cell
toxicity. These findings were comparable with those of other
studies, where inorganic arsenite compounds were more toxic than
inorganic arsenite compounds (36–38).
Cell cycle regulation is crucial in mammalian cells,
and cell cycle checkpoints, including G1 and
G2/M, are key regulators that ensure appropriate DNA
replication and division (39). The
G2/M checkpoint represents the acute response to DNA
damage. Subsequently, abnormal G2/M arrest can trigger
cell apoptosis (40). It has been
reported that arsenite is involved in cell cycle disruption in
myelomonocytic leukemia cells (41).
In the present study, the cell number in the G2/M phase
was significantly increased 24 h following treatment with arsenic
compounds. A previous study indicated that arsenic trioxide can
induce G2/M arrest through p53 phosphorylation and
increased p21 expression in TM4 Sertoli cells (6). Arsenic-induced MA-10 cell apoptosis may
therefore be associated with abnormal cell cycle distribution, and
it would be interesting to further investigate the mechanism of
cell cycle redistribution induced by arsenic compounds in MA-10
cells.
Previous studies have reported that arsenic trioxide
induces apoptosis through caspase-9 activation in cultured myeloma
(42) and ovarian cancer cells
(43). Furthermore, arsenic trioxide
induces apoptosis of human keratinocytes via the Fas/FasL pathway
(21). In MA-10 cells, caspase-8 and
−9 activation was observed following treatment with DMA and sodium
arsenite for 12 or 24 h, respectively. In addition, caspase
inhibition reversed arsenicinduced MA-10 cell apoptosis. However,
caspase-3 cleavage was re-activated when the cleavage of caspase-8
and −9 was inhibited following treatment with 0.1–10 µM Z-VAD-fmk.
This observation, at present, is difficult to explain, and
therefore merits being investigated in further studies. Treatment
with 100 µM Z-VAD-fmk decreased cleavedcaspase-8, −9 and −3 protein
levels. These results therefore demonstrated that both arsenic
compounds activated the intrinsic and extrinsic apoptotic pathways
and induced MA-10 cell apoptosis, which had been reported in
previous studies (21,42,43).
Taken together, the results from the present study
indicated that arsenic compounds induce apoptosis in vitro.
These observations, particularly the higher potency of sodium
arsenite, suggest that arsenic compounds may have potential
anti-tumorigenic therapeutic application in order to improve the
outcome of patients with testicular cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Ministry of
Science and Technology (grant no. MOST 105-2320-B-006-001).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YFM and YHC contributed to conduct the experiments,
figure generations and statistical analysis. MMC designed the
experiment and wrote the manuscript. YCC and BMH contributed to
experimental design, data interpretation, writing of the
manuscript, and ensuring the accuracy and the integrity of the
work. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interest.
References
|
1
|
Styblo M, Del Razo LM, Vega L, Germolec
DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR and Thomas
DJ: Comparative toxicity of trivalent and pentavalent inorganic and
methylated arsenicals in rat and human cells. Arch Toxicol.
74:289–299. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosen BP: Biochemistry of arsenic
detoxification. FEBS Lett. 529:86–92. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J,
Cai X, Han ZG, Ni JH, Shi GY, Jia PM, et al: Use of arsenic
trioxide (As2O3) in the treatment of acute
promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual
effects on APL cells. Blood. 89:3345–3353. 1997.PubMed/NCBI
|
|
4
|
Singh ZN, Duong VH, Koka R, Zou Y, Sawhney
S, Tang L, Baer MR, Ambulos N, El Chaer F and Emadi A: High-risk
acute promyelocytic leukemia with unusual T/Myeloid immunophenotype
successfully treated with ATRA and arsenic trioxide-based regimen.
J Hematop. 11:67–74. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Uslu R, Sanli UA, Sezgin C, Karabulut B,
Terzioglu E, Omay SB and Goker E: Arsenic trioxide-mediated
cytotoxicity and apoptosis in prostate and ovarian carcinoma cell
lines. Clin Cancer Res. 6:4957–4964. 2000.PubMed/NCBI
|
|
6
|
Kim YJ, Chung JY, Lee SG, Kim JY, Park JE,
Kim WR, Joo BS, Han SH, Yoo KS, Yoo YH and Kim JM: Arsenic
trioxide-induced apoptosis in TM4 Sertoli cells: The potential
involvement of p21 expression and p53 phosphorylation. Toxicology.
285:142–151. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang H, Jin Y, Jin S, Tan Z, Peng Z and
Kuang Y: Arsenite inhibits the function of CD133+ CD13+ liver
cancer stem cells by reducing PML and Oct4 protein expression.
Tumour Biol. 37:14103–14115. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dilda PJ and Hogg PJ: Arsenical-based
cancer drugs. Cancer Treat Rev. 33:542–564. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Don AS, Kisker O, Dilda P, Donoghue N,
Zhao X, Decollogne S, Creighton B, Flynn E, Folkman J and Hogg PJ:
A peptide trivalent arsenical inhibits tumor angiogenesis by
perturbing mitochondrial function in angiogenic endothelial cells.
Cancer Cell. 3:497–509. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duzkale H, Jilani I, Orsolic N, Zingaro
RA, Golemovic M, Giles FJ, Kantarjian H, Albitar M, Freireich EJ
and Verstovsek S: In vitro activity of dimethylarsinic acid against
human leukemia and multiple myeloma cell lines. Cancer Chemother
Pharmacol. 51:427–432. 2003.PubMed/NCBI
|
|
11
|
Velloso FJ, Bianco AF, Farias JO, Torres
NE, Ferruzo PY, Anschau V, Jesus-Ferreira HC, Chang TH, Sogayar MC,
Zerbini LF and Correa RG: The crossroads of breast cancer
progression: Insights into the modulation of major signaling
pathways. Onco Targets Ther. 10:5491–5524. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Derakhshan A, Chen Z and Van Waes C:
Therapeutic small molecules target inhibitor of apoptosis proteins
in cancers with deregulation of extrinsic and intrinsic cell death
pathways. Clin Cancer Res. 23:1379–1387. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lam M, Lawrence DA, Ashkenazi A and Walter
P: Confirming a critical role for death receptor 5 and caspase-8 in
apoptosis induction by endoplasmic reticulum stress. Cell Death
Differ. 25:1530–1531. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zimmermann KC, Bonzon C and Green DR: The
machinery of programmed cell death. Pharmacol Ther. 92:57–70. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Taylor RC, Cullen SP and Martin SJ:
Apoptosis: Controlled demolition at the cellular level. Nat Rev.
9:231–241. 2008. View
Article : Google Scholar
|
|
16
|
Cory S and Adams JM: The Bcl2 family:
Regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Antonsson B, Montessuit S, Sanchez B and
Martinou JC: Bax is present as a high molecular weight
oligomer/complex in the mitochondrial membrane of apoptotic cells.
J Biol Chem. 276:11615–11623. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang DC and Strasser A: BH3-Only
proteins-essential initiators of apoptotic cell death. Cell.
103:839–842. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Andreu-Fernández V, García-Murria MJ,
Bañó-Polo M, Martin J, Monticelli L, Orzáez M and Mingarro I: The
C-terminal domains of apoptotic BH3-only proteins mediate their
insertion into distinct biological membranes. J Biol Chem.
291:25207–25216. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Morales AA, Gutman D, Lee KP and Boise LH:
BH3-only proteins Noxa, Bmf, and Bim are necessary for arsenic
trioxide-induced cell death in myeloma. Blood. 111:5152–5262. 2009.
View Article : Google Scholar
|
|
21
|
Liao WT, Chang KL, Yu CL, Chen GS, Chang
LW and Yu HS: Arsenic induces human keratinocyte apoptosis by the
FAS/FAS ligand pathway, which correlates with alterations in
NF-kappaB and AP-1 activity. J Invest Dermatol. 122:125–129. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huyghe E, Matsuda T and Thonneau P:
Increasing incidence of testicular cancer worldwide: A review. J
Urol. 170:5–11. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bertram KA, Bratloff B, Hodges GF and
Davidson H: Treatment of malignant Leydig cell tumor. Cancer.
68:2324–2329. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chang MM, Lai MS, Hong SY, Pan BS, Huang
H, Yang SH, Wu CC, Sun HS, Chuang JI, Wang CY and Huang BM:
FGF9/FGFR2 increase cell proliferation by activating ERK1/2,
Rb/E2F1, and cell cycle pathways in mouse Leydig tumor cells.
Cancer Sci. 109:3503–3518. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Segaloff DL, Ascoli M and Puett D:
Characterization of the desensitized state of Leydig tumor cells.
Biochim Biophys Acta. 675:351–358. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu WC, Hsiao JR, Lian YY, Lin CY and Huang
BM: The apoptotic effect of cordycepin on human OEC-M1 oral cancer
cell line. Cancer Chemother Pharmacol. 60:103–111. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Green LM, Reade JL and Ware CF: Rapid
colorimetric assay for cell viability: Application to the
quantitation of cytotoxic and growth inhibitory lymphokines. J
Immunol Methods. 70:257–268. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Riccardi C and Nicoletti I: Analysis of
apoptosis by propidium iodide staining and flow cytometry. Nat
Protoc. 1:1458–1461. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kang FC, Wang SC, Chang MM, Pan BS, Wong
KL, Cheng KS, So EC and Huang BM: Midazolam activates caspase,
MAPKs and ER stress pathways, and inhibits cell cycle and Akt
pathway, to induce apoptosis in TM3 mouse Leydig progenitor cells.
Onco Targets Ther. 11:1475–1490. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
So EC, Chen YC, Wang SC, Wu CC, Huang MC,
Lai MS, Pan BS, Kang FC and Huang BM: Midazolam regulated caspase
pathway, endoplasmic reticulum stress, autophagy, and cell cycle to
induce apoptosis in MA-10 mouse Leydig tumor cells. Onco Targets
Ther. 9:2519–2533. 2016.PubMed/NCBI
|
|
31
|
Lowry OH, Rosebrough NJ, Farr AL and
Randall RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
32
|
Chen YH, Leu SF, Jen CY and Huang BM:
Effects of sesamol on apoptosis and steroidogenesis in MA-10 mouse
Leydig tumor cells. J Agric Food Chem. 59:9885–9891. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pao HY, Pan BS, Leu SF and Huang BM:
Cordycepin stimulated steroidogenesis in MA-10 mouse Leydig tumor
cells through the protein kinase C Pathway. J Agric Food Chem.
60:4905–4913. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen YC, Chen YH, Pan BS, Chang MM and
Huang BM: Functional study of Cordyceps sinensis and cordycepin in
male reproduction: A review. J Food Drug Anal. 25:197–205. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mullen P: PARP cleavage as a means of
assessing apoptosis. Methods Mol Med. 88:171–181. 2004.PubMed/NCBI
|
|
36
|
Hassani S, Khaleghian A, Ahmadian S,
Alizadeh S, Alimoghaddam K, Ghavamzadeh A and Ghaffari SH:
Redistribution of cell cycle by arsenic trioxide is associated with
demethylation and expression changes of cell cycle related genes in
acute promyelocytic leukemia cell line (NB4). Ann Hematol.
97:83–93. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Snow ET: Metal carcinogenesis: Mechanistic
implications. Pharmacol Ther. 53:31–65. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Styblo M, Del Razo LM, LeCluyse EL,
Hamilton GA, Wang C, Cullen WR and Thomas DJ: Metabolism of arsenic
in primary cultures of human and rat hepatocytes. Chem Res Toxicol.
12:560–565. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hong SK, Wu PK and Park JI: A cellular
threshold for active ERK1/2 levels determines Raf/MEK/ERK-mediated
growth arrest versus death responses. Cell Signal. 42:11–20. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Diepart C, Karroum O, Magat J, Feron O,
Verrax J, Calderon PB, Gregoire V, Leveque P, Stockis J, Dauguet N,
et al: Arsenic trioxide treatment decreases the oxygen consumption
rate of tumor cells and radiosensitizes solid tumors. Cancer Res.
72:482–490. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
McCabe MJ, Singh KP, Reddy SA, Chelladurai
B, Pounds JG, Reiners JJ and States JC: Sensitivity of
myelomonocytic leukemia cells to arsenite-induced cell cycle
disruption, apoptosis, and enhanced differentiation is dependent on
the inter-relationship between arsenic concentration, duration of
treatment, and cell cycle phase. J Pharmacol Exp Ther. 295:724–733.
2000.PubMed/NCBI
|
|
42
|
Hayashi T, Hideshima T, Akiyama M,
Richardson P, Schlossman RL, Chauhan D, Munshi NC, Waxman S and
Anderson KC: Arsenic trioxide inhibits growth of human multiple
myeloma cells in the bone marrow microenvironment. Mol Cancer Ther.
1:851–860. 2002.PubMed/NCBI
|
|
43
|
Yuan Z, Wang F, Zhao Z, Zhao X, Qiu J, Nie
C and Wei Y: BIM-mediated AKT phosphorylation is a key modulator of
arsenic trioxide-induced apoptosis in cisplatin-sensitive and
-resistant ovarian cancer cells. PLoS One. 6:e205862011. View Article : Google Scholar : PubMed/NCBI
|