Introduction
Non-small cell lung cancer (NSCLC) is the leading
cause of cancer-associated mortality worldwide, and adenocarcinoma
(ADC) is its most common histological subtype (1). Despite multiple treatment modalities,
NSCLC is commonly associated with unfavorable outcomes, and has a
5-year survival rate of <20% (1).
Several factors are known to contribute to the poor prognosis of
patients with NSCLC, including late diagnosis of disease and a lack
of effective drugs (2). NSCLCs are a
clinically and molecularly heterogeneous group of diseases, and
survival outcome or treatment response varies among individuals
(3). Therefore, the absence of
clinically informative biomarkers for stratifying different risk
subgroups or guiding targeted treatment decisions is also notable.
Efforts to identify potential biomarkers have been made, with a
focus on genetic alterations including somatic mutations, copy
number variations and gene expression; however, few are suitable
for routine use in the field of NSCLC treatment (3–5).
Epigenetic changes, and particularly those at the
DNA methylation level, are implicated in tumor initiation and
progression (6). Hypermethylation of
CpG islands (CGI) at the promoter regions of tumor-suppressor genes
and consequent transcriptional silencing represents the best-known
epigenetic event in cancer biology (6). As a novel molecular candidate for
cancer biomarker discovery, DNA methylation has numerous advantages
over the genetic alteration- or gene expression-based biomarkers
for clinical application, including reliable DNA samples, stable
methylation changes, informative biological relevance and
drug-induced reversibility (7).
Early efforts with candidate-gene approaches have identified a
number of useful prognostic biomarkers based on the CGI methylation
status of key genes, including Ras association domain family 1
isoform A (RASSF1A), runt-related transcription factor 3,
and deleted in esophageal and lung cancer 1, in NSCLC (8). Unfortunately, these single-gene
methylation events were unable to demonstrate consistent prognostic
ability in independent validation studies, and therefore have not
effected a real change in routine practice (8). High-throughput genome-wide DNA
methylation profiling techniques have been increasingly used for
the detection of the cancer genome markers. These methods may
provide a comprehensive and unbiased identification of prognostic
DNA methylation events throughout the epigenome, eventually leading
to the improvement of personalized medicine for NSCLC (3).
The present study aimed to identify clinically
useful epigenetic biomarkers from lung ADC-specific CGI methylation
changes at different gene regions using genome-wide DNA methylation
microarray data of lung ADCs and matched normal tissues from 6
publically available datasets. Accordingly, a 9-CpG CGI methylation
panel and hypomethylation/overexpression of ribosomal protein 39
like (RPL39L) were identified, which may be of potential
value for optimizing the risk stratification and personalized
management of lung ADCs.
Materials and methods
Public datasets
The Cancer Genome Atlas (TCGA)
Genome-wide DNA methylation data and corresponding
clinical information were retrieved from TCGA data portal
(https://tcga-data.nci.nih.gov/tcga/,
accessed March 2016), including a dataset of 65 lung ADCs
[female/male, 35/30; Tumor-Node-Metastasis (TNM) staging, I to IV
(1); median age, 67 years; age
range, 38–84 years] and 24 matched non-tumor lung samples assayed
using an Illumina Infinium 27k BeadChip system (TCGA-27k set) and a
dataset of 456 tumor samples [female/male, 244/212; TNM staging, I
to IV (1); median age, 66 years; age
range, 33–88 years] and 29 matched normal samples assayed using a
Illumina Infinium 450k BeadChip system (TCGA-450k set) (3). There were Infinium 27k and 450k DNA
methylation data for 6 tumor samples. For the transcriptome data,
Level 3 Illumina HiSeq_RNASeqV2 data were obtained for all tumor
samples from the TCGA-27k set, and for 452 tumor samples and 58
matched normal samples from the TCGA-450k set. Among the
aforementioned TCGA datasets, Level 2 IlluminaGA_DNASeq data were
also available for 490 samples, and Level 3 Affymetrix
Genome_Wide_SNP_6 data for 512 samples. Somatic copy number data
were analyzed within the GISTIC2.0 module on GenePattern
(http://genepattern.broadinstitute.org/gp/; accessed
March 2016). An amplitude threshold of ±0.2 was used.
Gene Expression Omnibus (GEO)
Genome-wide DNA methylation microarray data were
also obtained from 4 GEO series (https://www.ncbi.nlm.nih.gov/geo/; access at March
2016), including: i) A dataset of 59 matched lung ADCs
[female/male, 45/14; TNM stage I to IV (1); median age, 68 years; age range, 39–86
years] and non-tumor lung samples [accession no. GSE32861; Selamat
et al set (9)]; ii) a dataset
of 26 matched tumor [female/male, 14/12; TNM stage I to IV
(1); median age, unknown] and normal
lung samples [accession no. GSE32866; Ontario Tumor Bank set
(9)]; iii) a dataset of 28 matched
tumor [female/male, 22/6; TNM stage I to IV (1); median age, 65 years; age range,
unknown] and normal lung samples of never-smokers [accession no.
GSE62948; Mansfield et al set (10)]; and iv) a dataset of 35 matched
tumors [female/male, 19/16; TNM stage I to II (1); median age, 63 years; age range, 47–88
years] and normal lung samples of patients with lung ADCs
[accession no. GSE63384; Robles et al set (11)].
Ethical approval
All procedures performed in studies involving humans
were conducted in accordance with the ethical standards of the
institutional research committees and with the 1964 Declaration of
Helsinki and its later amendments or comparable ethical standards.
Informed consent was obtained from all individual participants as
reported by included datasets (3,9–11).
Microarray data processing
For the Level 3 DNA methylation microarray data
(Infinum BeadChips, Illumina Inc.), the methylation level of each
interrogated CpG locus was summarized as a β-value, providing a
continuous and quantitative index of DNA methylation, ranging from
0 (completely unmethylated) to 1 (completely methylated). To ensure
that β-values were comparable across each dataset/platform, batch
effects were adjusted by a non-parametric empirical Bayes approach
(ber R package; version 3.2.5; http://www.r-project.org/) (12–14). The
empirical Bayes correction was demonstrated to effectively remove
batch effects following initial microarray data normalization
(12,13). M-value transformation was applied
prior to the batch effect adjustment to avoid a negative β-value,
as described previously (15). For
the gene-level analysis of the Level 3 Illumina HiSeq_RNASeqV2
data, expression values of 0 were set as the overall minimum value,
and all data were log2 transformed and standardized to
z-scores within each gene. All missing values were imputed by
nearest neighbor averaging (impute R package) (3).
Cancer-specific CGI methylation loci
and their correlation with gene expression
The CpG probes interrogated by the Infinium 27k and
450k platforms were maintained for analysis, and were annotated
using the Infinium Human Methylation 450k annotation file. Prior
selection of CpGs probes was performed by removal of those that: i)
Targeted the X and Y chromosomes; ii) contained a single-nucleotide
polymorphism within 5 base pairs of and including the targeted
CpGs; and iii) were not located at CGI regions of a gene; CGI was
defined by the UCSC genome reference (http://genome.ucsc.edu/; accessed March 2016). For
CpGs corresponding to multiple annotation terms, the first one in
the 450k annotation file were used in the present study, to
simplify data interpretation. Finally, a total of 9,270 CpG probes
were included for additional analysis. Differentially methylated
CpGs were computed by two-sample Wilcoxon test (samr R
package). Lung ADC-specific CpGs were defined as those having a
median β difference ≥0.2 between matched tumor and non-tumor lung
samples and a false discovery rate (FDR) q-value ≤0.05 in at least
4 of the 6 datasets. Methylation and expression data were paired
based on each Entrez Gene ID (https://www.ncbi.nlm.nih.gov/gene/; accessed March
2016). The correlation between methylation and expression level of
each gene was evaluated by Pearson's correlation analysis, and
those having an absolute Pearson correlation coefficient (r)≥0.3,
0.2–0.3, or 0.1–0.2 and P≤0.05 were defined as strong, moderate or
weak correlations, respectively.
Construction and validation of a CGI
methylation-based risk score signature
The training-validation approach was used to
construct a prognostic CGI methylation signature. The training
phase was performed using the TCGA-450k set, where the methylation
levels of lung ADC-specific CpGs were correlated with overall
survival (OS) time by univariate Cox regression analysis with
permutation correction within the Biometric Research Branch-Array
Tools (http://brb.nci.nih.gov/BRB-ArrayTools, accessed March
2016). Those that exhibited significant correlation with OS
(permutation P≤0.05), and high variability [standard deviation
(SD)≥0.10] were finally selected as prognostic methylation
candidates. Probes with a higher SD variability indicated that the
interrogated CpGs loci may have more opportunities to be
dysregulated across tumors. These CpGs may therefore be more likely
to serve roles in tumor biology, and the alterations in those CpGs
may be easier to detect (16). The
prognostic model was established by risk-score analysis, where each
patient was assigned a risk score that is a linear combination of
the methylation levels of each CpG weighted by their corresponding
Cox regression coefficients (17).
The median risk score (3.08) from the training set was
pre-specified as cut-off for stratifying low-risk and high-risk
subgroups. The validation phase was performed on the aforementioned
TCGA-27k (3) and Robles et al
(11) datasets. An additional
dataset of patients with lung ADC [female/male, 127/125; TNM stage
I to IV (1); median age, 65 years;
age range, 40–90 years] with relapse-free survival (RFS) time was
also included for independent validation [accession no. GSE39279;
n=252; Sandoval et al set (2)].
Database for Annotation, Visualization
and Integrated Discovery (DAVID) annotation clustering
analysis
DAVID (version 6.7; https://david.ncifcrf.gov/; accessed March 2016)
(18) was used to create functional
annotation for genes corresponding to cancer-specific
differentially-methylated CpGs with Gene Ontology (19), BioCarta (20) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway tools (21).
Statistical analysis
Survival data were estimated by the Kaplan-Meier
method, and compared using the log-rank test. Survival data were
summarized as median OS time or RFS time. The associations between
variables and survival data were evaluated using the univariate Cox
regression model. A multivariate Cox regression model was used to
evaluate the independence of each potential prognostic indicator by
incorporating those significant variables from the univariate Cox
model. Pooled survival data were analyzed by meta-analysis with the
inverse-variance method, where either fixed- or random-effect
models were used on the basis of the intra-dataset heterogeneity.
Heterogeneity was analyzed using the χ2 test and
I2 statistic, with Pheterogeneity <0.1 or
I2 value >50% being considered significant. When
integrating the DNA methylation and gene expression data of
RPL39L, the optimal cut-off values to segregate patients
into poor and good prognostic subgroups were determined by the
maximally selected rank statistics, as described previously
(22). All calculations were
performed with SPSS v19.0 (SPSS Software, Inc., Chicago, IL, USA)
and R software version 3.2.5, and P≤0.05 was considered to indicate
a statistically significant difference.
Results
Identification of cancer-specific CGI
methylation loci in lung ADCs
By comparing the genome-wide DNA methylation data of
matched lung ADCs and non-tumor tissues from the 6 included
datasets, a total of 134 CGI loci (corresponding to 119 genes) that
met the study criteria of lung ADCs-specific methylation changes
were identified. Almost all of these CGI CpGs gained DNA
methylation, whereas only 3 loci ([cg07693270 (RPL39L);
cg24898753 (ferritin heavy chain 1); and cg06038133 (CORO6)]
were hypomethylated in lung ADCs (Table
I). DAVID annotation analysis (18) revealed that those cancer-specific
methylation changes often affected genes with roles in the
regulation of transcription (49 genes, P=3.60×10−10),
cell-cell signaling (17 genes, P=1.29×10−5) and cell
surface receptor linked signal transduction (22 genes, P=0.042).
Furthermore, by integrating TCGA gene expression data, it was
identified that the methylation levels of 27 (20%), 23 (17%) and 45
(34%) CpGs exhibited strong, moderate and weak correlations with
their gene expressions, respectively. Accordingly, among those that
were strongly associated with DNA methylation (n=82), 64 genes
(78%) were differentially expressed between tumor and non-tumor
tissues. In summary, ADC-specific CGI loci, and those with
corresponding gene expression aberrations in particular, may serve
as potential biomarker candidates with diagnostic and prognostic
possibilities.
 | Table I.Characteristics of the identified
lung ADC-specific CpGs at CpG island regions. |
Table I.
Characteristics of the identified
lung ADC-specific CpGs at CpG island regions.
| Probes | Chr. | Symbols | Gene ID | Association with
CpG island | Association with
gene region | Methylation status
between tumor and normal tissues | Pearson
coefficients between DNA methylation and gene
expressiona | Log2
fold change between tumor and normal tissuesb |
|---|
| cg18335068 | 19 | ZNF677 | 342926 | Island | 5′UTR |
Hypermethylation | −0.603 | −0.860 |
| cg08089301 | 17 | HOXB4 | 3214 | Island | 1stExon |
Hypermethylation | −0.536 | −0.326 |
| cg04317399 | 7 | HOXA4 | 3201 | Island | 1stExon |
Hypermethylation | −0.492 | −1.446 |
| cg07533148 | 1 | TRIM58 | 25893 | Island | 1stExon |
Hypermethylation | −0.475 | −1.407 |
| cg07703401 | 16 | HBQ1 | 3049 | Island | 1stExon |
Hypermethylation | −0.461 | −0.552 |
| cg23432345 | 7 | HOXA7 | 3204 | Island | 1stExon |
Hypermethylation | −0.436 | −0.648 |
| cg12880658 | 5 | CDO1 | 1036 | Island | 1stExon |
Hypermethylation | −0.414 | −1.599 |
| cg02919422 | 8 | SOX17 | 64321 | Island | 5′UTR |
Hypermethylation | −0.410 | −1.527 |
| cg25875213 | 19 | ZNF781 | 163115 | Island | 5′UTR |
Hypermethylation | −0.402 | −1.279 |
| cg14458834 | 17 | HOXB4 | 3214 | Island | 1stExon |
Hypermethylation | −0.389 | −0.326 |
| cg10088985 | 4 | CXCL5 | 6374 | Island | 1stExon |
Hypermethylation | −0.375 | −0.392 |
| cg04048259 | 20 | EDN3 | 1908 | Island | TSS200 |
Hypermethylation | −0.363 | −1.415 |
| cg04062391 | 19 | ZNF560 | 147741 | Island | 5′UTR |
Hypermethylation | −0.341 | Not
Significant |
| cg16428251 | 3 | SOX14 | 8403 | Island | TSS200 |
Hypermethylation | −0.341 | Not
Significant |
| cg07621046 | 10 |
C10orf82 | 143379 | Island | TSS200 |
Hypermethylation | −0.337 | −0.355 |
| cg18536148 | 17 | TBX4 | 9496 | Island | 5′UTR |
Hypermethylation | −0.332 | −1.540 |
| cg23290344 | 8 | NEFM | 4741 | Island | TSS1500 |
Hypermethylation | −0.328 | −0.336 |
| cg21233722 | 5 | DOCK2 | 1794 | Island | Body |
Hypermethylation | −0.325 | −0.946 |
| cg14384532 | 15 | NTRK3 | 4916 | Island | TSS1500 |
Hypermethylation | −0.322 | −1.120 |
| cg02008154 | 7 | TBX20 | 57057 | Island | 1stExon |
Hypermethylation | −0.320 | Not
Significant |
| cg21546671 | 17 | HOXB4 | 3214 | Island | 1stExon |
Hypermethylation | −0.319 | −0.326 |
| cg19885761 | 5 | CPLX2 | 10814 | Island | 5′UTR |
Hypermethylation | −0.318 | −0.476 |
| cg03734874 | 14 | TMEM179 | 388021 | Island | TSS1500 |
Hypermethylation | −0.313 | 0.530 |
| cg17525406 | 1 | AJAP1 | 55966 | Island | Body |
Hypermethylation | −0.295 | −0.943 |
| cg20616414 | 9 | WNK2 | 65268 | Island | 1stExon |
Hypermethylation | −0.288 | 0.946 |
| cg10235817 | 4 | ADRA2C | 152 | Island | 1stExon |
Hypermethylation | −0.269 | −1.000 |
| cg10141715 | 12 | SLC5A8 | 160728 | Island | 1stExon |
Hypermethylation | −0.254 | −0.829 |
| cg00015770 | 4 | QRFPR | 84109 | Island | 1stExon |
Hypermethylation | −0.246 | Not
Significant |
| cg07536847 | 1 | PAX7 | 5081 | Island | TSS1500 |
Hypermethylation | −0.237 | 0.777 |
| cg25484904 | 4 | CWH43 | 80157 | Island | TSS1500 |
Hypermethylation | −0.237 | −0.971 |
| cg13870866 | 7 | TBX20 | 57057 | Island | 1stExon |
Hypermethylation | −0.235 | Not
Significant |
| cg06092815 | 2 | SPHKAP | 80309 | Island | TSS200 |
Hypermethylation | −0.233 | −0.977 |
| cg23710218 | 8 | MSC | 9242 | Island | 1stExon |
Hypermethylation | −0.225 | 0.735 |
| cg12111714 | 13 | ATP8A2 | 51761 | Island | Body |
Hypermethylation | −0.220 | −1.184 |
| cg00548268 | 7 | NPTX2 | 4885 | Island | TSS1500 |
Hypermethylation | −0.215 | 0.843 |
| cg21376883 | 1 | ACTN2 | 88 | Island | Body |
Hypermethylation | −0.213 | −1.639 |
| cg08441806 | 10 | NKX6-2 | 84504 | Island | 1stExon |
Hypermethylation | −0.212 | −0.576 |
| cg20959866 | 1 | AJAP1 | 55966 | Island | TSS1500 |
Hypermethylation | −0.211 | −0.943 |
| cg00662556 | 18 | GALR1 | 2587 | Island | Body |
Hypermethylation | −0.211 | −0.322 |
| cg20792062 | 12 | KCNA5 | 3741 | Island | 5′UTR |
Hypermethylation | −0.211 | −1.276 |
| cg10556064 | 16 | SMPD3 | 55512 | Island | 5′UTR |
Hypermethylation | −0.206 | −0.405 |
| cg20291049 | 2 | POU3F3 | 5455 | Island | 1stExon |
Hypermethylation | −0.200 | 0.385 |
| cg12614105 | 7 | NPY | 4852 | Island | 5′UTR |
Hypermethylation | −0.195 | Not
Significant |
| cg09619146 | 10 | CPXM2 | 119587 | Island | 1stExon |
Hypermethylation | −0.193 | Not
Significant |
| cg04490714 | 16 | SLC6A2 | 6530 | Island | 1stExon |
Hypermethylation | −0.190 | −0.375 |
| cg13929328 | 10 | FOXI2 | 399823 | Island | 1stExon |
Hypermethylation | −0.189 | −0.781 |
| cg18081258 | 14 | NDRG2 | 57447 | Island | TSS1500 |
Hypermethylation | −0.188 | −1.450 |
| cg15343119 | 18 | GALR1 | 2587 | Island | TSS1500 |
Hypermethylation | −0.187 | −0.322 |
| cg00891541 | 16 | SMPD3 | 55512 | Island | 5′UTR |
Hypermethylation | −0.187 | −0.405 |
| cg10486998 | 18 | GALR1 | 2587 | Island | TSS1500 |
Hypermethylation | −0.187 | −0.322 |
| cg21245652 | 2 | MAL | 4118 | Island | TSS1500 |
Hypermethylation | −0.181 | −1.204 |
| cg06675478 | 13 | SOX1 | 6656 | Island | TSS200 |
Hypermethylation | −0.178 | 0.352 |
| cg26721264 | 18 | GALR1 | 2587 | Island | TSS1500 |
Hypermethylation | −0.178 | −0.322 |
| cg18952647 | 15 | BNC1 | 646 | Island | TSS1500 |
Hypermethylation | −0.177 | −0.498 |
| cg01683883 | 16 | CMTM2 | 146225 | Island | TSS1500 |
Hypermethylation | −0.175 | −1.336 |
| cg06722633 | 1 | GRIK3 | 2899 | Island | Body |
Hypermethylation | −0.175 | Not
Significant |
| cg25942450 | 5 | TLX3 | 30012 | Island | TSS200 |
Hypermethylation | −0.173 | 0.474 |
| cg27009703 | 7 | HOXA9 | 3205 | Island | 1stExon |
Hypermethylation | −0.170 | Not
Significant |
| cg04534765 | 18 | GALR1 | 2587 | Island | 1stExon |
Hypermethylation | −0.170 | −0.322 |
| cg19064258 | 16 | HS3ST2 | 9956 | Island | 1stExon |
Hypermethylation | −0.163 | −0.265 |
| cg02164046 | 3 | SST | 6750 | Island | 1stExon |
Hypermethylation | −0.159 | Not
Significant |
| cg12768605 | 19 | LYPD5 | 284348 | Island | TSS200 |
Hypermethylation | −0.153 | −0.346 |
| cg25720804 | 5 | TLX3 | 30012 | Island | 1stExon |
Hypermethylation | −0.153 | 0.474 |
| cg10883303 | 7 | HOXA13 | 3209 | Island | 1stExon |
Hypermethylation | −0.150 | 0.831 |
| cg12457773 | 6 | NRSN1 | 140767 | Island | 5′UTR |
Hypermethylation | −0.150 | −0.521 |
| cg14008883 | 10 | SLC18A3 | 6572 | Island | 1stExon |
Hypermethylation | −0.148 | 0.725 |
| cg03544320 | 4 | CRMP1 | 1400 | Island | 1stExon |
Hypermethylation | −0.147 | −0.610 |
| cg24199834 | 4 | POU4F2 | 5458 | Island | 1stExon |
Hypermethylation | −0.145 | Not
Significant |
| cg19456540 | 14 | SIX6 | 4990 | Island | 1stExon |
Hypermethylation | −0.144 | 0.392 |
| cg08572611 | 7 | ACTL6B | 51412 | Island | Body |
Hypermethylation | −0.142 | Not
Significant |
| cg00489401 | 5 | FLT4 | 2324 | Island | Body |
Hypermethylation | −0.133 | −1.340 |
| cg05373457 | 8 | KCNS2 | 3788 | Island | 5′UTR |
Hypermethylation | −0.133 | Not
Significant |
| cg14991487 | 2 | HOXD9 | 3235 | Island | TSS200 |
Hypermethylation | −0.123 | Not
Significant |
| cg02774439 | 4 | HAND2 | 9464 | Island | 5′UTR |
Hypermethylation | −0.122 | −0.364 |
| cg02757432 | 10 | GPR26 | 2849 | Island | 1stExon |
Hypermethylation | −0.114 | Not
Significant |
| cg25044651 | 5 | LVRN | 206338 | Island | 1stExon |
Hypermethylation | −0.112 | Not
Significant |
| cg01354473 | 7 | HOXA9 | 3205 | Island | 1stExon |
Hypermethylation | −0.112 | Not
Significant |
| cg08109815 | 6 | NMBR | 4829 | Island | 5′UTR |
Hypermethylation | −0.107 | −0.506 |
| cg10303487 | 8 | DPYS | 1807 | Island | 1stExon |
Hypermethylation | −0.107 | −0.816 |
| cg18555440 | 11 | MYOD1 | 4654 | Island | 1stExon |
Hypermethylation | −0.094 | Not
Significant |
| cg09936561 | 4 | DRD5 | 1816 | Island | 1stExon |
Hypermethylation | −0.085 | Not
Significant |
| cg14859460 | 5 | GRM6 | 2916 | Island | TSS200 |
Hypermethylation | −0.079 | Not
Significant |
| cg18722841 | 11 | PHOX2A | 401 | Island | 1stExon |
Hypermethylation | −0.079 | 0.428 |
| cg09229912 | 12 | CUX2 | 23316 | Island | 1stExon |
Hypermethylation | −0.076 | Not
Significant |
| cg20404387 | 1 | FAM43B | 163933 | Island | 1stExon |
Hypermethylation | −0.072 | 0.314 |
| cg12782180 | 7 | LEP | 3952 | Island | TSS1500 |
Hypermethylation | −0.070 | 0.987 |
| cg15489294 | 5 | LVRN | 206338 | Island | TSS1500 |
Hypermethylation | −0.068 | Not
Significant |
| cg25993718 | 20 | CBLN4 | 140689 | Island | TSS200 |
Hypermethylation | −0.067 | −0.431 |
| cg16787600 | 10 | SORCS3 | 22986 | Island | 1stExon |
Hypermethylation | −0.062 | Not
Significant |
| cg07307078 | 18 | TUBB6 | 84617 | Island | TSS1500 |
Hypermethylation | −0.059 | −1.308 |
| cg08832227 | 12 | KCNA1 | 3736 | Island | Body |
Hypermethylation | −0.058 | −0.405 |
| cg01381846 | 7 | HOXA9 | 3205 | Island | 1stExon |
Hypermethylation | −0.055 | Not
Significant |
| cg02332525 | 3 | GRM7 | 2917 | Island | 1stExon |
Hypermethylation | −0.050 | −0.368 |
| cg15748507 | 10 | PRLHR | 2834 | Island | Body |
Hypermethylation | −0.049 | Not
Significant |
| cg15191648 | 18 | SALL3 | 27164 | Island | TSS200 |
Hypermethylation | −0.048 | 0.690 |
| cg26609631 | 13 | GSX1 | 219409 | Island | 5′UTR |
Hypermethylation | −0.048 | Not
Significant |
| cg13302823 | 8 | SCRT1 | 83482 | Island | 1stExon |
Hypermethylation | −0.033 | Not
Significant |
| cg01839464 | 18 | DCC | 1630 | Island | Body |
Hypermethylation | −0.029 | −1.162 |
| cg25691167 | 7 | FERD3L | 222894 | Island | 1stExon |
Hypermethylation | −0.025 | Not
Significant |
| cg05345286 | 6 | MDFI | 4188 | Island | Body |
Hypermethylation | −0.024 | 0.960 |
| cg25574024 | 11 | IGF2AS | 51214 | Island | Body |
Hypermethylation | −0.020 | Not
Significant |
| cg11525285 | 14 | VSX2 | 338917 | Island | 1stExon |
Hypermethylation | −0.019 | −0.269 |
| cg22187630 | 19 | CACNA1A | 773 | Island | 1stExon |
Hypermethylation | −0.016 | 0.290 |
| cg21296230 | 15 | GREM1 | 26585 | Island | 5′UTR |
Hypermethylation | −0.010 | 1.327 |
| cg13791131 | 11 | IGF2AS | 51214 | Island | Body |
Hypermethylation | −0.009 | Not
Significant |
| cg01295203 | 8 | PRDM14 | 63978 | Island | TSS1500 |
Hypermethylation | 0.002 | Not
Significant |
| cg26252167 | 6 | GPR6 | 2830 | Island | 1stExon |
Hypermethylation | 0.004 | Not
Significant |
| cg13547644 | 1 | ACTA1 | 58 | Island | 5′UTR |
Hypermethylation | 0.012 | Not
Significant |
| cg22881914 | 14 | NID2 | 22795 | Island | TSS1500 |
Hypermethylation | 0.028 | 0.667 |
| cg23207990 | 4 | SFRP2 | 6423 | Island | TSS1500 |
Hypermethylation | 0.041 | 0.729 |
| cg13323752 | 12 | SLC2A14 | 144195 | Island | TSS200 |
Hypermethylation | 0.054 | −0.701 |
| cg09643544 | 19 | ZNF177 | 7730 | Island | 1stExon |
Hypermethylation | 0.064 | −0.725 |
| cg08575537 | 7 | EPO | 2056 | Island | Body |
Hypermethylation | 0.064 | −0.262 |
| cg15107670 | 11 | WT1 | 7490 | Island | 1stExon |
Hypermethylation | 0.067 | 0.569 |
| cg26186727 | 18 | NETO1 | 81832 | Island | 1stExon |
Hypermethylation | 0.086 | 1.372 |
| cg06958829 | 17 | ACSF2 | 80221 | Island | Body |
Hypermethylation | 0.091 | −0.500 |
| cg04907257 | 5 | ADCY2 | 108 | Island | TSS1500 |
Hypermethylation | 0.097 | −0.578 |
| cg21591742 | 2 | HOXD10 | 3236 | Island | TSS1500 |
Hypermethylation | 0.114 | 0.507 |
| cg03958979 | 6 | NR2E1 | 7101 | Island | TSS1500 |
Hypermethylation | 0.123 | 1.200 |
| cg02245378 | 2 | PAX3 | 5077 | Island | Body |
Hypermethylation | 0.126 | Not
Significant |
| cg14144305 | 11 | ALX4 | 60529 | Island | Body |
Hypermethylation | 0.129 | Not
Significant |
| cg25902889 | 19 | FSD1 | 79187 | Island | Body |
Hypermethylation | 0.141 | 0.842 |
| cg22660578 | 17 | LHX1 | 3975 | Island | TSS1500 |
Hypermethylation | 0.151 | 0.784 |
| cg22341310 | 19 | ZNF541 | 84215 | Island | Body |
Hypermethylation | 0.172 | −0.621 |
| cg13462129 | 7 | DLX5 | 1749 | Island | Body |
Hypermethylation | 0.193 | 0.678 |
| cg11376198 | 1 | AKR7L | 246181 | Island | TSS200 |
Hypermethylation | 0.243 | 0.531 |
| cg26316946 | 6 | GRIK2 | 2898 | Island | 1stExon |
Hypermethylation | 0.246 | 0.659 |
| cg03874199 | 2 | HOXD12 | 3238 | Island | TSS200 |
Hypermethylation | 0.283 | 0.501 |
| cg23130254 | 2 | HOXD12 | 3238 | Island | 1stExon |
Hypermethylation | 0.317 | 0.501 |
| cg00767581 | 2 | HOXD4 | 3233 | Island | TSS1500 |
Hypermethylation | 0.353 | Not
Significant |
| cg18702197 | 2 | HOXD3 | 3232 | Island | TSS1500 |
Hypermethylation | 0.355 | 0.422 |
| cg07693270 | 3 | RPL39L | 116832 | Island | 5′UTR |
Hypomethylation | −0.668 | 1.296 |
| cg24898753 | 11 | FTH1 | 2495 | Island | TSS1500 |
Hypomethylation | 0.053 | −0.589 |
| cg06038133 | 17 | CORO6 | 84940 | Island | Body |
Hypomethylation | 0.054 | −0.849 |
Identification of a novel CGI
methylation signature that is a potent prognostic indicator for OS
time in lung ADCs
Within the univariate Cox regression model
incorporating methylation data of those ADCs-specific CGI loci, a
total of 9 CGI CpGs were identified from the training set
(TCGA-450k set) that were significantly associated with OS
(permutation P≤0.05), and that had higher variability (SD≥0.10) in
lung ADCs. Characteristics of the 9 CGI CpGs are summarized in
Table II. Methylation data of 7 and
2 CpGs exhibited negative and positive associations with OS,
respectively (Table II).
Accordingly, as aforementioned, the risk score formula for the CpGs
of the MyoD family inhibitor (MDFI), homeobox D3
(HOXD3), CKLF like MARVEL transmembrane domain containing 2
(CMTM2), paired box 3 (PAX3), LY6/PLAUR domain
containing 5 (LYPD5), laeverin (LVRN), RPL39L,
glutamate ionotropic receptor kainite type subunit 2 (GRIK2)
and complexin 2 (CPLX2) genes was established as follows:
Risk score=[(1.403 × β-value of cg05345286 (MDFI)) + (1.564 ×
β-value of cg18702197 (HOXD3)) + (1.646 × β-value of
cg01683883 (CMTM2)) + (1.526 × β-value of cg02245378
(PAX3)) + (0.984 × β-value of cg12768605 (LYPD5)) +
(1.316 × β-value of cg25044651 (Laeverin (LVRN)) + (−1.130 ×
β-value of cg07693270 (RPL39L)) + (1.088 × β-value of
cg26316946 (GRIK2)) + (−0.835 × β-value of cg19885761
(CPLX2))]. On the basis of the risk formula, each patient
from the TCGA-450k set was assigned a risk score, and then
classified into low-risk or high-risk groups using the median score
as a cut-off (3.08). Survival analysis indicated that in the
TCGA-450k set, the low-risk group was associated with increased OS
times compared with the high-risk group [54.4 vs. 42.3 months,
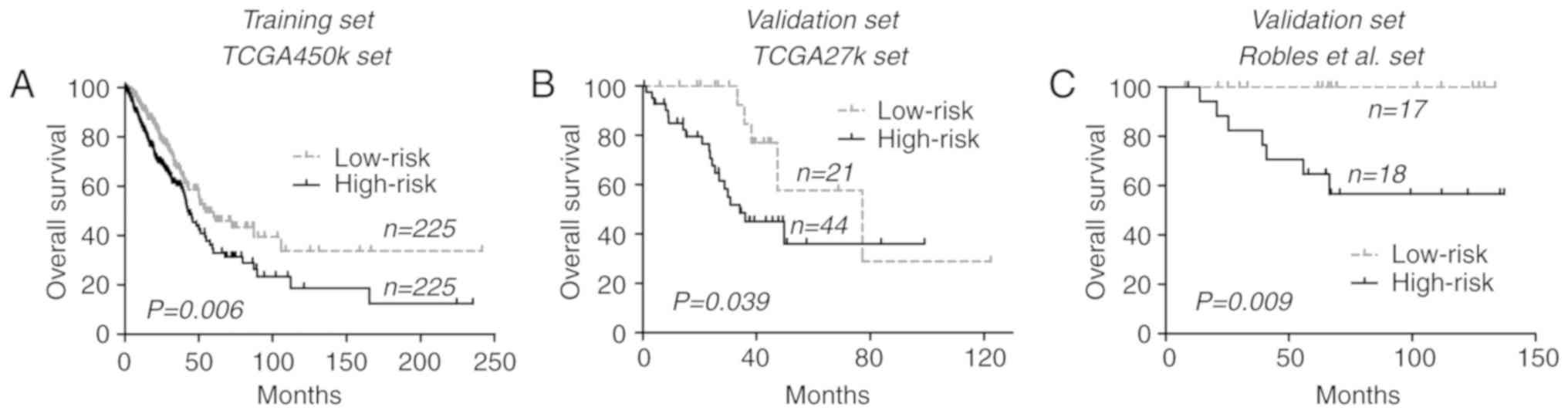
respectively; P=0.006 (log-rank test); Fig. 1A].
| Table II.Characteristics of the 9-CpG CpG
island methylation panel. |
Table II.
Characteristics of the 9-CpG CpG
island methylation panel.
| Probe ID | Symbol | Association with
gene region | Chr. | Methylation status
between tumor and normal tissuesa | Expression status
between tumor and normal tissuesb | Pearson
coefficients between DNA methylation and gene
expressionc | Univariate Cox
coefficientsd | Permutation
P-valued |
|---|
| cg05345286 | MDFI | Body | 6 | Hyper | Up | −0.024 | 1.403 | 0.003 |
| cg18702197 | HOXD3 | TSS1500 | 2 | Hyper | Up | 0.355 | 1.564 | 0.004 |
| cg01683883 | CMTM2 | TSS1500 | 16 | Hyper | Down | −0.175 | 1.646 | 0.012 |
| cg02245378 | PAX3 | Body | 2 | Hyper | NS | 0.126 | 1.526 | 0.017 |
| cg12768605 | LYPD5 | TSS200 | 19 | Hyper | Down | −0.153 | 0.984 | 0.024 |
| cg25044651 | LVRN | 1stExon | 5 | Hyper | NS | −0.112 | 1.316 | 0.030 |
| cg07693270 | RPL39L | 5′UTR | 3 | Hypo | Up | −0.668 | −1.130 | 0.043 |
| cg26316946 | GRIK2 | 1stExon | 6 | Hyper | Up | 0.246 | 1.088 | 0.043 |
| cg19885761 | CPLX2 | 5′UTR | 5 | Hyper | Down | −0.318 | −0.835 | 0.049 |
To confirm its prognostic relevance, the CGI
methylation signature in an additional 2 datasets, the TCGA-27k and
Robles et al (11) datasets,
were analyzed. By directly applying the risk formula and using
cut-off points, the TCGA-27k set was divided into a low-risk group
(n=21) and a high-risk group (n=44). In concordance with the
training set, patients within the low-risk group exhibited
increased OS times compared with those within the high-risk group
[77.3 vs. 34.2 months; P=0.039 (log-rank test); Fig. 1B]. Similar results were also observed
within the Robles et al (11)
set, where low-risk patients were associated with improved OS
compared with the high-risk patients [median time not reached for
either group; P=0.009 (log-rank test); Fig. 1C]. Pooled analysis at dataset level
confirmed the prognostic relevance of the CGI methylation signature
for lung ADCs [hazard ratio (HR)=1.61, 95% confidence interval
(CI), 1.20–2.17; P=0.002; I2=29%, P=0.25].
Univariate Cox regression analysis of all patients
from TCGA datasets (combined TCGA-27k and TCGA-450k sets) indicated
that only tumor stages and the CGI methylation signature were
significantly associated with OS, while patient age, sex, tumor
stages, smoking status, MET proto-oncogene, receptor tyrosine
kinase amplification and mutations in key genes including KRAS
proto-oncogene, GTPase, Epithelial growth factor receptor, tumor
protein P53 and B-Raf proto-oncogene, serine/threonine kinase were
not. Finally, the multivariate Cox regression analysis demonstrated
the prognostic significance of the CGI methylation signature of the
present study in lung ADCs (Table
III).
| Table III.Results from Cox regression models
within all The Cancer Genome Atlas samples. |
Table III.
Results from Cox regression models
within all The Cancer Genome Atlas samples.
|
| Univariate Cox
model | Multivariate Cox
model |
|---|
|
|
|
|
|---|
| Variables | HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Tumor stage | 1.651
(1.441–1.893) | <0.001 | 1.611
(1.405–1.847) | <0.001 |
| CGI methylation
signature | 1.606
(1.199–2.152) | 0.001 | 1.449
(1.078–1.947) | 0.014 |
| Sex | 1.057
(0.794–1.407) | 0.705 | – | – |
| Smoking status | 0.915
(0.611–1.371) | 0.666 | – | – |
| Age | 1.009
(0.993–1.024) | 0.271 | – | – |
| BRAF
mutations | 0.707
(0.402–1.246) | 0.231 | – | – |
| EGFR
mutations | 1.230
(0.830–1.824) | 0.302 | – | – |
| KRAS
mutations | 1.176
(0.858–1.610) | 0.314 | – | – |
| TP53
mutations | 1.332
(0.990–1.793) | 0.058 | – | – |
| MET
amplification | 1.027
(0.833–1.268) | 0.801 | – | – |
CGI methylation signature is not a
strong prognostic indicator of RFS in lung ADCs
To investigate the association of the CGI
methylation signature of the present study with RFS, it was
analyzed within the TCGA-450k set, which yielded a marginally
significant difference in RFS between each risk group [33.9 vs.
27.0 months; P=0.049 (log-rank test); Fig. 2A]. Then, in the TCGA-27k set,
low-risk patients appeared to exhibit longer RFS compared with the
high-risk patients, but the difference did not reach significance
(68.2 vs. 17.0 months; log-rank test P=0.072; Fig. 2B). An additional large cohort of lung
ADCs was finally introduced into the validation phase, where the
CGI methylation signature also failed to significantly stratify
patients into subgroups with distinct RFS outcomes [62.6 vs. 55.6
months; P=0.492 (log-rank test); Fig.
2C]. Despite that, the pooled analysis of the 3 datasets
yielded a significant difference in RFS between the risk groups
(HR, 1.30; 95% CI, 1.04–2.62; P=0.020; I2=0%; P=0.38).
The inconsistent results from different analysis levels indicated
that the CGI methylation signature is not a robust indicator for
RFS in lung ADCs.
Novel classification approach based on
the integration of DNA methylation and gene expression of RPL39L in
lung ADCs
By characterizing each member of the CGI methylation
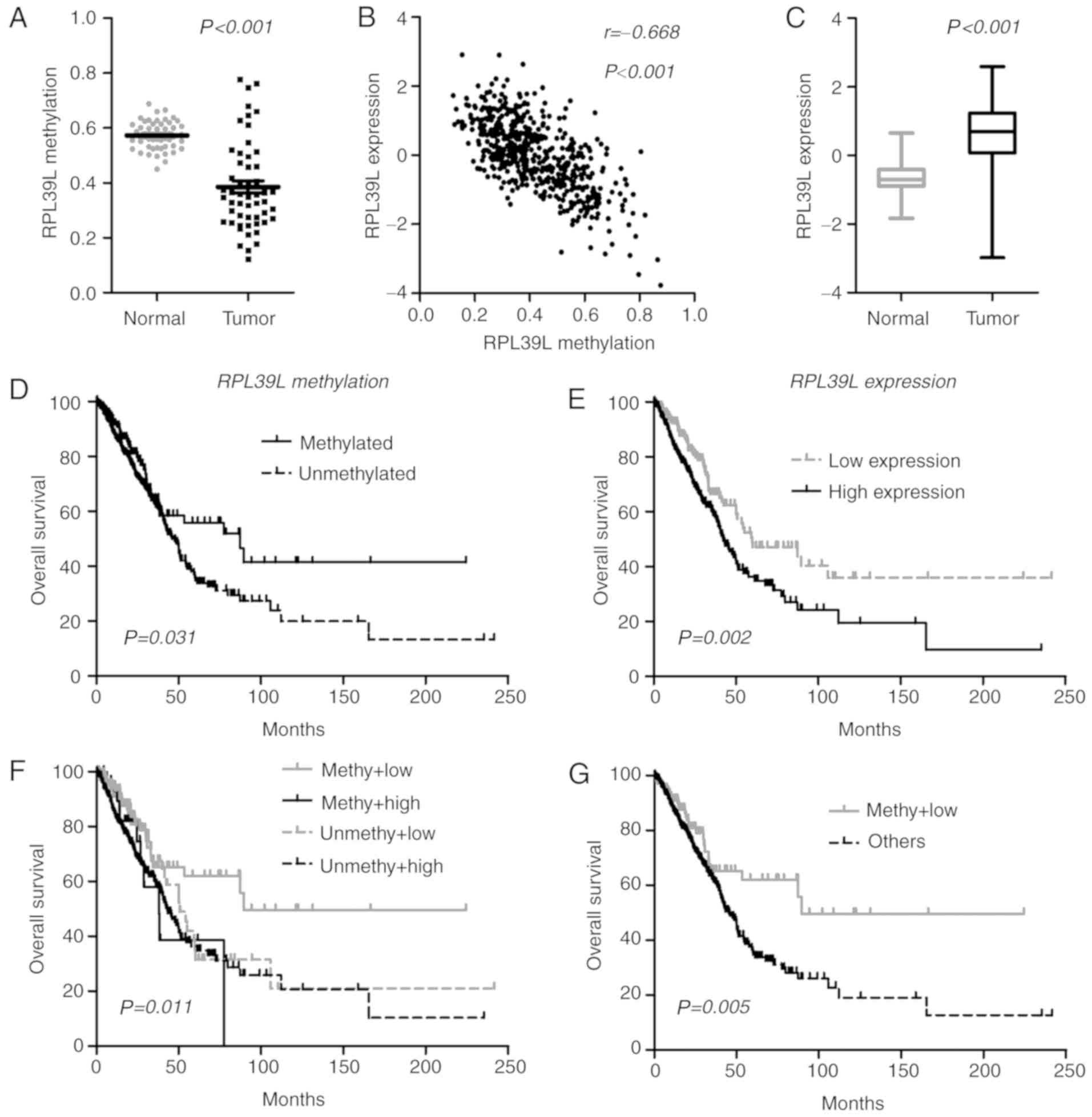
panel, it was identified that one CGI locus (cg07693270) was
consistently hypomethylated in lung ADCs (Fig. 3A), and the methylation data were
closely correlated with gene expression (RPL39L, Pearson
coefficient r=−0.668; P<0.0001; Fig.
3B), indicating a methylation-dependent transcriptional
regulatory mechanism for RPL39L. In line with its epigenetic
status, RPL39L was upregulated in lung ADCs, indicating a
tumor-promoting role (Fig. 3C). At
the initiation of the present study, the methylation level of
RPL39L was positively correlated with OS. Therefore, the
present study attempted to prognostically classify patients by
single-locus methylation status of RPL39L, and it was
identified that tumors with methylated CGI of RPL39L were
associated with increased OS compared with the unmethylated tumors
within TCGA samples (Fig. 3D).
Additionally, it was identified that based on RPL39L
expression levels, patients may also be classified into distinct
prognostic subgroups, in which tumors exhibiting decreased
RPL39L expression levels were associated with increased OS
time compared with those with increased expression levels [59.7 vs.
42.7 months; P=0.002 (log-rank test); Fig. 3E]. These data indicated the
possibility of a promising classification approach based on the
integration of the DNA methylation and gene expression of
RPL39L. Consequently, the present study identified that
tumors with increased methylation and decreased expression of
RPL39L exhibited the best OS among all cases (Fig. 3F and G). The multivariate Cox model
demonstrated the prognostic independence of the integrated approach
(HR, 0.54; 95% CI, 0.36–0.81; P=0.003) as compared with tumor
stages (HR, 1.66; 95% CI, 1.45–1.91; P<0.001) within TCGA
samples. These data indicated that RPL39L may serve
oncogenic roles in the progression of lung ADCs, and may represent
a novel promising therapeutic target for this disease. The
integrated epigenetic and transcriptional assessment of
PRL39L may be useful for optimizing the risk stratification
of patients with lung ADC, and for identifying the appropriate
subgroups sensitive to targeted drugs against RPL39L.
Discussion
The study of epigenetic markers, particularly DNA
methylation, represents one of the most promising and fastest
expanding areas in cancer biomarker identification (23). Similar to other tumors, lung ADCs are
characterized by distinct genome-wide DNA methylation landscapes,
where the global hypomethylation of DNA repeats occurs
concomitantly with CGI hypermethylation of gene regions (8). Among those cancer-specific DNA
methylation aberrations, the promoter-specific CGI de novo
methylation of tumor suppressor genes is the best-known epigenetic
abnormality in lung cancer patients (8). Studies using candidate gene approaches
have identified a large number of known tumor suppressors,
including cyclin-dependent kinase inhibitor 2A (24), RAS association domain family member 1
(25), O-6-methylguanine-DNA
methyltransferase (26) and APC, WNT
signaling pathway regulator (27),
to be consistently methylated in lung ADCs. A number of those
epigenetic alterations were identified to serve crucial roles in
tumorigenesis via the regulation of gene expression and to exhibit
promise in the diagnosis and prognostication of patients with lung
cancer (24–27). Previously, efforts have been made to
comprehensively assess cancer epigenomes using genome-wide DNA
methylation profiling techniques, including Illumina array-based
assays, restriction landmark genome scanning gel-based analysis,
and next-generation sequencing-based analysis (23,28). The
application of those high-throughput detection approaches may
provide an unbiased and clear view of the lung cancer epigenome,
and assist in identifying useful DNA methylation events for
diagnostic and prognostic purposes.
The reproducibility of results from genome-wide DNA
methylation analysis may be an issue for making definitive
conclusions from these types of studies, as false-positive data are
common in microarray analysis where the number of interrogated loci
within each tumor is larger compared with the number of
participants (29).
Batch effects appear to be a common phenomenon in
high-throughput microarray data, particularly for the Infinium
Methylation BeadChip (13). In the
present study, the effective empirical Bayes method was adopted to
remove the potential non-biological difference of methylation data
across each dataset. Genome-wide DNA methylation data of lung ADCs
and matched control tissues from 6 publically available datasets
were then independently re-analyzed, and stricter criteria were
adopted to identify robust cancer-specific CGI methylation loci in
lung ADCs. In total, 134 cancer-specific CpGs were consistently
observed in at least 4 of the 6 datasets examined in the present
study, 11 of which had been described by previous studies with
other DNA methylation detection approaches, for example genes in
HOX clusters (30) including
HOXB4, HOXA7 and HOXA9, TRIM58 (31) and GALR1 (32) (Table
I). The methylation status of these genes exhibited promise for
the early detection and risk prediction for lung cancer (30–32). In
addition, by integrating gene expression data, it was identified
that a considerable proportion of these cancer-specific CGI
methylation changes may have significant effects on their relevant
gene expression, and indicate potential functional value in
tumorigenesis of lung ADCs. Well-studied examples are the de
novo CGI methylation of zinc finger protein 677 (33), cysteine dioxygenase type 1 (34,35),
SRY-box 1 (SOX1) (36) and
SOX17 (37) in NSCLCs. The
data from the present study were corroborated by the validation of
the identified CGI methylation candidates in the literature
(30–34). In addition, the present study also
identified a panel of previously unknown cancer-specific CGI
methylation loci that may have potential roles in determining the
fate of patients with lung cancer, which will warrant future
investigation.
Clinically or functionally characterizing each CGI
candidate is beyond the scope of the present study. Instead, by
applying a univariate Cox regression model and permutation
correction, a panel of 9 CGI CpGs that were significantly
associated with OS time was identified in a large cohort of
patients with lung ADCs (TCGA-450k set; n=450). The detection of a
panel of biomarkers, compared with single markers, may have a
higher sensitivity and specificity for specific clinical purposes
(38). Therefore, a risk score-based
prognostic classifier was established based on the methylation
patterns of the 9 CpGs to assist in stratifying patients into
distinct prognostic subgroups. The novel methylation signature
indicated consistent prognostic ability in different patient
cohorts. Finally, a multivariate Cox model demonstrated its
prognostic significance in the context of different tumor stages.
However, with respect to the RFS data, which is an additional
notable clinical outcome, the novel methylation signature
demonstrated limited value for risk stratification, and future
validation is required for justifying a definitive conclusion. In
summary, the data in the present study indicated that the CGI
methylation signature of the present study may be a potent
prognostic indicator for OS outcome in patients with all-stage lung
ADCs. Additional supporting evidence for this novel CGI methylation
signature may support its potential biological relevance in cancer
biology. In the present study, it was identified that the
methylation levels of 8 CpG loci were significantly correlated with
gene expression (positively correlated: HOXD3, GRIK2 and
PAX3; and negative correlation: PRL39L, CPLX2, CMTM2,
LYPD5 and LVRN). Accordingly, the majority of the genes
were differentially expressed between lung ADCs and normal tissues
(upregulated: RPL39L, GRIK2 and HOXD3; and
downregulated: CMTM2, CPLX2 and LYPD5). The majority
of these genes have been demonstrated to be abnormally methylated
and expressed in a number of human cancer types, including breast,
colorectal and prostate cancer, and were closely associated with
patient prognosis and tumor aggressiveness (39–42).
However, limited data had been acquired on their functional roles
in cancer biology. RPL39L was identified to confer drug
resistance in lacrimal gland adenoid cystic carcinoma (43) and the lung cancer A549 cell line
(44), but others have not been
fully characterized in cancer. Future functional investigation of
these genes will assist in developing understanding of the
biological implications of the CGI methylation signature of the
present study, and for identifying promising epigenetic therapeutic
targets in lung ADCs.
Unlike the cancer-specific de novo DNA
methylation at CGI regions of genes, the presence and functional
roles of CGI hypomethylation have been much less well characterized
in cancer biology. The present study identified that CGI
hypomethylation of RPL39L was consistently observed in lung
ADCs. This epigenetic event may have functional significance in the
initiation and progression of lung cancer, as it markedly affected
gene transcription and resulted in the upregulation of
RPL39L in tumor tissues. In line with the aforementioned
data, it was also demonstrated that within TCGA samples, either
epigenetic or transcriptional activations of RPL39L were
associated with poorer OS time in patients with lung ADCs.
Furthermore, the integration of DNA methylation and gene expression
data identified a refined subset of tumors with favorable prognoses
whose RPL39L gene was epigenetically and transcriptionally
repressed. RPL39L is a recently evolved ribosomal protein
paralog that exhibits highly specific tissue expression patterns in
mice and humans (45). This gene was
previously described to be highly expressed in the testis and to be
upregulated in multiple cancer cell lines (45). Wong et al (45) had demonstrated that RPL39L was
highly upregulated in mouse embryonic stem cells, and that its
expression was markedly associated with tumor aggressiveness and
vascular invasiveness of hepatocellular carcinomas (45). High expression of RPL39L may
also confer the drug-resistant phenotype of lung cancer A549 cell
lines (44). However, RPL39L
was demonstrated to be associated with hypermethylation and gene
inactivation in prostate cancer cell lines (39). Together, these results indicated that
epigenetic and transcriptional abnormalities in RPL39L were
commonly implicated in the initiation and progression of human
cancer. Notably, the data from the present study is of interest as
it provides novel evidence for the contributing roles of CGI
hypomethylation and gene re-activation in lung cancer. In addition,
the data also raise concerns surrounding the current non-specific
demethylating anticancer approach, as it may promote cancer
development via the exacerbation of cancer-specific
hypomethylation. Targeted epigenetic therapy that has distinct
effects on cancer-specific hypermethylation and hypomethylation may
be a promising option for the future development of anticancer
therapy. Unfortunately, the oncogenic roles of RPL39L have
not been studied extensively in lung ADCs. Future functional
studies may assist in developing targeted therapies against this
gene. Finally, the integrated assessment of RPL39L may be a
promising approach for optimizing risk stratification, and
improving personalized medicine in lung ADCs.
There were several limitations to the present study.
The incompleteness of certain important clinical information for
the included patients, including performance status and treatment
modality, compromised the prognostic robustness of the
study-specific methylation signature. The clinical and
methodological heterogeneity across each dataset may also introduce
uncertainty in data interpretation. Other limitations include the
relatively small sample size of the validation sets, and the lack
of functional validation of those CGI methylation candidates. The
results of the present study were preliminary and primarily derived
from microarray data analysis. Additional studies will be required
to validate these results in vivo, and in a clinical
setting.
In conclusion, by comparing genome-wide DNA
methylation and gene expression profiles of lung ADCs and matched
non-tumor tissues from multiple independent datasets, the present
study identified a number of cancer-specific CGI methylation
changes in lung ADCs, and characterized their associations with
gene expression. Those CGI methylation changes may be useful for
the identification of novel biomarkers for diagnostic and
prognostic purposes in lung ADCs. One example is the identification
of a 9-CpG methylation panel that was demonstrated to be a potent
prognostic indicator for OS time. Furthermore, the identification
of CGI hypomethylation and consequent gene re-activation of
RPL39L provides novel insights into treatment development
and risk stratification for lung ADCs.
Acknowledgements
The authors would like to thank Dr Juan Li
(Department of Neurosur-gery, Xijing Hospital, First Affiliated
Hospital of Fourth Military Medical University) for revising the
manuscript and providing training of biostatistic softwares.
Funding
The present study was partially supported by the
Division of Technology, Tongchuan, China (grant no. kj2015).
Availability of data and materials
The datasets generated and/or analyzed during the
present study are available in the following repositories: i) TCGA,
(https://tcga-data.nci.nih.gov/tcga/);
ii) GEO, (https://www.ncbi.nlm.nih.gov/geo/); iii) R software,
(https://www.r-project.org/); iv) UCSC
genome reference, (http://genome.ucsc.edu/); v) Entrez Gene ID,
(https://www.ncbi.nlm.nih.gov/gene/);
vi) Biometric Research Branch-Array Tools, (http://brb.nci.nih.gov/BRB-ArrayTools; vii) DAVID,
(https://david.ncifcrf.gov/), with
accession nos. GSE32861, GSE32866, GSE62948, GSE63384 and
GSE39279.
Authors contributions
PZY, XHY and HR conceived and designed the study.
PZY, XHY and JHW acquired the data. PZY, XHY and SCW analyzed and
interpreted the data. PZY and XHY wrote and revised the paper. JHW
and SCW provided administrative, technical, or material support.
XHY and HR supervised the study.
Ethics approval and consent to
participate
All procedures performed in studies involving humans
were conducted in accordance with the ethical standards of the
institutional research committees and with the 1964 Declaration of
Helsinki and its later amendments or comparable ethical standards.
Informed consent was obtained from all individual participants as
reported by included datasets.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ettinger DS, Wood DE, Akerley W, Bazhenova
LA, Camidge DR, Cheney RT, Chirieac LR, D'Amico TA, Dilling TJ,
Dobelbower MC, et al: NCCN Guidelines Insights: Non-small cell lung
cancer, version 4.2016. J Natl Compr Canc Netw. 14:255–264. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sandoval J, Mendez-Gonzalez J, Nadal E,
Chen G, Carmona FJ, Sayols S, Moran S, Heyn H, Vizoso M, Gomez A,
et al: A prognostic DNA methylation signature for stage I
non-small-cell lung cancer. J Clin Oncol. 31:4140–4147. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cancer Genome Atlas Research Network.
Comprehensive molecular profiling of lung adenocarcinoma. Nature.
511:543–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tang H, Xiao G, Behrens C, Schiller J,
Allen J, Chow CW, Suraokar M, Corvalan A, Mao J, White MA, et al: A
12-gene set predicts survival benefits from adjuvant chemotherapy
in non-small cell lung cancer patients. Clin Cancer Res.
19:1577–1586. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karlsson A, Jönsson M, Lauss M, Brunnström
H, Jönsson P, Borg Å, Jönsson G, Ringnér M, Planck M and Staaf J:
Genome-wide DNA methylation analysis of lung carcinoma reveals one
neuroendocrine and four adenocarcinoma epitypes associated with
patient outcome. Clin Cancer Res. 20:6127–6140. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rodríguez-Paredes M and Esteller M: Cancer
epigenetics reaches mainstream oncology. Nat Med. 17:330–339. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Issa JP: DNA methylation as a clinical
marker in oncology. J Clin Oncol. 30:2566–2568. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mehta A, Dobersch S, Romero-Olmedo AJ and
Barreto G: Epigenetics in lung cancer diagnosis and therapy. Cancer
Metastasis Rev. 34:229–241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Selamat SA, Chung BS, Girard L, Zhang W,
Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, et al:
Genome-scale analysis of DNA methylation in lung adenocarcinoma and
integration with mRNA expression. Genome Res. 22:1197–1211. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mansfield AS, Wang L, Cunningham JM, Jen
J, Kolbert CP, Sun Z and Yang P: DNA methylation and RNA expression
profiles in lung adenocarcinomas of never-smokers. Cancer Genet.
208:253–260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Robles AI, Arai E, Mathé EA, Okayama H,
Schetter AJ, Brown D, Petersen D, Bowman ED, Noro R, Welsh JA, et
al: An integrated prognostic classifier for stage I lung
adenocarcinoma based on mRNA, microRNA, and DNA methylation
biomarkers. J Thorac Oncol. 10:1037–1048. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Johnson WE, Li C and Rabinovic A:
Adjusting batch effects in microarray expression data using
empirical Bayes methods. Biostatistics. 8:118–127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun Z, Chai HS, Wu Y, White WM, Donkena
KV, Klein CJ, Garovic VD, Therneau TM and Kocher JP: Batch effect
correction for genome-wide methylation data with illumina infinium
platform. BMC Med Genomics. 4:842011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Team RC: R, . A language and environment
for statistical computing. R Foundation for Statistical Computing;
Vienna, Austria: https://www.R-project.org/2016
|
|
15
|
Du P, Zhang X, Huang CC, Jafari N, Kibbe
WA, Hou L and Lin SM: Comparison of Beta-value and M-value methods
for quantifying methylation levels by microarray analysis. BMC
Bioinformatics. 11:5872010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yin AA, Lu N, Etcheverry A, Aubry M,
Barnholtz-Sloan J, Zhang LH, Mosser J, Zhang W, Zhang X, Liu YH and
He YL: A novel prognostic six-CpG signature in glioblastomas. CNS
Neurosci Ther. 24:167–177. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang XQ, Sun S, Lam KF, Kiang KM, Pu JK,
Ho AS, Lui WM, Fung CF, Wong TS and Leung GK: A long non-coding RNA
signature in glioblastoma multiforme predicts survival. Neurobiol
Dis. 58:123–131. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang DW, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mi H, Huang X, Muruganujan A, Tang H,
Mills C, Kang D and Thomas PD: PANTHER version 11: Expanded
annotation data from Gene Ontology and Reactome pathways, and data
analysis tool enhancements. Nucleic Acids Res. 45:D183–D189. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
BioCarta Pathways, . http://www.biocarta.com/March. 2016
|
|
21
|
KEGG, . Kyoto encyclopedia of genes and
genomes. https://www.genome.jp/kegg/Release 77.1.
2016
|
|
22
|
Hothorn T and Zeileis A: Generalized
maximally selected statistics. Biometrics. 64:1263–1269. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Heller G, Zielinski CC and
Zöchbauer-Müller S: Lung cancer: From single-gene methylation to
methylome profiling. Cancer Metastasis Rev. 29:95–107. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brock MV, Hooker CM, Ota-Machida E, Han Y,
Guo M, Ames S, Glöckner S, Piantadosi S, Gabrielson E, Pridham G,
et al: DNA methylation markers and early recurrence in stage I lung
cancer. N Engl J Med. 358:1118–1128. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Burbee DG, Forgacs E, Zöchbauer-Müller S,
Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S,
et al: Epigenetic inactivation of RASSF1A in lung and breast
cancers and malignant phenotype suppression. J Natl Cancer Inst.
93:691–699. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Esteller M, Corn PG, Baylin SB and Herman
JG: A gene hypermethylation profile of human cancer. Cancer Res.
61:3225–3229. 2001.PubMed/NCBI
|
|
27
|
Virmani AK, Rathi A, Sathyanarayana UG,
Padar A, Huang CX, Cunnigham HT, Farinas AJ, Milchgrub S, Euhus DM,
Gilcrease M, et al: Aberrant methylation of the adenomatous
polyposis coli (APC) gene promoter 1A in breast and lung
carcinomas. Clin Cancer Res. 7:1998–2004. 2001.PubMed/NCBI
|
|
28
|
Heyn H and Esteller M: DNA methylation
profiling in the clinic: Applications and challenges. Nat Rev
Genet. 13:679–692. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang X, Sun S, Pu JK, Tsang AC, Lee D,
Man VO, Lui WM, Wong ST and Leung GK: Long non-coding RNA
expression profiles predict clinical phenotypes in glioma.
Neurobiol Dis. 48:1–8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pfeifer GP and Rauch TA: DNA methylation
patterns in lung carcinomas. Semin Cancer Biol. 19:181–187. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Diaz-Lagares A, Mendez-Gonzalez J, Hervas
D, Saigi M, Pajares MJ, Garcia D, Crujerias AB, Pio R, Montuenga
LM, Zulueta J, et al: A novel epigenetic signature for early
diagnosis in lung cancer. Clin Cancer Res. 22:3361–3371. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo S, Yan F, Xu J, Bao Y, Zhu J, Wang X,
Wu J, Li Y, Pu W, Liu Y, et al: Identification and validation of
the methylation biomarkers of non-small cell lung cancer (NSCLC).
Clin Epigenetics. 7:32015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heller G, Altenberger C, Schmid B, Marhold
M, Tomasich E, Ziegler B, Müllauer L, Minichsdorfer C, Lang G,
End-Pfützenreuter A, et al: DNA methylation transcriptionally
regulates the putative tumor cell growth suppressor ZNF677 in
non-small cell lung cancers. Oncotarget. 6:394–408. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wrangle J, Machida EO, Danilova L, Hulbert
A, Franco N, Zhang W, Glöckner SC, Tessema M, Van Neste L, Easwaran
H, et al: Functional identification of cancer-specific methylation
of CDO1, HOXA9, and TAC1 for the diagnosis of lung cancer. Clin
Cancer Res. 20:1856–1864. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brait M, Ling S, Nagpal JK, Chang X, Park
HL, Lee J, Okamura J, Yamashita K, Sidransky D and Kim MS: Cysteine
dioxygenase 1 is a tumor suppressor gene silenced by promoter
methylation in multiple human cancers. PLoS One. 7:e449512012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li N and Li S: Epigenetic inactivation of
SOX1 promotes cell migration in lung cancer. Tumour Biol.
36:4603–4610. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yin D, Jia Y, Yu Y, Brock MV, Herman JG,
Han C, Su X, Liu Y and Guo M: SOX17 methylation inhibits its
antagonism of Wnt signaling pathway in lung cancer. Discov Med.
14:33–40. 2012.PubMed/NCBI
|
|
38
|
Yin A, Etcheverry A, He Y, Aubry M,
Barnholtz-Sloan J, Zhang L, Mao X, Chen W, Liu B, Zhang W, et al:
Integrative analysis of novel hypomethylation and gene expression
signatures in glioblastomas. Oncotarget. 8:89607–89619. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Devaney JM, Wang S, Funda S, Long J,
Taghipour DJ, Tbaishat R, Furbert-Harris P, Ittmann M and
Kwabi-Addo B: Identification of novel DNA-methylated genes that
correlate with human prostate cancer and high-grade prostatic
intraepithelial neoplasia. Prostate Cancer Prostatic Dis.
16:292–300. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fang WJ, Zheng Y, Wu LM, Ke QH, Shen H,
Yuan Y and Zheng SS: Genome-wide analysis of aberrant DNA
methylation for identification of potential biomarkers in
colorectal cancer patients. Asian Pac J Cancer Prev. 13:1917–1921.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Litovkin K, Joniau S, Lerut E, Laenen A,
Gevaert O, Spahn M, Kneitz B, Isebaert S, Haustermans K, Beullens
M, et al: Methylation of PITX2, HOXD3, RASSF1 and TDRD1 predicts
biochemical recurrence in high-risk prostate cancer. J Cancer Res
Clin Oncol. 140:1849–1861. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shaoqiang C, Yue Z, Yang L, Hong Z, Lina
Z, Da P and Qingyuan Z: Expression of HOXD3 correlates with shorter
survival in patients with invasive breast cancer. Clin Exp
Metastasis. 30:155–163. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ye Q, Ding SF, Wang ZA, Feng J and Tan WB:
Influence of ribosomal protein L39-L in the drug resistance
mechanisms of lacrimal gland adenoid cystic carcinoma cells. Asian
Pac J Cancer Prev. 15:4995–5000. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu HS, Tan WB, Yang N, Yang YY, Cheng P,
Liu LJ, Wang WJ and Zhu CL: Effects of ribosomal protein l39-L on
the drug resistance mechanisms of lung cancer A549 cells. Asian Pac
J Cancer Prev. 15:3093–3097. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wong QW, Li J, Ng SR, Lim SG, Yang H and
Vardy LA: RPL39L is an example of a recently evolved ribosomal
protein paralog that shows highly specific tissue expression
patterns and is upregulated in ESCs and HCC tumors. RNA Biol.
11:33–41. 2014. View Article : Google Scholar : PubMed/NCBI
|