Introduction
Prostate cancer (PC) is a disease of the old age
(1). The specific underlying
mechanisms of prostate carcinogenesis have not been unraveled yet.
The only well-established risk factors for PC are older age, black
race/ethnicity, and a family history of the disease (2,3).
Therefore, future progress in combating PC will be highly dependent
upon an understanding of the mechanisms involved in the development
and steady progression into prostate malignancy.
Mitochondria are emerging as key players in the
tumorigenic process of cells by maintaining the biosynthetic and
energetic capabilities of cancer cells. Besides compartmentalizing
different metabolic pathways, the mitochondria is engaged in the
generation of much of the cellular energy, regulation of the redox
state of the cell, generation of reactive oxygen species (ROS),
buffering Ca2+ and initiating apoptosis (4). Mitochondria are involved in the final
stage of the cellular catabolism and maintain the redox homeostasis
at different levels. Through several enzymatic reactions,
carbohydrates, fats and proteins are degraded into smaller
molecules, which is further converted to pyruvate by glycolysis,
fatty acids and amino acids (Fig.
1). Mitochondria further transform these small molecules into
NADH and FADH2 (reduced energy equivalents), through
β-oxidation and TCA cycle or rerouted to biosynthetic pathways. The
reduced energy equivalents are then utilized by the mitochondrial
electron transport chain (ETC) through oxidative phosphorylation
(OXPHOS). The electrons liberated by the oxidation of NADH and
FADH2 are passed along a series of carriers of ETC
located in mitochondrial inner membrane. The electrons are
ultimately transferred to molecular oxygen to form water (Fig. 2). ETC consists of four enzyme
complexes (complexes I–IV), and two electron carriers (coenzyme Q
and cytochrome c). These complexes are composed of numerous protein
subunits encoded by nuclear and mitochondrial genes, except complex
II, which are encoded by nuclear genes only.
In addition, the electron-flavoprotein (ETF) system,
which involves ETFA/B and the ETF-QO proteins, connects
fatty-acid oxidation with coenzyme Q reduction. The
glycerol-3-phosphate dehydrogenase also participates in redox
homeostasis by oxidizing cytosolic NADH to reduce mitochondrial FAD
to FADH2. Finally, the malate-aspartate shuttle
(NADH-shuttling) also fuels OXPHOS and supports redox homeostasis
via the delivery of cytosolic NADH to the mitochondrial matrix. The
oxidation of NADH or FADH2 by complex I or complex II,
respectively, triggers the transfer of electrons from complex I (or
II) to complex IV. Meanwhile, protons are pumped from the matrix
into the inter-membrane space, thus generating an electrochemical
gradient of protons which is finally used by the
F1F0ATP synthase (i.e., complex V) to produce
adenosine triphosphate (ATP), the main energetic currency of the
cell (5).
Mitochondrial function, its genetics and energy
metabolism have important roles in tumor initiation and progression
and are considered emerging indicators of PC biology (6). There is increasing evidence that key
oncogenes and tumor suppressors modulate mitochondrial dynamics
through important signaling pathways. Thus, mitochondrial mass and
function vary between tumors and individuals, but, the significance
of these events for cancer is not fully appreciated (7). Therefore, in the present review, we
emphasize on the current knowledge on the mitochondrial
bioenergetics of normal prostate epithelial cells (PECs; normal
physiology) and as they transform from indolent tumor to aggressive
PC (pathology).
Intermediary metabolism of prostate
peripheral zone epithelium
The major characteristic of PECs of the peripheral
zone of the prostate is that they accumulate four times higher
concentrations of citrate compared to epithelial cells from other
mammalian tissues. PECs secrete citrate into the prostatic fluid at
a concentration of 3–10 times higher than the prostate tissue
levels itself (8). This metabolic
transformation of PECs during cellular differentiation is referred
to as ‘net citrate production’ (8).
In parallel, the prostate peripheral zone accumulates extremely
high levels of zinc in the range of 10–15 times higher compared to
other tissues (8,9). Current hypothesis, based on the
evidence, is that increased accumulation of citrate is due to the
inhibition of aconitase by high levels of zinc, which inhibits the
conversion of citrate to aconitate. This results in truncated Krebs
cycle and reduced OXPHOS (10).
Consequently, to meet the cellular energy demands and synthesize
large amounts of citrate, precursor requirements of PECs are
different and exceed from that of other normal cells.
In the citrate producing PECs, instead of being
oxidized, citrate becomes an end-product of TCA cycle (9). To maintain high levels of citrate, PECs
require substantial amounts of acetyl CoA and oxaloacetate for its
synthesis by citrate synthase. Therefore, under these metabolic
demands, availability of precursors for citrate synthesis is
crucial. Recent evidence indicates that PECs have highly flexible
metabolism (11,12). PECs could meet the high precursor
requirements through the utilization of five different substrates
such as glucose, pyruvate, aspartate, glutamine, and fatty acids.
Cells can utilize these substrates based on its availability.
However, glucose seems to be the preferred substrate. Studies have
shown that PECs consume high amounts of glucose (13–15) and
have higher aerobic glycolysis (10)
compared to other cell types (11,16),
such as normal breast epithelial cells or cells with rapid cell
turnover such as monocytes (17).
Acetyl CoA is mainly generated from pyruvate and β-oxidation of
fatty acids. Although, pyruvate is the end-product of glycolysis
and is the major source of pyruvate, it can also be generated from
other sources and could be utilized for the synthesis of acetyl
CoA. We have recently shown that RWPE-1 cells could efficiently
oxidize both endogenous as well as exogenous fatty acids
significantly (11). We have also
shown that glutamine could be utilized both as a substrate for
energy and citrate production by gaining access into the TCA cycle
by its conversion to α-ketoglutarate through the enzymatic action
of glutaminases followed by glutamate dehydrogenase (11). Consequently, glutamine could be
converted to OAA for citrate synthesis and at the same time
generate 9 ATP's in PECs with a truncated TCA cycle compared to 12
ATPs made when an acetyl CoA molecule completes one TCA cycle
(Fig. 1). Aspartate could be
converted by PEC's to produce OAA (8–10). It is
catalyzed by glutamate oxaloacetate transaminase (GOT). GOT
catalyzes the interconversion of aspartate and α-ketoglutarate to
oxaloacetate and glutamate (Fig. 1).
High aspartate levels were maintained by transporting it into the
cell from blood by taking advantage of a Na+-coupled
co-transporter EAAC1 (18). Another
anaplerotic pathway that may be important in providing OAA for
citrate synthesis is the conversion of pyruvate to OAA catalyzed by
pyruvate carboxylate (PyC). This pathway may be more efficient in
generating OAA as the cells are highly glycolytic producing large
amounts of pyruvate and part of it could be shunted for OAA
synthesis. However, the importance of PyC in PECs is still elusive.
Overall, it should be concluded that metabolic plasticity is an
inherent feature of citrate producing PECs and these cells may
acquire these characteristics as they functionally
differentiate.
Defining mitochondrial oncobioenergetic
profile and mitochondrial oncobioenergetic index
Metabolic profile of a cell is governed by the way
which the mitochondria functions in a cell. Any change in
mitochondrial function would alter the cellular metabolic profile.
Thus, under physiological conditions, the term mitochondrial
bioenergetics indicates how mitochondria functions or its
functional status/profile that allows the cells to meet the
metabolic demands. Several studies have already been conducted to
report the metabolic profile of cancer cells. However, how the
mitochondria functions or what is the mitochondrial bioenergetics
during the evolution of cancer that support its metabolic
requirements are still unexplored.
MOB may be simply stated as the mitochondrial
bioenergetics that is similar to that of a cancer cell. It is
defined as stable (at least in cell and ex vivo models),
measurable, change in the mitochondrial bioenergetics during the
course of cell transformation from normal to malignant tumor
(11). It indicates the
mitochondrial bioenergetics or the functional state of the
mitochondria that predisposes, promotes or favors carcinogenesis.
It is dynamic in nature (11) and
depends on the stage of the tumor (11,16),
tissue origin and microenvironment (19). It may be mathematically represented
as mitochondrial oncobioenergetic index (MOBI) (11) (see below). MOBI is not only
associated with cancer cells but also with the individual cellular
components of the tumor, such as fibroblasts, immune cells etc.,
that support cancer cell growth at each stage of the tumor
development. It also represents mitochondrial bioenergetics of
cancer tissues itself and the normal tissues that are highly
vulnerable to carcinogenesis. MOB of a tissue represents or is the
result of both the metabolic rewiring of the cancer and other tumor
associated cells induced during transformation and pathological
stress induced by tumor growth.
It is now evident that mitochondria are involved in
malignant transformation, cell proliferation, aggression, and
metastatic behavior of prostate cancer (6,20).
Recent comprehensive analysis of the oncobioenergetic profile of PC
cells using microplate-based high resolution respirometry has
clearly demonstrated that characteristic MOB changes occurs at
different stages of PC development (11). Modification of MOB significantly
inhibits the growth of malignant cells (21,22).
This observation suggests that dynamic changes in the MOB function
are a critical component in the development of malignant disease of
the prostate.
Different parameters of mitochondrial
oncobioenergetic (MOB) function could be established by using
microplate (23) or Oxygraph high
resolution respirometry (24).
Microplate assay provides a high-throughput approach, which provide
valuable information on the various aspects of mitochondrial
function through mitochondrial stress test (MiST) (25,26). The
function of mitochondria is directly related to the amount of
oxygen consumed and various parameters are derived from the oxygen
consumption rate (OCR) trace after interrogation with compounds
that can modulate the mitochondrial function (Mito-effectors;
Fig. 3). Major functional parameters
of a MiST are the following:
Basal oxygen consumption
It represents the amount of oxygen consumed by the
mitochondria, which allows the cells to support the fundamental
cellular functions under optimal physiological conditions. Basal
oxygen consumption contributes to the synthesis of ATP at complex V
and proton leak (see below). A change in basal rate in each cell
type indicates some bioenergetic change in the cell, which can be
estimated by performing MiST. Basal respiration can alter
dramatically, due to inherent properties of the cells, such as
metabolic rewiring, as described above. It can also be affected by
the presence or absence of different substrates (glucose, pyruvate,
lactate, fatty acids, glutamine, oxygen), growth factors and
hormones, pH and osmolality in the environment (26). Therefore, optimal concentrations of
these substrates should be employed for analysis of the
mitochondrial function. For example, glucose concentrations above
10 mM may significantly inhibit the basal respiration of different
cell types. The mean normal blood glucose level in humans is about
5.5 mmol/l (100 mg/dl). Therefore, it is always preferable to use
physiological concentration of glucose for cellular respiration
assays. Moreover, the availability of different substrates in the
surrounding environment may also influence the basal oxygen
consumption.
ATP-dependent oxygen consumption
Otherwise described as oligomycin sensitive oxygen
consumption, ATP-dependent oxygen consumption represents the amount
of oxygen consumed to generate ATP at complex V or ATP synthase. It
is determined by measuring the OCR after interrogating with
oligomycin. It is the difference between basal oxygen consumption
and the oxygen consumption due to oligomycin inhibition of complex
V (25). Inhibition of ATP synthase
would increase the proton gradient (mitochondrial
hyperpolarization), which in turn prevents the electron flow
through other complexes. This inhibition results in reduced oxygen
consumption. Therefore, any increase in ATP-dependent OCR implies a
higher ATP demand. On the other hand, a decrease in ATP-dependent
OCR represents a reduced ATP demand, a lack of substrate
availability and/or severe damage to OXPHOS (25).
Proton leak
Proton leak is a physiologic process, which
represents reentry of protons into the mitochondrial matrix through
facilitated diffusion without contributing to ATP synthesis and
energy dissipated as heat. Proton leak can be an intrinsic property
of the cell or induced. The intrinsic proton leak is unregulated,
cell-type specific and correlates with metabolic rate of the cell.
Intrinsic proton leak is mainly mediated by mitochondrial anion
carriers and is minimal through the lipid bilayer. Inducible leak
occurs through the adenine nucleotide translocase (ANT) and
uncoupling proteins (UCPs) and can be initiated by fatty acids,
superoxide, or peroxidation products (27). The physiological role of inducible
leak through UCP1 in mammalian brown adipose tissue is heat
production (27).
An increase in intrinsic leak could be attributed to
damage of the inner mitochondrial membrane or ETC complexes. This
disproportionate increase in oxygen consumption is termed ‘electron
slip,’ where electrons are transferred through the ETC complexes
(Complexes I, III and IV) without pumping protons into the
intermembrane space. There is also evidence to show that oxidative
stress capable of damaging the mitochondrial membrane or ETC
complexes could increase ATP-dependent oxygen consumption and
proton leak (28).
Maximum oxygen consumption
Use of uncouplers (e.g. FCCP or carbonyl cyanide
trifluoromethoxy phenylhydrazone) to generate artificial energy
demand would allow measuring the maximum possible mitochondrial
oxygen consumption under the experimental conditions where
substrate concentration is not limiting. Uncouplers disrupt the
electrochemical gradient across the membrane and inhibit the
coupling between the electron transport and phosphorylation
reactions. Thus, by uncoupling, electron flow and ATP synthase
activity are maintained and at the same time the oxygen consumption
is elevated without ATP synthesis. During artificially induced
uncoupling, glucose/pyruvate/glutamine/fatty acids are continuously
utilized to generate reducing equivalents (NADH and
FADH2) to run the ETC at its maximum rate as an attempt
to restore the electrochemical gradient across the membrane. A high
maximal respiration compared with basal OCR implies that the cells
require or uses only partial capability of the mitochondria to
support the energy demand even in the presence of ample amounts of
substrate.
A reduction in maximum oxygen consumption is a
strong indicator of potential mitochondrial dysfunction or
metabolic rewiring as in the case of cancer cells. A low maximal
capacity could also indicate decreased activity of metabolic
pathways that produce reducing equivalents, substrate availability,
mitochondrial mass or integrity is compromised.
Concept of reserve capacity
The spare or reserve capacity is a well-established
concept (25,29,30).
This value denotes the difference between the maximal and the basal
respiration. It represents the ability of the mitochondria to
respond to an increase in energy demand. Conceptually, reserve
capacity indicates how close to its bioenergetic limit a cell is
operating. Thus, the reserve or spare capacity describes an
estimate of the potential bioenergetic reserve the cell can call
upon in times of stress (25,29,30).
It has been shown that cancer cells under conditions of stress,
such as targeted thiol modification of mitochondrial proteins that
affect the mitochondrial protein function, the reserve capacity is
completely depleted and if the threshold for the basal respiration
is breached then cell death occurs (16,21,22).
Whether cells can utilize the maximal electron transport activity
for ATP synthesis will depend on the capacity of the components of
the ETC and oxidative phosphorylation system (25). From a translational point of view,
alterations in reserve capacity of tumor cells may be linked to
their changing biology during the progression of the disease
(11).
MOBI
Mitochondrial dysfunction, for example due to
stress, is characterized by a loss of efficiency in the electron
transport chain and reductions in the synthesis of high-energy
molecules, such as ATP (31).
Alternative to mitochondrial dysfunction, we introduce MOBI, which
denotes a mathematical representation or quantitative assessment of
the oncobioenergetics due to cell transformation. It indicates a
stable change in mitochondrial bioenergetics because of metabolic
rewiring that occurs at various stages of oncogenesis to meet
different cellular functional requirements, such as proliferation,
invasion and metastasis. It may represent a higher, or lower
cellular mitochondrial function compared to the normal cell
population. MOBI is derived from the parameters of mitochondrial
stress test (MiST) and is calculated from the following formula
modified from previously described (11).
MOBI=BasalMito.Resp.×log(NetMitochondrialEfficiency)
Where, the net mitochondrial efficiency
(Eq-2) is a constant for each cell type at a defined optimal
set of conditions (data unpublished). Under these conditions, it is
possible to compare the MOBI of a normal cell with another
transformed phenotype. Thus, higher net mitochondrial efficiency
indicates functionally efficient mitochondria and vice
versa.
The net mitochondrial efficiency is defined as
NetMitochondrialEfficiency=(ATPDep.Resp.+ReserveCap.)ProtonLeak
Where, basal mitochondrial respiration (Basal Mito.
Resp.), ATP dependent respiration (ATP Dep. Resp.), and reserve
capacity (Reserve Cap.) were used in the equations. MOBI was found
to be a valuable tool to distinguish the aggressiveness of
different PC cells of different stages (11) and possibly in other cancers too.
However, further studies are required to establish this notion.
Oncobioenergetic changes during prostate
carcinogenesis
Mitochondrial bioenergetic profile of
normal peripheral zone PECs
Basal mitochondrial function and ATP requirements of
PECs could be achieved efficiently by utilizing glucose, glutamine
or fatty acids as their energy substrate depending on their
bioavailability (11). This is
comparable with that of other cells such as breast epithelial cells
or cells with rapid turnover, such as monocytes. In presence of
glutamine alone, the basal respiration increases sharply to meet
the energy demands, because ATP generation through glycolysis is
significantly low or absent.
One of the major mitochondrial bioenergetic
functions of a cell is its reserve capacity. PECs demonstrate a
variable reserve capacity depending on the substrate utilization.
In presence of glucose, PECs have a very low reserve capacity.
However, in presence of glutamine, the cells have a very high
reserve capacity. This may be due to truncated Krebs cycle, where
in presence of glucose, much of the ATP generation is met through
glycolysis. Glutamine on the other hand fed into the Krebs cycle as
α-ketoglutarate, which simultaneously generates ATP and produce
metabolites for the synthesis of citrate such as OAA (Fig. 1). It seems that cells prefer glucose
over glutamine as combined use of both substrates did not elevate
reserve capacity like that of glutamine alone. However, further
studies are essential to establish substrate specificity of
PECs.
Other notable feature of PECs is the bioenergetic
phenotype which is distinct from other cell types. Majority of the
normal epithelial cells have either a resting or aerobic phenotype
(11,16,21).
PECs are specialized cells and are highly energetic that consume
large amount of energy producing substrates, especially glucose, to
maintain the metabolic needs of the cells and is necessary as they
accumulate and secrete enormous amounts of citrate.
Utilization of fatty acids to maintain the
mitochondrial respiration of PECs is poorly understood. Recent
studies show that PECs could significantly mobilize endogenous
fatty acids and can utilize both endogenous and exogenous fatty
acids to maintain the mitochondrial respiration (11). Our studies show that endogenous as
well as exogenous fatty acids could elevate reserve capacity
significantly even with a truncated TCA cycle through the
generation of NADH and FADH2 during β-oxidation and possibly
through glycerol-phosphate shuttle (Fig.
2). This reinforces the idea that PECs can utilize all energy
producing substrates efficiently to maintain its cellular metabolic
function or requirements that are unique to PECs.
Glycolytic profile of prostate
tumorigenesis
PECs are generally glycolytic. Glycolysis does not
change drastically at early non-malignant stages compared to PECs
and maintain the basal rate but changes as they become malignant.
As the cells become malignant, the glycolytic capacity (peak
glycolytic activity), and glycolytic reserve (available glycolytic
activity at any time when stressed) are all elevated (11). Deriving the cellular Oncoglycolytic
Index (OGI) from GlyST would be very useful to define the metabolic
rewiring during the transformation of PECs from indolent to
aggressive prostate cancer. Combining OGI and MOBI would be useful
parameters in defining the oncogenicity of the tumor. Our ongoing
studies are currently in this direction.
Factors affecting mitochondrial
function during prostate tumorigenesis
Prostate tumor growth and development into a
malignant form occurs over several years. During this period, tumor
undergoes various changes at tissue, cellular, genetic and
biochemical levels. Due to reduced tumor vasculature and increase
in size of the tumor, both PC cells as well as stromal cells must
‘functionally adapt’ to the microenvironment within the tumor mass
and mutually support their growth in a hostile environment
(32). There are factors that
permanently or transiently alter the mitochondrial bioenergetics of
PECs as they progress from indolent to invasive and metastatic
cancer. In addition to nuclear and mitochondrial genetic mutations,
the major environmental factor that molds the biology of cancer
cells and stromal components is low O2 tension as well
as availability of nutrients within the tumor. The key cellular
organelle that is functionally affected by changes in O2
tensions and supply of energy substrates is the mitochondria.
Therefore, modulation of mitochondrial function or
oncobioenergetics of tumor at each stage of the tumor development
is crucial for the successful growth, progression and dissemination
of the tumor.
The most common known genomic alterations in PCa
involve four pathways/genes: the androgen receptor pathway, PI3K
pathway, rearrangements that place members of the ETS transcription
factor family under control of androgen responsive promoter
TMPRSS2, and loss of function of the prostate tumor suppressor
NKX3.1 (33). These pathways can
contribute directly or indirectly to changes in mitochondrial
function. For example, PI3K/AKT activation leads to increased
mitochondrial respiration by increasing substrate supply to
mitochondria, enhancing the mitochondrial catalytic machinery such
as pyruvate dehydrogenase activity and up-regulation of
mitochondrial electron transfer and energy-transduction capacity
through complex I and phosphorylating ATP synthase (34).
The only human cellular cytoplasmic organelle that
possess their own functional DNA are the mitochondria (35). Most mtDNA copies are identical at
birth (homoplasmy). However, mtDNA molecule has no adequate
proofreading and repair mechanisms, as it is exposed to an
environment with high concentrations of ROS. Thus, mtDNA molecule
mutates at a rate that is about 10–100 times higher than nDNA.
Occasionally, a subpopulation of mtDNA molecules carries a
pathogenic mutation (heteroplasmy) and has been shown to promote
tumorigenesis (35,36). Many studies have shown that mtDNA
homoplasmic and heteroplasmic mutations in PC are very common,
perhaps due to increased ROS production (37–52) and
have been reviewed comprehensively elsewhere (6,46). In an
extensive well-controlled mitochondrial DNA sequence analysis
(6), it was concluded that in
addition to frequent somatic mtDNA mutations in PC cells, normal
appearing benign cells surrounding the tumor harbor somatic mtDNA
mutations (field cancerization effect) as well, (c) these mutations
demonstrate a progressive pattern of malignant disease and finally,
(d) some of these mutations are linked to PCa development. Thus,
these studies convey the idea that PCa development involves changes
in the mitochondrial oncobioenergetics through mtDNA mutations and
may allow the cells to meet the metabolic demands during
transformation.
Another major factor that may rapidly influence the
mitochondrial function is the tumor microenvironment.
Microenvironment consists of insoluble extracellular matrix
(proteins, glycoproteins, proteoglycans and polysaccharides), a
stroma (non-cancerous cells such as fibroblasts, adipose immune
cells and endothelial cells), and several growth factors and
cytokines secreted by stromal components. Cancer cells are embedded
in the tumor encapsulated by stromal cells. Microenvironment
provides a metabolically friendly environment for cancer cells to
grow and multiply within the tumor. They supply O2 and
energy producing substrates (glucose, fatty acids and glutamine)
required to meet high energy demands of cancer cells. As the tumor
grows, their energy demands are so high, and stroma induces
abnormal angiogenesis and nutrients are delivered through aberrant
blood vessels. This generates hypoxia and nutrient deprivation
within the tumor. It has been reported that many prostate cancers
are hypoxic, and the distribution of oxygen values is similar to
other human tumors (53). The major
organelle that is affected by low oxygen and nutrient deprivation
is the mitochondria and it must adapt to the ever-changing
microenvironment for the survival of cancer cells under these
severe conditions. Hypoxia can cause changes in mitochondrial
structure and dynamics such as impairment of mitochondrial fusion,
cause mitochondrial depolarization, and loss mitochondrial DNA
within the cells. Hypoxia can cause changes in the activity of
metabolic enzymes such as citrate synthase, respiratory complexes
such as inhibition of cytochrome oxidases by nitric oxide generated
through the activation of nitric oxide synthases, enhance
super-complex disassembly and inhibition of complex V through ROS
generation (54). Therefore, one of
our hypotheses is that energy flow within the tumor tissue
dynamically changes at each stage of the tumor development to
support the tumor growth and progression into an aggressive
tumor.
Mitochondrial oncobioenergetics of
prostate epithelial cells upon transformation and at different
stages of malignancy
To study the oncobioenergetic profile of PC cells at
different stages of cancer development, a model with genetically
identical clones of cells is required that recapitulate the
multi-stage carcinogenesis process. Recently, using RWEP1 and
malignant clones derived from this cell line, we demonstrated that
PC cells at each stage show characteristic oncobioenergetic
features as they grow from an indolent tumor to an aggressive
tumor. Our studies showed that immediately after transformation,
the cells achieve significantly higher OXPHOS and decrease further
as the cells achieve malignancy. This early switch in bioenergetics
may be due to reduced zinc accumulation, restoration of aconitase
activity, and functioning of TCA cycle without truncation (8–10). These
changes are also reflected in various mitochondrial bioenergetic
parameters such as basal, maximal, and ATP-dependent respiration as
they are elevated during early non-malignant stages and decreases
with increasing malignancy irrespective of the substrates used,
such as glucose or glutamine. Although oncobioenergetics changes
occur at each stage of tumor progression, the amount of ATP
generated is comparable with that of the normal. On the other hand,
both endogenous and exogenous fatty acid oxidation decreases with
increasing malignancy suggesting that much of the fatty acids may
be utilized for lipid synthesis for the biomass. All these data
indicate that in prostate tumorigenesis, the mitochondrial
bioenergetics of the tumor cells changes dynamically to
functionally adapt to the stage of the tumor.
Mitochondrial oncobioenergetic index of
prostate tumorigenesis
Different cellular bioenergetic parameters could be
derived from MiST trace and these parameters are valuable to
compare genetically identical cells at various stages of
malignancy. However, tumor cells of varying degree of malignancy
from genetically different cells have overlapping mitochondrial
oncobioenergetic and metabolic phenotypes and each individual
parameter does not correlate with the degree of malignancy and is
less useful and has less clinical translational value. Recently we
have shown that calculating MOBI of various PC cell lines of
various malignant stages is very useful to differentiate one cell
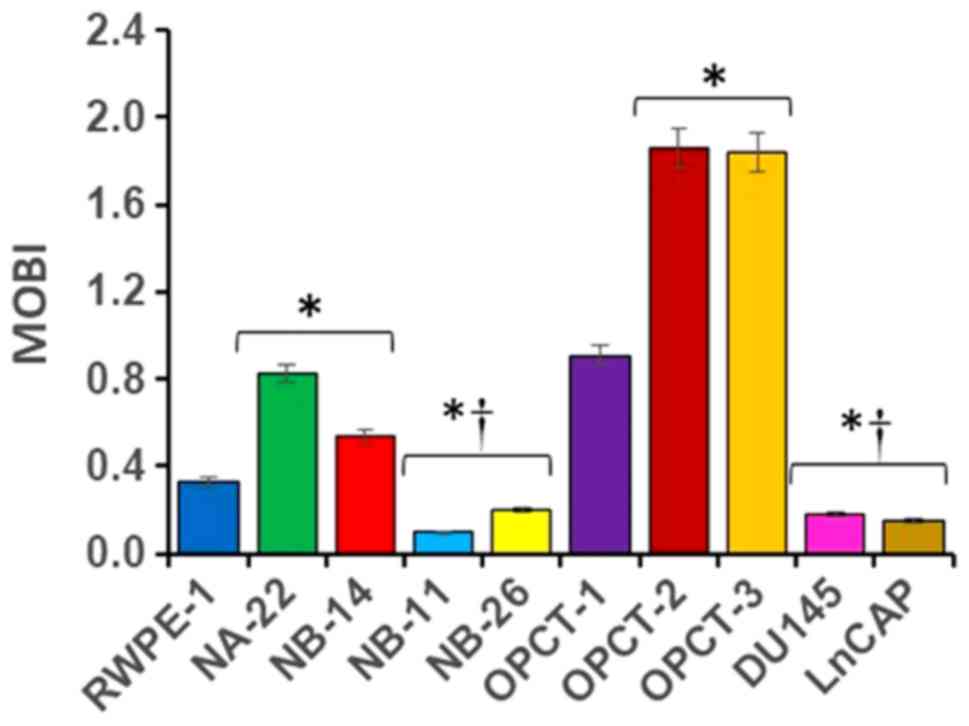
from one another based on their malignancy (Fig. 4). Our studies using ten PC cell lines
demonstrated for the first time that each cell line has a unique
MOBI, under standard assay conditions. It increases drastically as
the prostate epithelial cells transform into localized
pre-malignant stages of tumor compared to the normal PECs and it
falls below the normal as soon as they become aggressive or
malignant. MOBI seems to be independent of genetic similarity,
androgen sensitivity or mitochondrial content determined recently
for many PC cells (12), and is more
related to their malignant stage. Although yet to define, MOBI
specific for individual substrates and under stressed conditions or
both would also provide valuable information that would allow
defining the oncobioenergetics of cancer cells much more accurately
at various stages of carcinogenic process. Therefore, these
observations emphasize the importance of studying mitochondrial
function of tumors within the context of the physiological
environment to understand the mechanisms by which mitochondria
contribute to the tumor pathology.
MOBI could be a valuable tool for assessing the
aggressiveness of the tumor as well as detect whether an indolent
tumor may become aggressive in the future. It is also a valuable
tool to test the metabolic health of prostate. Any abnormality in
the mitochondrial bioenergetics of prostate tissue may represent a
pathological condition. However, suitable techniques are not
available to assess MOBI in the physiological environment without
disrupting the interaction between the cell environment and the
mitochondria in the tissues. Ex vivo approaches allow
analysis of mitochondrial bioenergetics by rigorously controlling
the assay conditions. Recent developments in technologies to
measure mitochondrial bioenergetics have shown that tissue slices
could be used for assessing the oncobioenergetics ex-vivo
(55–57). Tissue slices essentially are viable
explants of the tissue that can be cultured ex vivo and are
widely used by many researchers for several years as a model of the
organ under study including metabolism. Now digitally controlled
tissue slicers are commercially available to prepare 200 µm thick
precision-cut tissue slices (58–60).
Thus, it is possible to obtain tissue cores from specific locations
and cut slices from specific depths within the tissues and could be
compared with another slice obtained from same location and depth
with minimum variations (58–60).
However, some of the major issues confronted during the measurement
of mitochondrial bioenergetics in tissue slices needs to be
resolved. These include poor penetration of mito-effectors into the
mitochondria and slow diffusion and depletion of substrates and
oxygen during measurements. Hyperoxia and increasing substrate
concentration is an option but may result in non-physiological
response. Moreover, new algorithms should be developed by taking
into consideration of all these impeding factors and corrected for
accurate measurements.
Non-invasive techniques, such as imaging approaches
for the evaluation of mitochondrial bioenergetics of cancer, are
still in its infancy (61–64). It was mainly designed to characterize
different aspects of mitochondrial dysfunction. It permits
quantitative measurement of fluxes of ATP and oxygen, which are
controlled by mitochondria. Thus, it allows determining the
mitochondrial coupling efficiency (ATP/O2) as well as
the tissue capacities for phosphorylation and oxidation. By these
approaches, it has been shown to reveal important aspects of
mitochondrial dysfunction with disease and age. However, the
utility of these methods for quantifying the oncobioenergetics of
tumors in vivo is yet to be discovered.
Limitations of XF technology
There are several limitations to consider when
analyzing mitochondrial respiration using the Seahorse XF. They are
given below (Table I).
 | Table I.Limitations of XF technology. |
Table I.
Limitations of XF technology.
| Item | Limitations |
|---|
| Injectable | Cost of optimizing
injectable concentrations and the subsequent cost per respiratory
assay are high |
|
| A maximum of four.
It will be necessary to use multiple plates when more than four
injectable reagents are required |
|
| Injectable
compounds may potentially interfere with sensor fluorescence or the
plastic plate and produce misleading data |
| Cell lines | The generation of
immortalized cell lines are limited due to loss of differentiated
functions, high glycolytic rates, and reduced respiration |
|
| Since individual
cell populations are analyzed, they lack direct comparisons with
whole - tissue measurements that may contain multiple cell
types. |
|
| Mitochondrial
responses to substrates such as ADP, creatine, or cytochrome c
cannot be investigated in intact preparations because they are
unable to penetrate the cellular membrane. |
| Pharmaceutical | Requires
optimization for each cell line. This is to ensure that they do not
interfere with other inhibitors and produce misleading data |
| Contamination | Contamination by
viruses, mycoplasma, and other cell lines |
Conclusion
Mitochondria play a significant role in prostate
tumorigenesis and are modulated at each stage of the tumor
development to meet the metabolic and/or bioenergetic requirements
of the cell. Thus, quantification of mitochondrial oncobioenergetic
function will be a good biomarker for predicting the tumor stage.
Future studies would allow novel approaches to measure
oncobioenergetics of tumor with minimal invasive procedures
effectively in men to determine the general prostate health, tumor
staging and predict whether a tumor will proceed to malignancy in
patients. Moreover, as prostate is a slow growing tumor, modulating
the mitochondrial bioenergetics at specific stages of tumor
development will be a novel approach to treatment or prevention of
prostate cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Department of
Defense through an Exploration-Hypothesis Development Award (grant
no. W81XWH-14-1-0255).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
PKV conceived and designed the review, researched
the literature and wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The author declares that they have no competing
interests.
References
|
1
|
American Cancer Society, . Cancer Facts
& Figures 2015American Cancer Society; Atlanta, GA: 2015
|
|
2
|
Baetke SC, Adriaens ME, Seigneuric R,
Evelo CT and Eijssen LM: Molecular pathways involved in prostate
carcinogenesis: Insights from public microarray datasets. PLoS One.
7:e498312012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Center MM, Jemal A, Lortet-Tieulent J,
Ward E, Ferlay J, Brawley O and Bray F: International variation in
prostate cancer incidence and mortality rates. Eur Urol.
61:1079–1092. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kornmann B: Quality control in
mitochondria: Use it, break it, fix it, trash it. F1000Prime Rep.
6:152014. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
van der Bliek AM, Sedensky MM and Morgan
PG: Cell biology of the mitochondrion. Genetics. 207:843–871. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Parr RL, Mills J, Harbottle A, Creed JM,
Crewdson G, Reguly B and Guimont FS: Mitochondria, prostate cancer,
and biopsy sampling error. Discov Med. 15:213–220. 2013.PubMed/NCBI
|
|
7
|
Boland ML, Chourasia AH and Macleod KF:
Mitochondrial dysfunction in cancer. Front Oncol. 3:2922013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Costello LC and Franklin RB: The clinical
relevance of the metabolism of prostate cancer; zinc and tumor
suppression: Connecting the dots. Mol Cancer. 5:172006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Costello LC, Guan Z, Kukoyi B, Feng P and
Franklin RB: Terminal oxidation and the effects of zinc in prostate
versus liver mitochondria. Mitochondrion. 4:331–338. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Costello LC and Franklin RB: The
intermediary metabolism of the prostate: A key to understanding the
pathogenesis and progression of prostate malignancy. Oncology.
59:269–282. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vayalil PK and Landar A: Mitochondrial
oncobioenergetic index: A potential biomarker to predict
progression from indolent to aggressive prostate cancer.
Oncotarget. 6:43065–43080. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Panov A and Orynbayeva Z: Bioenergetic and
antiapoptotic properties of mitochondria from cultured human
prostate cancer cell lines PC-3, DU145 and LNCaP. PLoS One.
8:e720782013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Andersen KF, Divilov V, Sevak K,
Koziorowski J, Lewis JS and Pillarsetty N: Influence of free fatty
acids on glucose uptake in prostate cancer cells. Nucl Med Biol.
41:254–258. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hofer C, Laubenbacher C, Block T, Breul J,
Hartung R and Schwaiger M: Fluorine-18-fluorodeoxyglucose positron
emission tomography is useless for the detection of local
recurrence after radical prostatectomy. Eur Urol. 36:31–35. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu Y, Zuckier LS and Ghesani NV: Dominant
uptake of fatty acid over glucose by prostate cells: A potential
new diagnostic and therapeutic approach. Anticancer Res.
30:369–374. 2010.PubMed/NCBI
|
|
16
|
Diers AR, Vayalil PK, Oliva CR, Griguer
CE, Darley-Usmar V, Hurst DR, Welch DR and Landar A: Mitochondrial
bioenergetics of metastatic breast cancer cells in response to
dynamic changes in oxygen tension: Effects of HIF-1α. PLoS One.
8:e683482013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kramer PA, Chacko BK, Ravi S, Johnson MS,
Mitchell T, Barnes S, Arabshahi A, Dell'Italia LJ, George DJ,
Steele C, et al: Hemoglobin-associated oxidative stress in the
pericardial compartment of postoperative cardiac surgery patients.
Lab Invest. 95:132–141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Franklin RB, Zou J, Yu Z and Costello LC:
EAAC1 is expressed in rat and human prostate epithelial cells;
functions as a high-affinity L-aspartate transporter; and is
regulated by prolactin and testosterone. BMC Biochem. 7:102006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mayers JR and Vander Heiden MG: Nature and
nurture: What determines tumor metabolic phenotypes? Cancer Res.
77:3131–3134. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Parr RL, Dakubo GD, Thayer RE, McKenney K
and Birch-Machin MA: Mitochondrial DNA as a potential tool for
early cancer detection. Hum Genomics. 2:252–257. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vayalil PK, Oh JY, Zhou F, Diers AR, Smith
MR, Golzarian H, Oliver PG, Smith RA, Murphy MP, Velu SE and Landar
A: A novel class of mitochondria-targeted soft electrophiles
modifies mitochondrial proteins and inhibits mitochondrial
metabolism in breast cancer cells through redox mechanisms. PLoS
One. 10:e01204602015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Smith MR, Vayalil PK, Zhou F, Benavides
GA, Beggs RR, Golzarian H, Nijampatnam B, Oliver PG, Smith RA,
Murphy MP, et al: Mitochondrial thiol modification by a targeted
electrophile inhibits metabolism in breast adenocarcinoma cells by
inhibiting enzyme activity and protein levels. Redox Biol.
8:136–148. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu M, Neilson A, Swift AL, Moran R,
Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J,
et al: Multiparameter metabolic analysis reveals a close link
between attenuated mitochondrial bioenergetic function and enhanced
glycolysis dependency in human tumor cells. Am J Physiol Cell
Physiol. 292:C125–C136. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schöpf B, Schäfer G, Weber A, Talasz H,
Eder IE, Klocker H and Gnaiger E: Oxidative phosphorylation and
mitochondrial function differ between human prostate tissue and
cultured cells. FEBS J. 283:2181–2196. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chacko BK, Kramer PA, Ravi S, Benavides
GA, Mitchell T, Dranka BP, Ferrick D, Singal AK, Ballinger SW,
Bailey SM, et al: The bioenergetic health index: A new concept in
mitochondrial translational research. Clin Sci (Lond). 127:367–373.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brand MD and Nicholls DG: Assessing
mitochondrial dysfunction in cells. Biochem J. 435:297–312. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jastroch M, Divakaruni AS, Mookerjee S,
Treberg JR and Brand MD: Mitochondrial proton and electron leaks.
Essays Biochem. 47:53–67. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hill BG, Benavides GA, Lancaster JR Jr,
Ballinger S, Dell'Italia L, Jianhua Z and Darley-Usmar VM:
Integration of cellular bioenergetics with mitochondrial quality
control and autophagy. Biol Chem. 393:1485–1512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dranka BP, Hill BG and Darley-Usmar VM:
Mitochondrial reserve capacity in endothelial cells: The impact of
nitric oxide and reactive oxygen species. Free Radic Biol Med.
48:905–914. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hill BG, Dranka BP, Zou L, Chatham JC and
Darley-Usmar VM: Importance of the bioenergetic reserve capacity in
response to cardiomyocyte stress induced by 4-hydroxynonenal.
Biochem J. 424:99–107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nicolson GL: Mitochondrial dysfunction and
chronic disease: Treatment with natural supplements. Integr Med
(Encinitas). 13:35–43. 2014.PubMed/NCBI
|
|
32
|
Fiaschi T, Marini A, Giannoni E, Taddei
ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P and
Chiarugi P: Reciprocal metabolic reprogramming through lactate
shuttle coordinately influences tumor-stroma interplay. Cancer Res.
72:5130–5140. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shtivelman E, Beer TM and Evans CP:
Molecular pathways and targets in prostate cancer. Oncotarget.
5:7217–7259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li C, Li Y, He L, Agarwal AR, Zeng N,
Cadenas E and Stiles B: PI3K/AKT signaling regulates bioenergetics
in immortalized hepatocytes. Free Radic Biol Med. 60:29–40. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lightowlers RN, Chinnery PF, Turnbull DM
and Howell N: Mammalian mitochondrial genetics: Heredity,
heteroplasmy and disease. Trends Genet. 13:450–455. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wallace DC and Chalkia D: Mitochondrial
DNA genetics and the heteroplasmy conundrum in evolution and
disease. Cold Spring Harb Perspect Biol. 5:a0212202013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jerónimo C, Nomoto S, Caballero OL, Usadel
H, Henrique R, Varzim G, Oliveira J, Lopes C, Fliss MS and
Sidransky D: Mitochondrial mutations in early stage prostate cancer
and bodily fluids. Oncogene. 20:5195–5198. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jessie BC, Sun CQ, Irons HR, Marshall FF,
Wallace DC and Petros JA: Accumulation of mitochondrial DNA
deletions in the malignant prostate of patients of different ages.
Exp Gerontol. 37:169–174. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen JZ, Gokden N, Greene GF, Mukunyadzi P
and Kadlubar FF: Extensive somatic mitochondrial mutations in
primary prostate cancer using laser capture microdissection. Cancer
Res. 62:6470–6474. 2002.PubMed/NCBI
|
|
40
|
Chen JZ, Gokden N, Greene GF, Green B and
Kadlubar FF: Simultaneous generation of multiple mitochondrial DNA
mutations in human prostate tumors suggests mitochondrial
hyper-mutagenesis. Carcinogenesis. 24:1481–1487. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen JZ and Kadlubar FF: Mitochondrial
mutagenesis and oxidative stress in human prostate cancer. J
Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 22:1–12.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Petros JA, Baumann AK, Ruiz-Pesini E, Amin
MB, Sun CQ, Hall J, Lim S, Issa MM, Flanders WD, Hosseini SH, et
al: mtDNA mutations increase tumorigenicity in prostate cancer.
Proc Natl Acad Sci USA. 102:719–724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dakubo GD, Parr RL, Costello LC, Franklin
RB and Thayer RE: Altered metabolism and mitochondrial genome in
prostate cancer. J Clin Pathol. 59:10–16. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gómez-Zaera M, Abril J, González L, Aguiló
F, Condom E, Nadal M and Nunes V: Identification of somatic and
germline mitochondrial DNA sequence variants in prostate cancer
patients. Mutat Res. 595:42–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Higuchi M, Kudo T, Suzuki S, Evans TT,
Sasaki R, Wada Y, Shirakawa T, Sawyer JR and Gotoh A: Mitochondrial
DNA determines androgen dependence in prostate cancer cell lines.
Oncogene. 25:1437–1445. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Parr RL, Dakubo GD, Crandall KA, Maki J,
Reguly B, Aguirre A, Wittock R, Robinson K, Alexander JS,
Birch-Machin MA, et al: Somatic mitochondrial DNA mutations in
prostate cancer and normal appearing adjacent glands in comparison
to age-matched prostate samples without malignant histology. J Mol
Diagn. 8:312–319. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Khandrika L, Kumar B, Koul S, Maroni P and
Koul HK: Oxidative stress in prostate cancer. Cancer Lett.
282:125–136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kloss-Brandstätter A, Schäfer G, Erhart G,
Hüttenhofer A, Coassin S, Seifarth C, Summerer M, Bektic J, Klocker
H and Kronenberg F: Somatic mutations throughout the entire
mitochondrial genome are associated with elevated PSA levels in
prostate cancer patients. Am J Hum Genet. 87:802–812. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ashtiani ZO, Heidari M, Hasheminasab SM,
Ayati M and Rakhshani N: Mitochondrial D-Loop polymorphism and
microsatellite instability in prostate cancer and benign
hyperplasia patients. Asian Pac J Cancer Prev. 13:3863–3868. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Arnold RS, Sun Q, Sun CQ, Richards JC,
O'Hearn S, Osunkoya AO, Wallace DC and Petros JA: An inherited
heteroplasmic mutation in mitochondrial gene COI in a patient with
prostate cancer alters reactive oxygen, reactive nitrogen and
proliferation. Biomed Res Int. 2013:2392572013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Arnold RS, Fedewa SA, Goodman M, Osunkoya
AO, Kissick HT, Morrissey C, True LD and Petros JA: Bone metastasis
in prostate cancer: Recurring mitochondrial DNA mutation reveals
selective pressure exerted by the bone microenvironment. Bone.
78:81–86. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dong YL, Luan ZM, Xue ZY and Li YJ:
Complete mitochondrial genome sequence and mutations of the
prostate cancer model inbred Sprague-Dawley strain. Mitochondrial
DNA A DNA Mapp Seq Anal. 27:2266–2267. 2016.PubMed/NCBI
|
|
53
|
Milosevic M, Warde P, Ménard C, Chung P,
Toi A, Ishkanian A, McLean M, Pintilie M, Sykes J, Gospodarowicz M,
et al: Tumor hypoxia predicts biochemical failure following
radiotherapy for clinically localized prostate cancer. Clin Cancer
Res. 18:2108–2114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Solaini G, Baracca A, Lenaz G and Sgarbi
G: Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys
Acta. 1797:1171–1177. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Feeley KP, Westbrook DG, Bray AW and
Ballinger SW: An ex-vivo model for evaluating bioenergetics in
aortic rings. Redox Biol. 2:1003–1007. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Schuh RA, Clerc P, Hwang H, Mehrabian Z,
Bittman K, Chen H and Polster BM: Adaptation of microplate-based
respirometry for hippocampal slices and analysis of respiratory
capacity. J Neurosci Res. 89:1979–1988. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Perry CG, Kane DA, Lanza IR and Neufer PD:
Methods for assessing mitochondrial function in diabetes. Diabetes.
62:1041–1053. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
de Graaf IA, Olinga P, de Jager MH, Merema
MT, de Kanter R, van de Kerkhof EG and Groothuis GM: Preparation
and incubation of precision-cut liver and intestinal slices for
application in drug metabolism and toxicity studies. Nat Protoc.
5:1540–1551. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Martin C, Uhlig S and Ullrich V:
Videomicroscopy of methacholine-induced contraction of individual
airways in precision-cut lung slices. Eur Respir J. 9:2479–2487.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Poosti F, Pham BT, Oosterhuis D, Poelstra
K, van Goor H, Olinga P and Hillebrands JL: Precision-cut kidney
slices (PCKS) to study development of renal fibrosis and efficacy
of drug targeting ex vivo. Dis Model Mech. 8:1227–1236. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lanza IR and Nair KS: Mitochondrial
metabolic function assessed in vivo and in vitro. Curr Opin Clin
Nutr Metab Care. 13:511–517. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Marcinek DJ: Mitochondrial dysfunction
measured in vivo. Acta Physiol Scand. 182:343–352. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Amara CE, Marcinek DJ, Shankland EG,
Schenkman KA, Arakaki LS and Conley KE: Mitochondrial function in
vivo: Spectroscopy provides window on cellular energetics. Methods.
46:312–318. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Campbell MD and Marcinek DJ: Evaluation of
in vivo mitochondrial bioenergetics in skeletal muscle using NMR
and optical methods. Biochim Biophys Acta. 1862:716–724. 2016.
View Article : Google Scholar : PubMed/NCBI
|