Introduction
The mortality rate of ovarian cancer ranks as the
highest among gynecological tumors in the western world, and its
incidence is increasing on a yearly basis (1). This is due to a lack of specific
symptoms, which impedes its early diagnosis and results in high
recurrence rates following radical surgery and chemotherapy
(1). Although treatment outcomes
have greatly improved, the 5-year survival rate of patients with
ovarian cancer remains low, at 46.5% in 2017 (2), whereas the survival rate of patients
with distant metastases is worse (29%). Out of all cases, ~70% are
diagnosed at an advanced stage, and have poor prognosis (3). The 5-year survival rate for patients
with advanced ovarian cancer is only ~20%; however, if diagnosed
early, this can increase to 85–90% (4). Among the different pathological types,
serous epithelial ovarian cancer is the most common (5). Therefore, an early diagnosis of serous
epithelial ovarian cancer may greatly improve prognosis.
At present, the standard method for the early
diagnosis and monitoring of ovarian cancer is ultrasound
examination combined with serum tumor marker detection (6). However, the specificity of this
diagnostic method is low, and the 5-year survival rate after
diagnosis using this approach is only 30% (7). The occurrence and development of tumors
are associated with accumulated molecular genetic or genomic
alterations (8). For instance,
high-grade serous ovarian cancer cases frequently exhibit tumor
protein p53 mutations and alterations in BRCA1/2 DNA repair
associated and related homologous recombination genes, either by
mutation, promoter methylation or loss of heterozygosity (9). Therefore, it is important to
investigate the molecular mechanisms underpinning the malignant
behavior of serous epithelial ovarian cancer cells to develop more
effective methods for early diagnosis, and to identify more
reliable molecular markers that may be used either as novel
therapeutic targets or to assess prognosis. Gene expression
microarray analysis is an efficient and large-scale technique for
obtaining genetic data (10). It has
been widely used to explore gene expression profiles in numerous
types of human cancer (11).
Microarray data have become increasingly available in the public
domain over the last few years, in platforms such as the National
Center for Biotechnology Information (NCBI) Gene Expression Omnibus
(GEO) database. The large volume of data that has been published in
these public databases, and the integration of multiple databases,
allow for an exhaustive study of underlying molecular mechanisms.
The integration and analysis of microarray data from several gene
expression profiles may enable investigators to obtain more
reliable molecular markers. However, since these data originate
from different microarray products from a wide range of experiments
using different reagents, which have also been performed by
operators of varying proficiencies, a large degree of variability
exists among datasets (12). This
problem may be solved using batch normalization programs available
in R software.
In the present study, we employed an integrated
bioinformatics approach to identify potential molecular markers for
the early detection and prognosis of serous epithelial ovarian
cancer. Furthermore, the markers obtained may be targets for the
development of novel therapies for serous epithelial ovarian
cancer.
Materials and methods
Microarray data
The GEO database (www.ncbi.nlm.nih.gov/geo) is an international public
repository that archives and distributes high-throughput gene
expression data and other functional genomics datasets (13). The keywords ‘ovarian cancer gene
expression’ were used to search the GEO database, and the CEL files
of GSE14407 (14), GSE54388
(15) and GSE38666 (16) datasets were downloaded for subsequent
analysis. The quality of the gene chips was detected by RNA
degradation mapping (17). Only gene
chips with a proper degradation slope in RNA degradation mapping
were included in the subsequent analysis.
Data pre-treatment and identification
of differentially expressed genes (DEGs)
All data were processed using R software (www.r-project.org). The Affy package (version 3.9;
www.bioconductor.org/packages/release/bioc/html/affy.html)
was used to extract expression data from CEL files, and the Robust
Multi-Array Average method in R was used to perform quartile data
normalization of the three expression datasets (18). Following normalization, data from the
three microarray datasets were merged to form a new gene expression
profile. The sva package (version 3.9; bioconductor.org/packages/release/bioc/html/sva.html.)
in R was used to identify, estimate and remove unwanted sources of
variation in high-throughput experiments to eliminate the batch
effect (19). The DEGs between
serous epithelial ovarian cancer and normal ovarian surface
epithelial tissue from the three microarray datasets were analyzed
using the Limma package (version 3.9; http://www.bioconductor.org/packages/release/bioc/html/limma.html).
Values of |log fold change (FC)|>1.0 and adjusted P<0.05 were
selected as the cut-off criteria for DEG selection.
GO and KEGG pathway enrichment
analyses of DEGs
The DAVID database is an important online tool for
gene function analysis. Gene Ontology (GO) analysis of the DEGs was
performed using DAVID 6.7 (david.ncifcrf.gov). Kyoto Encyclopedia of Genes and
Genomes (KEGG) is an online encyclopedia that assigns functions to
genes and genomes at molecular and higher levels (20). KEGG pathway analysis of DEGs was
performed using KEGG Orthology-Based Annotation System (KOBAS) 3.0
(kobas.cbi.pku.edu.cn), an online
analysis tool. GO functional enrichment was assessed using the
criteria of P<0.05 and false discovery rate (FDR) <0.05.
P<0.05 was used to identify statistically KEGG pathways.
Subsequently, the GOplot package (version 1.0.2; wencke.github.io)
was used to construct the Chord diagram, and the clusterProfiler
package (version 3.9; www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
was used to create the bar plot.
Protein-protein interaction (PPI)
networks
PPI networks may be used to understand normal cell
function and to study disease pathogenesis (21). In the present study, the STRING
database (string-db.org) was used to explore the
PPIs of the DEGs, with a cut-off criterion set at an interaction
score >0.99. PPI networks were constructed using Cytoscape
software (version 3.6.1; http://cytoscape.org), which is a bioinformatics
program for the visualization of molecular interaction networks.
Each node in the PPI network represents a gene, protein or other
molecule, and the connections between the nodes represent the
interactions between these biomolecules. The most closely
associated nodes may indicate core proteins or key genes with
important physiological regulatory functions (22). Therefore, the interactions and
pathway associations among proteins encoded by the DEGs in serous
epithelial ovarian cancer were assessed in this manner.
Oncomine analysis of hub genes
Oncomine (www.oncomine.org) is a bioinformatics program designed
to collect, standardize and analyze cancer transcriptome data. It
integrates RNA- and DNA-sequencing data from various sources,
including GEO, The Cancer Genome Atlas (TCGA) (https://cancergenome.nih.gov) and published literature
(23). A meta-analysis of the
selected hub genes in ovarian cancer compared with normal ovarian
tissue was performed using Oncomine to compare these genes
expression across different studies.
Kaplan-Meier (KM) survival
analysis
The KM estimate is a nonparametric statistic used to
measure the percentage of patients living for a certain period of
time following a specific treatment. The hub genes were analyzed
using an online tool, KM Plotter (updated on 2/20/2019; kmplot.com/analysis), which was used to assess overall
and progression-free survival of patients with serous epithelial
ovarian cancer by the log-rank test. This tool was constructed
using the gene expression and survival data of 1,232 patients with
serous epithelial ovarian cancer, which were downloaded from the
GEO and TCGA databases (24).
Results
Details of the datasets
CEL files of the datasets GSE14407, GSE54388 and
GSE38666 were downloaded from the GEO database. The platform used
to generate data for all three datasets was the Human Genome U133
Plus 2.0 array (GPL570; HG-U133_Plus_2; Affymetrix; Thermo Fisher
Scientific, Inc.). These datasets are stored in a public repository
(doi.org/10.6084/m9.figshare.8148608.v2) and are easily
obtained. The GSE14407 dataset included data from 12 healthy
ovarian surface epithelial samples and 12 laser-capture
microdissected serous ovarian cancer epithelial samples. The
GSE54388 dataset included data from 6 human ovarian surface
epithelial samples and 16 serous ovarian cancer epithelial samples.
The GSE38666 dataset included data from 12 normal ovarian surface
epithelial samples and 18 serous cancer epithelial samples. The
characteristics of these datasets are shown in Table I. The data from the three microarray
datasets were merged to form a novel gene expression profile, and
the detailed results are available in a public repository
(doi.org/10.6084/m9.figshare.8148617.v1). Gene chips of
good quality from each dataset were selected for subsequent
analysis. Additionally, the RNA degradation maps for the three
datasets are shown in Fig. S1.
 | Table I.Characteristics of the three
datasets. |
Table I.
Characteristics of the three
datasets.
| GSE accession
number | GPL | Organism | Control samples,
n | Cancer samples,
n | Country |
|---|
| GSE14407 | GPL570 | Homo
sapiens | 12 | 12 | USA |
| GSE54388 | GPL570 | Homo
sapiens | 6 | 16 | USA |
| GSE38666 | GPL570 | Homo
sapiens | 12 | 18 | USA |
Identification of DEGs
The novel gene expression profile created by merging
the three original microarray datasets was subsequently analyzed
using the Limma package. According to the criteria of |log
FC|>1.0 and adjusted P<0.05, 2,212 DEGs were identified,
comprising 1,300 upregulated and 912 downregulated genes. The
detailed results are shown in Table
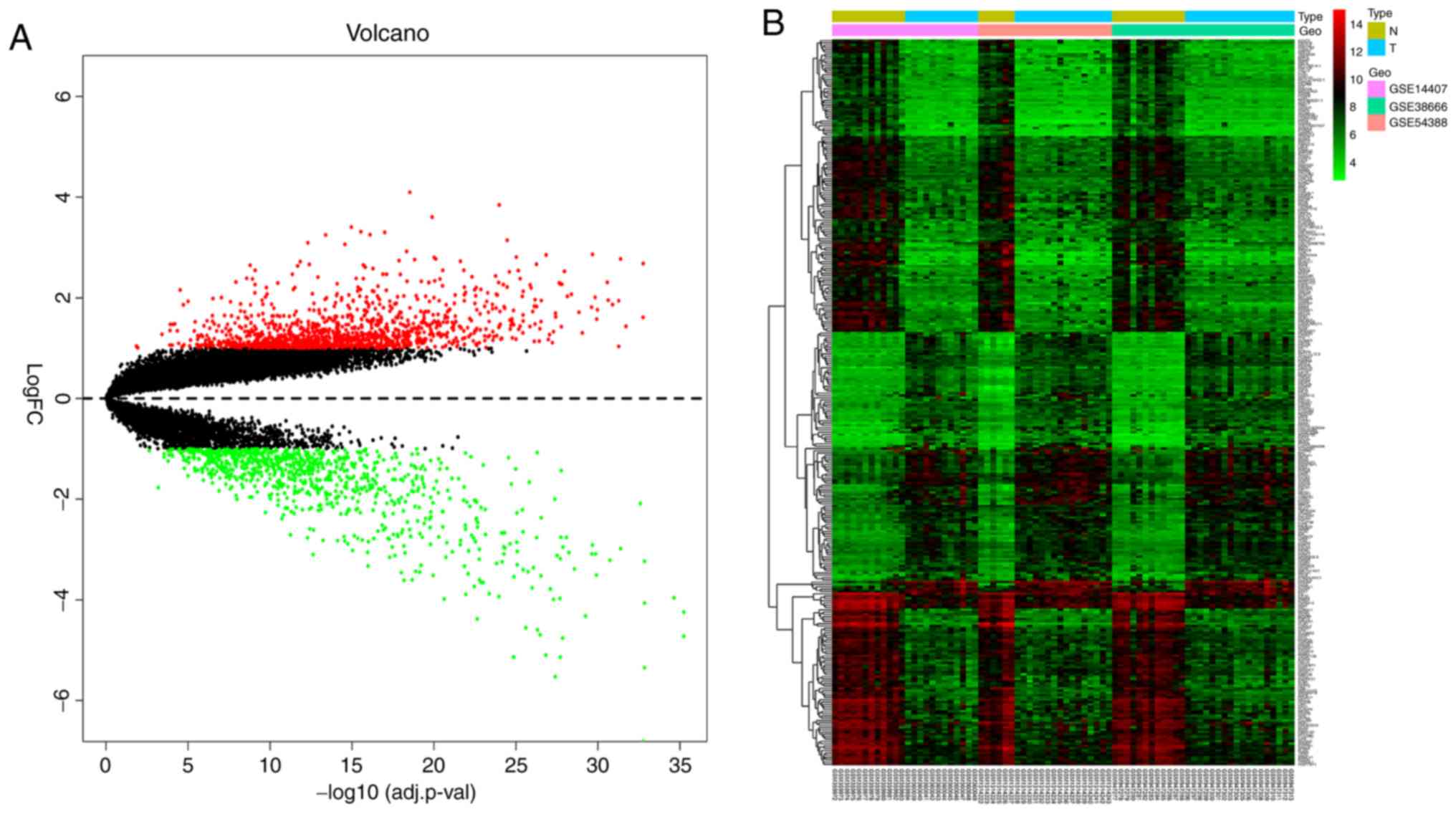
SI. Heat and volcano maps, illustrating the trends in DEG
expression, are shown in Fig. 1.
GO term enrichment analysis
GO enrichment analysis was performed using the DAVID
online analysis tool. GO enrichment with FDR <0.05 is shown in
Fig. 2A. GO enrichment with
P<0.05 was divided into three functional groups, including
molecular function, biological processes and cellular components.
The parsed results are shown in Fig.
2B and Tables II–IV. The distribution of certain DEGs in
serous epithelial ovarian cancer for different GO enriched
functions is shown in Fig. 3. The
detailed results are shown in Table
SII. The results revealed that these DEGs were mainly involved
in the tumor-associated biological processes such as cell cycle,
cell division, mitosis and others.
| Table II.Enrichment of biological
processes. |
Table II.
Enrichment of biological
processes.
| Term | Description | Gene count, n | P-value |
|---|
| GO:0000280 | Nuclear
division | 63 |
1.52×10−16 |
| GO:0007067 | Mitosis | 63 |
1.52×10−16 |
| GO:0048285 | Organelle
fission | 63 |
1.65×10−15 |
| GO:0051301 | Cell division | 71 |
3.59×10−14 |
| GO:0007059 | Chromosome
segregation | 27 |
5.61×10−9 |
| GO:0051726 | Regulation of cell
cycle | 62 |
6.58×10−8 |
| GO:0006323 | DNA packaging | 31 |
1.24×10−7 |
| GO:0007051 | Spindle
organization | 18 |
1.71×10−7 |
| GO:0065004 | Protein-DNA complex
assembly | 24 |
4.68×10−6 |
| GO:0007017 | Microtubule-based
process | 44 |
4.44×10−5 |
| GO:0006260 | DNA
replication | 35 |
9.68×10−5 |
| GO:0007155 | Cell adhesion | 89 | 0.001049 |
| GO:0042127 | Regulation of cell
proliferation | 98 | 0.001138 |
| GO:0016477 | Cell migration | 42 | 0.001186 |
| GO:0006259 | DNA metabolic
process | 67 | 0.001746 |
| GO:0001525 | Angiogenesis | 26 | 0.001804 |
| GO:0008283 | Cell
proliferation | 59 | 0.002059 |
| Table IV.Enrichment of molecular function. |
Table IV.
Enrichment of molecular function.
| Term | Description | Gene count, n | P-value |
|---|
| GO:0004857 | Enzyme inhibitor
activity | 43 |
1.20×10−4 |
| GO:0001871 | Pattern
binding | 28 |
2.80×10−4 |
| GO:0030247 | Polysaccharide
binding | 28 |
2.80×10−4 |
| GO:0030246 | Carbohydrate
binding | 51 |
3.27×10−4 |
| GO:0005509 | Calcium ion
binding | 109 |
4.52×10−4 |
| GO:0005539 | Glycosaminoglycan
binding | 25 |
8.32×10−4 |
| GO:0008083 | Growth factor
activity | 24 | 0.01087 |
| GO:0016538 | Cyclin-dependent
protein kinase regulator activity | 6 | 0.01207 |
| GO:0003777 | Microtubule motor
activity | 14 | 0.01373 |
| GO:0005201 | Extracellular
matrix structural constituent | 15 | 0.01470 |
| GO:0004859 | Phospholipase
inhibitor activity | 5 | 0.01543 |
| GO:0051287 | NAD or NADH
binding | 10 | 0.01735 |
| GO:0046915 | Transition metal
ion transmembrane transporter activity | 7 | 0.02473 |
| GO:0042813 | Wnt receptor
activity | 4 | 0.03605 |
| GO:0019887 | Protein kinase
regulator activity | 13 | 0.04381 |
KEGG pathway analysis
The most significantly enriched pathways of the DEGs
were identified using the KOBAS database. The results of this
analysis are shown in Table V and
Fig. 4. The signaling pathways of
DEGs were predominantly enriched in ‘Wnt signaling pathway’, ‘viral
carcinogenesis’, ‘pathways in cancer’, ‘PI3K-Akt signaling
pathway’, ‘cell cycle’, ‘extracellular matrix (ECM)-receptor
interaction’, ‘p53 signaling pathway’ and ‘focal adhesion’.
| Table V.Kyoto Encyclopedia of Genes and
Genomes pathways of differentially expressed genes in ovarian
cancer. |
Table V.
Kyoto Encyclopedia of Genes and
Genomes pathways of differentially expressed genes in ovarian
cancer.
| ID | Term | Gene count, n | P-value |
|---|
| hsa01100 | Metabolic
pathways | 124 |
1.58×10−16 |
| hsa04110 | Cell cycle | 35 |
2.86×10−16 |
| hsa05200 | Pathways in
cancer | 52 |
2.32×10−11 |
| hsa05203 | Viral
carcinogenesis | 36 |
2.33×10−11 |
| hsa04114 | Oocyte meiosis | 24 |
8.04×10−9 |
| hsa04151 | PI3K-Akt signaling
pathway | 42 |
1.02×10−8 |
| hsa04310 | Wnt signaling
pathway | 24 |
1.00×10−7 |
| hsa05205 | Proteoglycans in
cancer | 28 |
4.20×10−7 |
| hsa04512 | ECM-receptor
interaction | 16 |
2.40×10−6 |
| hsa04115 | p53 signaling
pathway | 14 |
6.84×10−6 |
| hsa00350 | Tyrosine
metabolism | 10 |
1.14×10−5 |
| hsa05217 | Basal cell
carcinoma | 12 |
1.63×10−5 |
| hsa04914 |
Progesterone-mediated oocyte
maturation | 16 |
1.76×10−5 |
| hsa04510 | Focal adhesion | 24 |
2.43×10−5 |
| hsa04974 | Protein digestion
and absorption | 14 |
9.22×10−5 |
| hsa04550 | Signaling pathways
regulating pluripotency of stem cells | 18 | 0.00011 |
| hsa04014 | Ras signaling
pathway | 24 | 0.00012 |
| hsa05222 | Small cell lung
cancer | 13 | 0.00020 |
| hsa04150 | mTOR signaling
pathway | 18 | 0.00027 |
| hsa03030 | DNA
replication | 8 | 0.00037 |
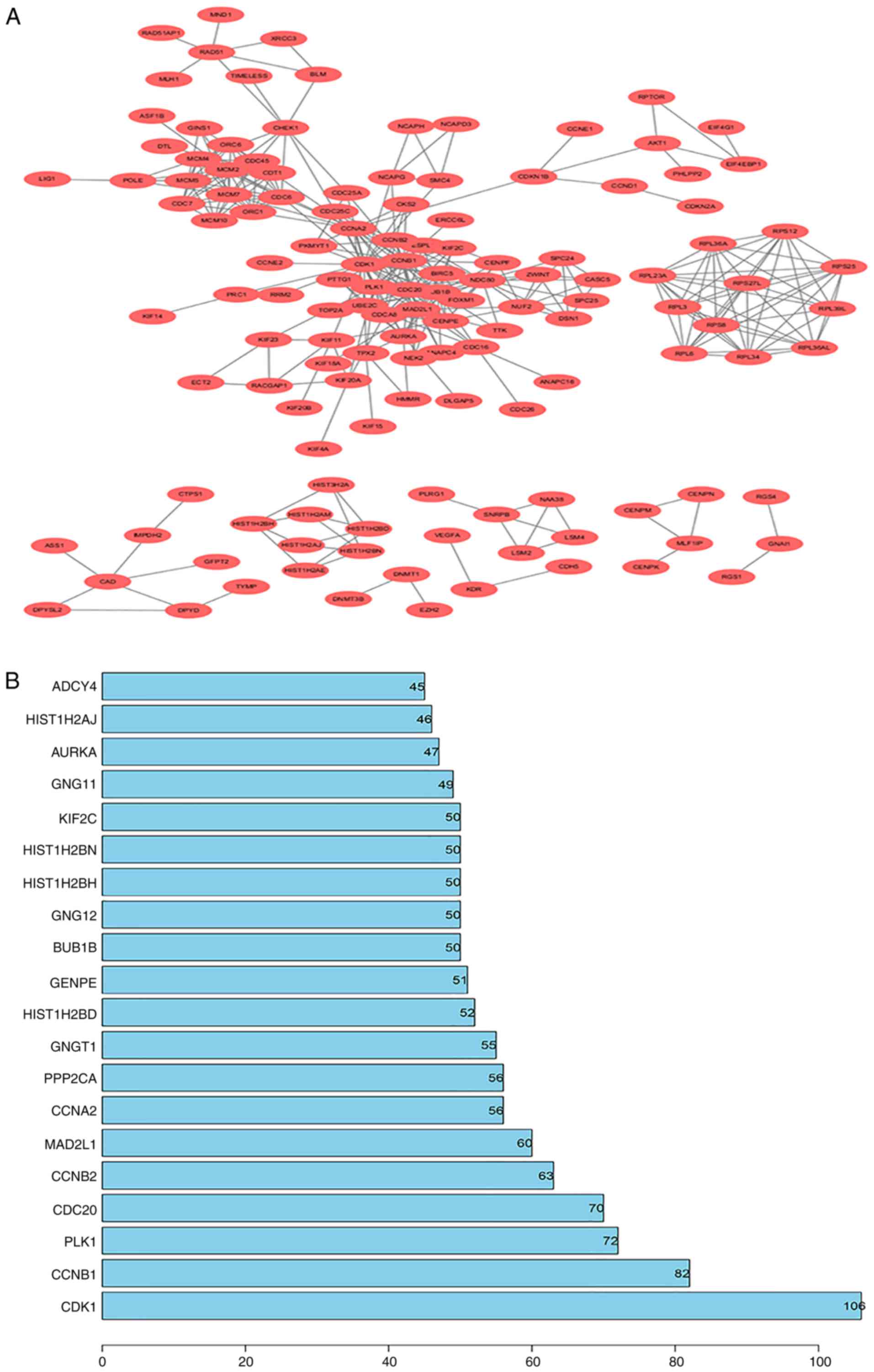
PPI network construction
All DEGs were screened using the STRING database to
further investigate their properties and the interactions among
them. The PPI network of DEGs, with a criterion of interaction
score >0.99, was built using Cytoscape software, and the results
are shown in Fig. 5A. To identify
core genes, the number of connections were counted for each gene.
The detailed results are shown in Table
SIII. The top 20 genes with the most connections, which
represent the most important DEGs, are presented in Fig. 5B. Among the 20 closely associated
genes, CDK1 exhibited the highest node degree of 106.
Oncomine analysis of hub genes
An Oncomine database analysis of cancer tissue
compared with normal tissue was performed for the 20 core genes
identified for serous epithelial ovarian cancer. These
meta-analysis results revealed that cell division control protein 1
(CDC1), cyclin B1 (CCNB1), polo like kinase 1
(PLK1), cell division cycle 20 (CDC20), cyclin B2
(CCNB2), mitotic arrest deficient 2 like 1 (MAD2L1),
cyclin A2 (CCNA2), histone cluster 1 H2B family member d
(HIST1H2BD), centromere protein E (CENPE), BUB1
mitotic checkpoint serine/threonine kinase B (BUB1B),
histone cluster 1 H2B family member h (HIST1H2BH), kinesin
family member 2C (KIF2C) and aurora kinase A (AURKA)
were upregulated, whereas G protein subunit γ 12 (GNG12) and
G protein subunit γ 11 (GNG11) were downregulated, among the
different datasets. The results of this analysis are shown in
Fig. 6.
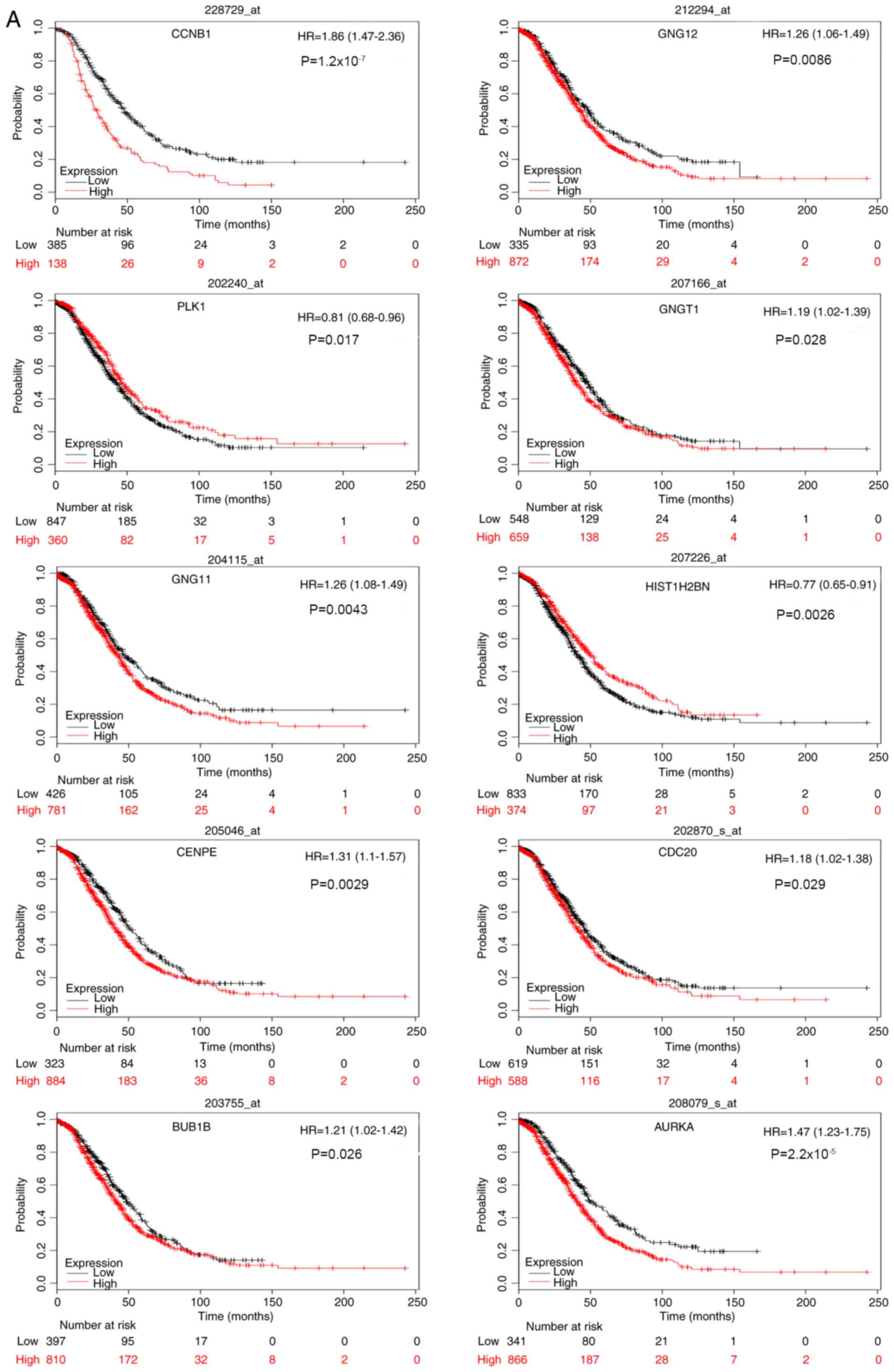
KM survival analysis
Survival analysis of the 20 core genes for serous
epithelial ovarian cancer was performed by constructing a KM curve
using the KM Plotter package. This analysis revealed that high
expression levels of CCNB1, GNG12 and G protein subunit g
transducin 1 (GNGT1), and low expression levels of
PLK1 were associated with poor overall and progression-free
survival in patients with serous ovarian cancer. In addition, high
expression levels of AURKA, BUB1B, CDC20, CENPE and
GNG11, and low expression levels of HIST1H2BN, were
associated with poor overall survival, whereas high expression
levels of adenylate cyclase 4 (ADCY4) and protein
phosphatase 2 catalytic subunit α (PPP2CA) were associated
with poor progression-free survival. The results for overall and
progression-free survival analysis are shown in Fig. 7A and B, respectively.
Discussion
Ovarian cancer is the most prevalent gynecological
cancer, and 75% of patients are diagnosed with advanced disease, of
which only 20% survive for 5 years after diagnosis (25). The majority of patients with ovarian
cancer are initially responsive to conventional chemotherapy, and
enter clinical remission following initial treatment (26). However, tumor metastasis and
recurrence occur in >70% of patients with ovarian cancer,
despite treatment, and lead to mortality (27). Among the various types of ovarian
cancer, serous epithelial ovarian cancer is the most common
pathological type (5). Therefore,
exploring the molecular mechanisms of serous epithelial ovarian
cancer development is important to identify novel molecular markers
and therapeutic targets. Identifying effective methods for
preventing the progression of ovarian cancer is particularly
important for improving the overall and progression-free survival
of patients with serous epithelial ovarian cancer.
Previous research has suggested that molecular
biomarkers may be used for the accurate diagnosis of cancer
(28). These molecular markers may
be more sensitive and specific than traditional screening methods,
and they are easier to use (24).
Microarray and high-throughput sequencing technologies, capable of
detecting the expression levels of tens of millions of human genes,
have been widely used to identify molecular biomarkers and
potential targets for the diagnosis and treatment of cancer
(29). Thus far, numerous basic
research papers on the mechanisms of ovarian cancer have been
published, but the 5-year survival rate of patients with ovarian
cancer remains relatively low. Furthermore, no biomarkers for
predicting the prognosis or monitoring the effectiveness of
treatments have been identified, since the majority of studies have
focused on simple genetic events or the results of a single
experimental study (30). In the
present study, three gene expression datasets from different
experiments were combined and batch-corrected using the sva
package. They were subsequently analyzed using R software and other
bioinformatics tools. A total of 2,212 DEGs were identified in the
present study using the Limma package. This included 1,300
upregulated and 912 downregulated genes. These were further divided
into three groups through GO functional annotation, including
molecular functions, biological processes and cellular components.
The molecular functions included ‘Calcium ion binding’,
‘polysaccharide binding’, ‘enzyme inhibitor activity’, ‘growth
factor activity’, ‘cyclin-dependent protein kinase regulator
activity’, ‘microtubule motor activity’, ‘Wnt receptor activity’
and ‘protein kinase regulator activity’. The biological processes
included ‘regulation of cell cycle’, ‘mitosis’, ‘DNA packaging’,
‘DNA replication’, and ‘Chromosome segregation’, whereas the
cellular components included ‘extracellular region part’,
‘chromosome’, ‘extracellular matrix’, ‘microtubule cytoskeleton’,
‘nucleosome’, ‘spindle’ and ‘condensed chromosome kinetochore’. The
majority of these enrichment functions are associated with
tumorigenesis and development. For instance, growth factor activity
in various types of cancer is able to regulate cell proliferation,
differentiation and apoptosis, thus affecting the ability of cells
to self-renew, migrate, senesce or undergo apoptosis (31). Cyclin-dependent kinases
(CDKs/cyclins) form a family of heterodimeric kinases that serve
important roles in regulating cell cycle progression, transcription
and other major biological processes (32). Alterations in CDK activity affect the
proliferation of cancer cells, and abnormal activities of these
proteins have been reported in various types of human cancer, such
as pancreatic cancer (32,33). Wnt signaling regulates an
evolutionarily conserved pathway that serves an important role in
numerous cellular activities, including cell proliferation, calcium
homeostasis and cellular polarity (34). Wnt receptor activity is upregulated
in a variety of cancer types, such as colorectal and gastric cancer
(34–36). Microtubules are dynamic structures
that are involved in cell movement, intracellular trafficking and
mitosis (37). Alterations in
microtubule activity have been reported in a range of cancer types,
such as breast and non-small cell lung cancer (37). These alterations have been associated
with poor prognosis and chemotherapy resistance in solid and
hematological types of cancer (37).
Nucleosome assembly following DNA replication, DNA repair and gene
transcription is critical for the maintenance of genome stability
and epigenetic information (38).
Alterations or mutations that affect nucleosome assembly have also
been implicated in certain types of cancer, such as cervical cancer
(38,39).
In addition, the enriched KEGG pathways of DEGs
identified in the present study included the ‘cell cycle’,
‘pathways in cancer’, ‘PI3K-Akt signaling pathway’, ‘Wnt signaling
pathway’, ‘ECM-receptor interaction’, ‘mTOR signaling pathway’ and
‘focal adhesion’. The significance of the PI3K-Akt signaling
pathway in ovarian cancer has been reported previously (40). In a copy number analysis on 93
primary ovarian tumors using array comparative genomic
hybridization, Huang et al (40) identified that the PI3K-Akt signaling
pathway was the most frequently altered cancer-associated signaling
pathway. The Wnt/β-catenin signaling pathway regulates a variety of
fundamental cellular functions, including proliferation, polarity,
adhesion and motility during development, differentiation and adult
tissue homeostasis (41).
Furthermore, the Wnt/β-catenin signaling pathway has been
demonstrated to be essential for the growth and progression of
ovarian cancer (42). Bodnar et
al (43) demonstrated that
activation of the Wnt/β-catenin signaling pathway may facilitate
the proliferation and differentiation of ovarian cancer cells,
inhibit apoptosis and promote ovarian cancer growth (43). The mTOR signaling pathway regulates
several major physiological processes, including protein synthesis,
macromolecular biosynthesis, cytoskeleton remodeling, angiogenesis,
survival, metabolism, autophagy and response to stress (44). Due to its pivotal role in cell growth
and differentiation, its dysregulation is associated with
pathological conditions, including tumor transformation and
progression in breast, gastrointestinal, liver and prostate cancer
(45). The detection of components
of these signaling pathways, and their expression levels, may help
predict the occurrence and development of serous epithelial ovarian
cancer, and provide potential therapeutic targets.
In the present study, 20 closely associated genes
were identified by constructing a PPI network of proteins encoded
by DEGs. Oncomine analysis further revealed that the following 15
genes were core serous epithelial ovarian cancer-associated genes
among the different datasets: CDC1, CCNB1, PLK1, CDC20, CCNB2,
MAD2L1, CCNA2, HIST1H2BD, CENPE, BUB1B, HIST1H2BH, KIF2C, AURKA,
GNG12 and GNG11. Among the 20 closely associated genes,
CDK1 exhibited the highest node degree of 106. CDK1 is an important
cell cycle-regulating protein that serves key roles in the cell
cycle G2/M-phase regulation network (46). Upregulated protein expression levels
of CDK1 have been detected in numerous human malignant tumor
tissues, and have been found to be closely associated with the
malignant prognosis (47,48). Yang et al (49) demonstrated that high expression
levels of cytoplasmic CDK1 promote the growth of epithelial ovarian
cancer cells, indicating a poor overall survival rate (49). Therefore, CDK1 is expected to be an
effective therapeutic target for epithelial ovarian cancer by
disrupting the ovarian cancer cell cycle. Survival analysis
identified CCNB1, PLK1, GNG12 and GNGT1 as being
associated with the overall and progression-free survival of
patients with serous epithelial ovarian cancer. In addition, high
expression levels of AURKA, BUB1B, CDC20, CENPE and
GNG11, and low expression levels of HIST1H2BN, were
associated with poor overall survival of serous epithelial ovarian
cancer, whereas high expression levels of ADCY4 and PPP2CA were
associated with poor progression-free survival.
CCNB1 is a mitotic cyclin, due to its crucial role
in modulating G2/M-phase progression in the cell cycle
(50). It has been demonstrated to
be involved in cell growth, differentiation, apoptosis and
metastasis in numerous types of cancer such as lung cancer
(51–53). Previous studies have indicated that
CCNB1 is associated with malignancy, and upregulation of CCNB1 has
been identified as a marker of poor prognosis in patients with
non-small cell lung cancer, head and neck squamous cell carcinoma,
breast cancer and hepatocellular carcinoma (54–57).
Therefore, CCNB1 has the potential to also be a molecular marker of
ovarian cancer prognosis. PLK1 is a member of the polo subfamily of
serine/threonine protein kinases (collectively referred to as
PLKs), which serve key roles in a variety of cellular processes,
including cell cycle progression, differentiation and survival
(58). Overexpression of PLK1 in
breast cancer cells is able to initiate transcriptional programs
required for mitosis by phosphorylating the transcription factor
forkhead box M1, overriding the DNA damage checkpoint, contributing
to the induction of invasiveness by phosphorylating vimentin and
impairing mitotic integrity, which lead to aneuploidy and are
associated with tumor formation (59). PLK1 is upregulated in various types
of human cancer, including glioma, thyroid cancer, head and neck
squamous cell carcinoma, melanoma, and colorectal, esophageal,
ovarian, breast and prostate cancer (60). Weichert et al (61) reported that PLK1 is frequently
upregulated in malignant epithelial ovarian tumors, and that this
upregulation is associated with mitosis and poor prognosis in
patients (61). However, a recently
published study revealed that overexpression of PLK1 could act as a
tumor suppressor by disrupting mitotic progression and cytokinesis
in vitro and in vivo, and an increase in PLK1 levels
in patients with breast cancer was associated with an improved
prognosis (62). In the present
study, high expression levels of PLK1 were associated with an
improvement in overall and progression-free survival of patients
with serous epithelial ovarian cancer. However, further research is
required to explore the association between PLK1 and survival in
such patients.
GNG12 is a member of the G-protein family,
corresponding to the G-protein γ12 subunit (63). Larson et al (64) revealed that GNG12 is a negative
regulator of the response to lipopolysaccharide, and may be a
critical factor in the overall inflammatory signaling cascade
(64). Proteomic analysis has
demonstrated that GNG12 regulates cell growth and casein synthesis
by activating the Leu-mediated mTOR complex 1 signaling pathway
(65). However, at present, a
limited number of studies have been published regarding GNG12, and
therefore further studies are required to determine its role in
cancer. CENPE is a kinesin motor protein found in kinetochore
protein complexes, whose motility is required for medium-term
correct chromosomal alignment (66).
Balamuth et al (67) reported
that CENPE may be a novel target for neuroblastoma. In addition,
CENPE has been revealed to be upregulated in invasive breast tumors
compared with normal breast tissue (68). BUB1B exerts an important role in
spindle assembly checkpoint signaling and the stable attachment of
kinetochore and spindle microtubules (69–71).
Therefore, the disruption of BUB1B function often leads to abnormal
mitosis. A growing body of evidence suggests that BUB1B serves a
key role in several types of cancer, including breast, stomach,
colorectal and prostate cancer (72–75).
AURKA, a member of the serine/threonine kinase
family, is localized on centrosomes and mitotic spindles, where it
mediates mitotic progression and chromosomal stability (76). The AURKA gene is upregulated
in numerous types of malignancies, including bladder, breast,
colon, liver, ovarian, pancreatic, gastric and esophageal cancer
(77). Several previously published
studies have revealed that upregulation of AURKA in clinical
head and neck squamous cell carcinoma (HNSCC) specimens is
associated with invasion, advanced stage and poor prognosis
(78,79). Mignogna et al (80) revealed that AURKA may be used
to predict resistance to platinum-based chemotherapy, and as a
prognostic factor in ovarian cancer. Therefore, AURKA
warrants further investigation in prospective clinical trials, and
may have prognostic and therapeutic value in ovarian cancer.
In conclusion, the present study integrated multiple
microarray datasets from the NCBI GEO database into one dataset,
which was subsequently subjected to bioinformatics analysis. DEGs
were identified, GO and KEGG analyses were performed and a PPI
network of DEGs in serous epithelial ovarian cancer was
constructed. DEGs were revealed to be mainly enriched in pathways
associated with tumor formation and development, such as ‘Wnt
signaling pathway’, ‘PI3K-Akt signaling pathway’, ‘pathways in
cancer’ and ‘mTOR signaling pathway’, which provide a theoretical
basis for studying the biological processes of serous ovarian
cancer. In addition, the Oncomine database was used to compare the
identified candidate genes across multiple databases. Finally, the
effect of these genes on survival rate was investigated. Overall,
the results obtained in the present study enhanced the
understanding of the pathogenesis of serous epithelial ovarian
cancer and provided novel avenues for investigating the potential
molecular mechanisms. The present study had important clinical
implications for the early diagnosis, prognosis and development of
more precise molecular therapies of ovarian cancer, although
further studies are required to validate the identified candidate
genes.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the [FIGSHARE] repository
(https://figshare.com/authors/Yubo_Zhang/6712286).
Authors' contributions
YBZ designed the study, analyzed the data, and wrote
the manuscript. YJ collected the data and drafted the manuscript.
JW and JM designed the study and analyzed the data. SH designed the
study and revised the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Emmings E, Mullany S, Chang Z, Landen CN
Jr, Linder S and Bazzaro M: Targeting mitochondria for treatment of
chemoresistant ovarian cancer. Int J Mol Sci. 20(pii): E2292019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou Y, Layton O and Hong L:
Identification of genes and pathways involved in ovarian epithelial
cancer by bioinformatics analysis. J Cancer. 9:3016–3022. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cortez AJ, Tudrej P, Kujawa KA and
Lisowska KM: Advances in ovarian cancer therapy. Cancer Chemother
Pharmacol. 81:17–38. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Szabova L, Bupp S, Kamal M, Householder
DB, Hernandez L, Schlomer JJ, Baran ML, Yi M, Stephens RM,
Annunziata CM, et al: Pathway-specific engineered mouse allograft
models functionally recapitulate human serous epithelial ovarian
cancer. PLoS One. 9:e956492014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rojas V, Hirshfield KM, Ganesan S and
Rodriguez-Rodriguez L: Molecular characterization of epithelial
ovarian cancer: Implications for diagnosis and treatment. Int J Mol
Sci. 17(pii): E21132016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancer. Nature. 396:643–649. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Previs RA, Sood AK, Mills GB and Westin
SN: The rise of genomic profiling in ovarian cancer. Expert Rev Mol
Diagn. 16:1337–1351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sanchez-Pena ML, Isaza CE, Perez-Morales
J, Rodriguez-Padilla C, Castro JM and Cabrera-Rios M:
Identification of potential biomarkers from microarray experiments
using multiple criteria optimization. Cancer Med. 2:253–265. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Su LJ, Hsu SL, Yang JS, Tseng HH, Huang SF
and Huang CY: Global gene expression profiling of
dimethylnitrosamine-induced liver fibrosis: From pathological and
biochemical data to microarray analysis. Gene Expr. 13:107–132.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Muller C, Schillert A, Rothemeier C,
Trégouët DA, Proust C, Binder H, Pfeiffer N, Beutel M, Lackner KJ,
Schnabel RB, et al: Removing batch effects from longitudinal gene
Expression-Quantile normalization plus combat as best approach for
microarray transcriptome data. PLoS One. 11:e01565942016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Clough E and Barrett T: The gene
expression omnibus database. Methods Mol Biol. 1418:93–110. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bowen NJ, Walker LD, Matyunina LV, Logani
S, Totten KA, Benigno BB and McDonald JF: Gene expression profiling
supports the hypothesis that human ovarian surface epithelia are
multipotent and capable of serving as ovarian cancer initiating
cells. BMC Med Genomics. 2:712009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yeung TL, Leung CS, Wong KK,
Gutierrez-Hartmann A, Kwong J, Gershenson DM and Mok SC: ELF3 is a
negative regulator of epithelial-mesenchymal transition in ovarian
cancer cells. Oncotarget. 8:16951–16963. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lili LN, Matyunina LV, Walker LD, Benigno
BB and McDonald JF: Molecular profiling predicts the existence of
two functionally distinct classes of ovarian cancer stroma. Biomed
Res Int. 2013:8463872013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fasold M and Binder H: Estimating
RNA-quality using GeneChip microarrays. BMC Genomics. 13:1862012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leek JT, Johnson WE, Parker HS, Jaffe AE
and Storey JD: The Sva package for removing batch effects and other
unwanted variation in high-throughput experiments. Bioinformatics.
28:882–883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanehisa M, Furumichi M, Tanabe M, Sato Y
and Morishima K: KEGG: New perspectives on genomes, pathways,
diseases and drug. Nucleic Acids Res. 45:D353–D361. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Braun P and Gingras AC: History of
protein-protein interactions: From egg-white to complex networks.
Proteomics. 12:1478–1498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang K, Kong X, Feng G, Xiang W, Chen L,
Yang F, Cao C, Ding Y, Chen H, Chu M, et al: Investigation of
hypoxia networks in ovarian cancer via bioinformatics analysis. J
Ovarian Res. 11:162018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rhodes DR, Kalyana-Sundaram S, Mahavisno
V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ,
Kincead-Beal C, Kulkarni P, et al: Oncomine 3.0: Genes, pathways,
and networks in a collection of 18,000 cancer gene expression
profiles. Neoplasia. 9:166–180. 2017. View Article : Google Scholar
|
|
24
|
Gyorffy B, Lánczky A and Szállási Z:
Implementing anonline tool for genome-wide validation of
survival-associated biomarkers in ovarian-cancer using microarray
data from 1287 patients. Endocr Relat Cancer. 19:197–208. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Drakes ML, Mehrotra S, Aldulescu M, Potkul
RK, Liu Y, Grisoli A, Joyce C, O'Brien TE, Stack MS and Stiff PJ:
Stratification of ovarian tumor pathology by expression of
programmed cell Death-1 (Pd-1) and Pd-Ligand-1 (Pd-L1) in ovarian
cancer. J Ovarian Res. 11:432018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moufarrij S, Dandapani M, Arthofer E,
Gomez S, Srivastava A, Lopez-Acevedo M, Villagra A and Chiappinelli
KB: Epigenetic therapy for ovarian cancer: Promise and progress.
Clin Epigenetics. 11:72019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hennessy BT, Coleman RL and Markman M:
Ovarian cancer. Lancet. 374:1371–1382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baron JA: Screening for cancer with
molecular markers: Progress comes with potential problem. Nat Rev
Cancer. 12:368–371. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Soon WW, Hariharan M and Snyder MP:
High-throughput sequencing for biology and medicine. Mol Syst Biol.
9:6402013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Duffy MJ: Use of biomarkers in screening
for cancer. Adv Exp Med Biol. 867:27–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vlasova-St Louis I and Bohjanen PR:
Post-transcriptional regulation of cytokine and growth factor
signaling in cancer. Cytokine Growth Factor Rev. 33:83–93. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Peyressatre M, Prevel C, Pellerano M and
Morris MC: Targeting cyclin-dependent kinases in human cancers:
From small molecules to Peptide inhibitors. Cancers (Basel).
7:179–237. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Garcia-Reyes B, Kretz AL, Ruff JP, von
Karstedt S, Hillenbrand A, Knippschild U, Henne-Bruns D and Lemke
J: The emerging role of Cyclin-dependent kinases (CDKs) in
pancreatic ductal adenocarcinoma. Int J Mol Sci. 19(pii):
E32192018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mohammed MK, Shao C, Wang J, Wei Q, Wang
X, Collier Z, Tang S, Liu H, Zhang F, Huang J, et al: Wnt/β-Catenin
signaling plays an ever-expanding role in stem cell self-renewal,
tumorigenesis and cancer chemoresistance. Genes Dis. 3:11–40. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhan T, Rindtorff N and Boutros M: Wnt
signaling in cancer. Oncogene. 36:1461–1473. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Clements WM, Wang J, Sarnaik A, Kim OJ,
MacDonald J, Fenoglio-Preiser C, Groden J and Lowy AM: Beta-Catenin
mutation is a frequent cause of Wnt pathway activation in gastric
cancer. Cancer Res. 62:3503–3506. 2002.PubMed/NCBI
|
|
37
|
Parker AL, Kavallaris M and McCarroll JA:
Microtubules and their role in cellular stress in cancer. Front
Oncol. 4:1532014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Burgess RJ and Zhang Z: Histone chaperones
in nucleosome assembly and human disease. Nat Struct Mol Biol.
20:14–22. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Polo SE, Theocharis SE, Grandin L,
Gambotti L, Antoni G, Savignoni A, Asselain B, Patsouris E and
Almouzni G: Clinical significance and prognostic value of chromatin
assembly factor-1 overexpression in human solid tumours.
Histopathology. 57:716–724. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang J, Zhang L, Greshock J, Colligon TA,
Wang Y, Ward R, Katsaros D, Lassus H, Butzow R, Godwin AK, et al:
Frequent genetic abnormalities of the PI3K/AKT pathway in primary
ovarian cancer predict patient outcome. Genes Chromosomes Cancer.
50:606–618. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chien AJ, Conrad WH and Moon RT: A Wnt
survival guide: From flies to human disease. J Invest Dermatol.
129:1614–1627. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bitler BG, Nicodemus JP, Li H, Cai Q, Wu
H, Hua X, Li T, Birrer MJ, Godwin AK, Cairns P and Zhang R: Wnt5a
suppresses epithelial ovarian cancer by promoting cellular
senescence. Cancer Res. 71:6184–6194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bodnar L, Stanczak A, Cierniak S, Smoter
M, Cichowicz M, Kozlowski W, Szczylik C, Wieczorek M and
Lamparska-Przybysz M: Wnt/β-catenin pathway as a potential
prognostic and predictive marker in patients with advanced ovarian
cancer. J Ovarian Res. 7:162014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Conciatori F, Ciuffreda L, Bazzichetto C,
Falcone I, Pilotto S, Bria E, Cognetti F and Milella M: mTOR
cross-talk in cancer and potential for combination therapy. Cancers
(Basel). 10:E232018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 169:361–371. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang R, Shi H, Ren F, Zhang M, Ji P, Wang
W and Liu C: The aberrant upstream pathway regulations of CDK1
protein were implicated in the proliferation and apoptosis of
ovarian cancer cells. J Ovarian Res. 10:602017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sung WW, Lin YM, Wu PR, Yen HH, Lai HW, Su
TC, Huang RH, Wen CK, Chen CY, Chen CJ and Yeh KT: High
nuclear/cytoplasmic ratio of Cdk1 expression predicts poor
prognosis in colorectal cancer patients. BMC Cancer. 14:9512014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yang W, Cho H, Shin HY, Chung JY, Kang ES,
Lee EJ and Kim JH: Accumulation of cytoplasmic Cdk1 is associated
with cancer growth and survival rate in epithelial ovarian cancer.
Oncotarget. 7:49481–49497. 2016.PubMed/NCBI
|
|
50
|
Ou Y, Ma L, Huang Z, Zhou W, Zhao C, Zhang
B, Song Y, Yu C and Zhan Q: Overexpression of cyclin B1 antagonizes
chemotherapeutic-induced apoptosis through PTEN/Akt pathway in
human esophageal squamous cell carcinoma cells. Cancer Biol Ther.
14:45–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bonnet ME, Gossart JB, Benoit E, Messmer
M, Zounib O, Moreau V, Behr JP, Lenne-Samuel N, Kedinger V, Meulle
A, et al: Systemic delivery of sticky siRNAs targeting the cell
cycle for lung tumor metastasis inhibition. J Control Release.
170:183–190. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kedinger V, Meulle A, Zounib O, Bonnet ME,
Gossart JB, Benoit E, Messmer M, Shankaranarayanan P, Behr JP,
Erbacher P and Bolcato-Bellemin AL: Sticky siRNAs targeting
survivin and cyclin B1 exert an antitumoral effect on melanoma
subcutaneous xenografts and lung metastases. BMC Cancer.
13:3382013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Matthess Y, Raab M, Sanhaji M, Lavrik IN
and Strebhardt K: Cdk1/cyclin B1 controls Fas-mediated apoptosis by
regulating caspase-8 activity. Mol Cell Biol. 30:5726–5740. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Soria JC, Jang SJ, Khuri FR, Hassan K, Liu
D, Hong WK and Mao L: Overexpression of cyclin B1 in early-stage
non-small cell lung cancer and its clinical implication. Cancer
Res. 60:4000–4004. 2000.PubMed/NCBI
|
|
55
|
Nozoe T, Korenaga D, Kabashima A, Ohga T,
Saeki H and Sugimachi K: Significance of cyclin B1 expression as an
independent prognostic indicator of patients with squamous cell
carcinoma of the esophagus. Clin Cancer Res. 8:817–822.
2002.PubMed/NCBI
|
|
56
|
Ding K, Li W, Zou Z, Zou X and Wang C:
CCNB1 is a prognostic biomarker for ER+ breast cancer. Med
Hypotheses. 83:359–364. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Weng L, Du J, Zhou Q, Cheng B, Li J, Zhang
D and Ling C: Identification of cyclin B1 and Sec 62 as biomarkers
for recurrence in patients with HBV-related hepatocellular
carcinoma after surgical resection. Mol Cancer. 11:392012.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lee KS, Burke TR Jr, Park JE, Bang JK and
Lee E: Recent Advances and new strategies in targeting Plk1 for
anticancer therapy. Trends Pharmacol Sci. 36:858–877. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Iyer RS, Nicol SM, Quinlan PR, Thompson
AM, Meek DW and Fuller-Pace FV: The RNA Helicase/Transcriptional
Co-regulator, P68 (DDX5), stimulates expression of oncogenic
protein kinase, Polo-like Kinase-1 (PLK1), and is associated with
elevated PLK1 levels in human breast cancers. Cell Cycle.
13:1413–1423. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu Z, Sun Q and Wang X: PLK1, A potential
target for cancer therapy. Transl Oncol. 10:22–32. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Weichert W, Denkert C, Schmidt M, Gekeler
V, Wolf G, Köbel M, Dietel M and Hauptmann S: Polo-like kinase
isoform expression is a prognostic factor in ovarian carcinoma. Br
J Cancer. 90:815–821. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
de Carcer G, Venkateswaran SV, Salgueiro
L, El Bakkali A, Somogyi K, Rowald K, Montañés P, Sanclemente M,
Escobar B, de Martino A, et al: Plk1 overexpression induces
chromosomal instability and suppresses tumor development. Nat
Commun. 9:30122018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yasuda H, Lindorfer MA, Myung CS and
Garrison JC: Phosphorylation of the G protein gamma12 subunit
regulates effector specificity. J Biol Chem. 273:21958–21965. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Larson K.C, Lipko M, Dabrowski M and
Draper MP: Gng12 is a novel negative regulator of LPS-induced
inflammation in the microglial cell line BV-2. Inflamm Res.
59:15–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Luo C, Zhao S, Dai W, Zheng N and Wang J:
Proteomic analyses reveal GNG12 regulates cell growth and casein
synthesis by activating the Leu-mediated mTORC1 signaling pathway.
Biochim Biophys Acta Proteins Proteom. 1866:1092–1101. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hao X and Qu T: Expression of CENPE and
its prognostic role in non-small cell lung cancer. Open Med (Wars).
14:497–502. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Balamuth NJ, Wood A, Wang Q, Jagannathan
J, Mayes P, Zhang Z, Chen Z, Rappaport E, Courtright J, Pawel B, et
al: Serial transcriptome analysis and cross-species integration
identifies centromere-associated protein E as a novel neuroblastoma
target. Cancer Res. 70:2749–2758. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bieche I, Vacher S, Lallemand F,
Tozlu-Kara S, Bennani H, Beuzelin M, Driouch K, Rouleau E,
Lerebours F, Ripoche H, et al: Expression analysis of mitotic
spindle checkpoint genes in breast carcinoma: Role of NDC80/HEC1 in
early breast tumorigenicity, and a two-gene signature for
aneuploidy. Mol Cancer. 10:232011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bolanos-Garcia VM and Blundell TL: BUB1
and BUBR1: Multifaceted kinases of the cell cycle. Trends Biochem
Sci. 36:141–150. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Karess RE, Wassmann K and Rahmani Z: New
insights into the role of BubR1 in mitosis and beyond. Int Rev Cell
Mol Biol. 306:223–273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Elowe S: Bub1 and BubR1: At the interface
between chromosome attachment and the spindle checkpoint. Mol Cell
Biol. 31:3085–3093. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hudler P, Britovsek NK, Grazio SF and
Komel R: Association between polymorphisms in segregation genes
BUB1B and TTK and gastric cancer risk. Radiol Oncol. 50:297–307.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Fu X, Chen G, Cai ZD, Wang C, Liu ZZ, Lin
ZY, Wu YD, Liang YX, Han ZD, Liu JC and Zhong WD: Overexpression of
BUB1B contributes to progression of prostate cancer and predicts
poor outcome in patients with prostate cancer. Onco Targets Ther.
9:2211–2220. 2016.PubMed/NCBI
|
|
74
|
Hahn MM, Vreede L, Bemelmans SA, van der
Looij E, van Kessel AG, Schackert HK, Ligtenberg MJ, Hoogerbrugge
N, Kuiper RP and de Voer RM: Prevalence of germline mutations in
the spindle assembly checkpoint gene BUB1B in individuals with
early-onset colorectal cancer. Genes Chromosomes Cancer.
55:855–863. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mansouri N, Movafagh A, Sayad A, Heidary
Pour A, Taheri M, Soleimani S, Mirzaei HR, Alizadeh Shargh S,
Azargashb E, Bazmi H, et al: Targeting of BUB1b gene expression in
sentinel lymph node biopsies of invasive breast cancer in iranian
female patients. Asian Pac J Cancer Prev. 17:317–321. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Do TV, Xiao F, Bickel LE, Klein-Szanto AJ,
Pathak HB, Hua X, Howe C, O'Brien SW, Maglaty M, Ecsedy JA, et al:
Aurora kinase A mediates epithelial ovarian cancer cell migration
and adhesion. Oncogene. 33:539–549. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zou Z, Yuan Z, Zhang Q, Long Z, Chen J,
Tang Z, Zhu Y, Chen S, Xu J, Yan M, et al: Aurora kinase a
inhibition-induced autophagy triggers drug resistance in breast
cancer cells. Autophagy. 8:1798–1810. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Reiter R, Gais P, Jutting U, Steuer-Vogt
MK, Pickhard A, Bink K, Rauser S, Lassmann S, Höfler H, Werner M
and Walch A: Aurora kinase a messenger RNA overexpression is
correlated with tumor progression and shortened survival in head
and neck squamous cell carcinoma. Clin Cancer Res. 12:5136–5141.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wan XB, Long ZJ, Yan M, Xu J, Xia LP, Liu
L, Zhao Y, Huang XF, Wang XR, Zhu XF, et al: Inhibition of Aurora-A
suppresses epithelial-mesenchymal transition and invasion by
downregulating MAPK in nasopharyngeal carcinoma cells.
Carcinogenesis. 29:1930–1937. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Mignogna C, Staropoli N, Botta C, De Marco
C, Rizzuto A, Morelli M, Di Cello A, Franco R, Camastra C, Presta
I, et al: Aurora Kinase A expression predicts platinum-resistance
and adverse outcome in high-grade serous ovarian carcinoma
patients. J Ovarian Res. 9:312016. View Article : Google Scholar : PubMed/NCBI
|