Introduction
A number of previous studies have identified lung
malignancy as one of the most common causes of cancer-associated
mortality (1–3). A previous study has characterized the
significant role of particulate matter (PM2.5) in increasing the
occurrence of lung cancer (4). PM2.5
is a variety of airborne particulate matter that is derived from
automobile exhausts, coal combustion and biomass burning (5). Previous studies have revealed that
PM2.5 can induce carcinogenesis and metastasis of lung cancer via
the inhibition of microRNA expression and the deregulation of
tumor-associated DNA methylation (6,7).
However, to the best of our knowledge, the role of PM2.5 in lung
cancer inflammation and angiogenesis has not been determined.
Macrophages enhance lung cancer invasion and
infiltration by secreting angiogenic cytokines, including vascular
endothelial growth factor A (VEGF), platelet-derived growth factor
(PDGF) and basic fibroblast growth factors (bFGFs) (8). VEGF has been demonstrated to be a key
regulator in angiogenesis, and is mainly produced by macrophages,
tumor cells and fibroblasts (9,10).
Studies have demonstrated that VEGF can promote the proliferation
and migration of endothelial cells by stimulating the release of
angiogenic factors, binding to VEGF receptor and initiating
downstream signaling pathways (11,12).
Additionally, PM2.5 has been indicated to activate a number of
signaling pathways that promote inflammatory responses and
oxidative stress in macrophages (13). Macrophage polarization has been
reported to be affected by PM2.5, which can lead to the induction
of the immune response and air pollutant-associated diseases
(14).
As a member of the immunoglobulin superfamily,
integrin associated protein, which is also known as CD47, is highly
expressed in lung cancer (15,16).
CD47 binds to signal regulatory protein α (Sirp-α), which is
located on the surface of macrophages (17). This induces the release of a signal
that prevents phagocytosis of these cells by macrophages (18,19).
Additionally, the CD47/Sirp-α signaling pathway has been
demonstrated to increase dendritic cell proliferation and T cell
activation, which results in the infiltration of inflammatory cells
into the tumor microenvironment (18). Furthermore, a previous study has
revealed that CD47/Sirp-α is associated with the inflammatory
response in the tumor microenvironment (20). Therefore, activation of macrophages
and the tumor-associated CD47/Sirp-α signaling pathway are
important in modulating lung cancer.
In the present study, it was hypothesized that
macrophages may be crucial in the PM2.5-induced angiogenesis of
lung cancer. To validate this hypothesis, angiogenic cytokines were
measured in Lewis lung carcinoma (LLC) cells following exposure to
PM2.5 or PM2.5-induced macrophage supernatant (RAW264.7). It was
hypothesized that the possible mechanisms may include
macrophage-induced VEGF release and activation of the CD47/Sirp-α
signaling pathway. Therefore, VEGF and CD47/Sirp-α expression was
determined in RAW264.7 cells following PM2.5 exposure.
Subsequently, a mouse polyvinyl alcohol (PVA) sponge implantation
model was established to investigate PM2.5-induced angiogenesis and
macrophage accumulation in an inflammatory model. Macrophage
recruitment and angiogenesis were also determined in mice that were
subcutaneously injected with LLC following exposure to PM2.5 or
PM2.5-induced RAW264.7 supernatant. The current study provided
novel insights into the potential mechanisms of PM2.5-induced lung
cancer angiogenesis.
Materials and methods
Animals
C57BL/6 mice (n=90; 6–8 weeks old; male; 23–25 g)
were purchased from Silaike Laboratory Animal Co., Ltd. All mice
were housed in a temperature controlled room (20-24°C) on a 12 h
light-dark cycle with unrestricted access to food and water. For
all procedures performed on live mice, animals were anesthetized
using sodium pentobarbital (80 mg/kg). All mice were sacrificed by
cervical dislocation.
Preparation of PM2.5
PM2.5 was collected using a PM2.5 sampler
(TishTE-6070D; Tisch Environmental, Inc.) at Zhengzhou University
(Zhengzhou, China) between April 2016 and December 2016. PM2.5
samples were suspended in a mixture of methanol, methylene chloride
and deionized water at a ratio of 8:4:1. PM2.5 was then placed into
an ultrasonic bath for 30 min. Finally, PM2.5 was obtained using
ultrafiltration, and stored at −20°C until subsequent use.
Cell culture
RAW264.7, LLC (ATCC® CRL-1642TM), and
human non-small lung cancer H1299 and A549 cell lines were
purchased from the Shanghai Institutes for Biological Sciences
(http://www.cellbank.org.cn/). RAW264.7
cells were cultured in 10% FBS and RPMI-1640 medium (both Thermo
Fisher Scientific, Inc.). LLC, H1299 and A549 cells were cultured
in 10% FBS and DMEM (both Thermo Fisher Scientific, Inc.). All cell
lines were supplemented with 1% penicillin-streptomycin (Thermo
Fisher Scientific, Inc.) and cultured in a humidified incubator
with 5% CO2 at 37°C.
MTT assays
Cell viability was evaluated using MTT assays. LLC,
RAW264.7, H1299 and A549 cells were seeded in 96-well plates
(5×103−1×104 cells/well). Cells were
subsequently exposed to different concentrations of PM2.5 (100,
200, 400, 600, 800 and 1,200 µg/ml) at 37°C for 48 h. The same
volume of PBS, as the volume of PM2.5 extract, were used in the
control group. A total of 20 µl MTT (5 g/l) was added to each well
for 4 h, followed by the replacement of the medium with 150 µl
DMSO. Optical density (OD) values were determined using a
microplate reader at an absorbance of 570 nm (BioTek Instruments,
Inc.). Cell viability was calculated based on the OD values.
Reverse transcription-quantitative PCR
(RT-qPCR)
LLC and RAW264.7 cells were treated with different
concentrations of PM2.5 (50, 500, 2,000, 4,000, 6,000, 8,000 µg/ml)
or 500 µg/ml PM2.5-induced RAW264.7 supernatant for 3 h at 37°C.
Total RNA was isolated from cells using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), and cDNA was
synthesized using a Takara reverse transcriptase kit (for 15 min at
37°C and 5 min at 85°C; Takara Biotechnology Co., Ltd.) and a
LabCycler PCR instrument (SensoQuest GmbH). The resulting cDNA
template was subjected to qPCR using Real Time PCR Easy™
(SYBR Green I; Takara Biotechnology Co., Ltd.) on a
LightCycler® 96 instrument (Roche Diagnostics), using
the following conditions: Initial denaturation for 30 sec at 98°C,
followed by 17 cycles of denaturation (10 sec at 98°C), annealing
(15 sec at 72°C; decreasing by 1°C/cycle), and extension (30 sec at
72°C). This step was followed by 18 cycles of denaturation (10 sec
at 98°C), annealing (15 sec at 54°C), initial extension (30 sec at
98°C), and a final extension (1 min at 72°C). The primer sequences
used were as follows: VEGF-A forward, 5′-GGAGATCCTTCGAGGAGCACTT-3′
and reverse, 5′-GGCGATTTAGCAGCAGATATAAGAA-3′; β-actin forward,
5′-GTGGGCCGCTCTAGGCACCAA-3′ and reverse,
5′-TGGCTTTAGGGTTCAGGGGG-3′; bFGF forward 5′-AGCGGCTCTACTGCAAGAAC-3′
and reverse, 5′-GCCGTCCATCTTCCTTCATA-3′; PDGF forward,
5′-CAAGACCAGGACGGTCATTT-3′ and reverse, 5′-ACTTTGGCCACCTTGACACT-3′;
CD47 forward, 5′-TGGTATCCAGCAAGCCTTAG-3′ and reverse,
5′-AAGACACCAGTGCCATCAAT-3′; Sirp-α forward,
5′-ACCACCGTGAACCCTAGTGGAA-3′ and reverse,
5′-GGTGGGTGAAACTCGGATGAAG-3′. Alterations in cytokine expression
were determined by normalizing levels to those of β-actin. RT-qPCR
were analyzed and fold changes were determined using the
2−ΔΔCq method (21).
ELISA
VEGF protein levels of RAW264.7 were measured in
supernatant following treatment with 500 µg/ml PM2.5 for 12 h at
37°C. VEGF protein levels were determined using a VEGF ELISA kit
(eBioscience; Thermo Fisher Scientific, Inc.), which was performed
according to the manufacturer's protocol. OD values were obtained
at 450 nm in triplicate using a microplate reader (Molecular
Devices, LLC).
Immunofluorescence
Sponge tissues and tumor tissues were fixed using 4%
paraformaldehyde (PFA) for 24 h (4°C) and embedded in paraffin (5
µm sections). For immunofluorescence staining of VEGF, F4/80+,
FITC-labeled lectin CD31 and α-smooth muscle actin (α-SMA), sponge
slides and tumor tissue slides were permeabilized with 0.3% Triton
X-100 in PBS for 20 min at room temperature and blocked with 5%
bovine serum albumin (Sigma-Aldrich; Merck KGaA) for 30 min, then
the slides were incubated with the primary antibodies at 4°C for 24
h. Primary antibodies are as follows: VEGF (1:300 dilution; cat.
no. sc-7269; Santa Cruz Biotechnology, Inc.), F4/80+ (1:350
dilution; cat. no. 14-4801-82; eBioscience; Thermo Fisher
Scientific, Inc.), FITC-labeled lectin (1:350 dilution; cat. no.
L9381; Sigma-Aldrich; Merck KGaA), CD31 (1:300 dilution; cat. no.
ab9498; Abcam) and α-SMA (1:300 dilution; cat. no. ab5694Abcam).
Subsequently, slides were washed with PBS and then incubated with
the appropriate secondary antibody at 37°C for 3 h. The secondary
antibodies were anti-mouse IgG (1:450 dilution; cat. no. 4410) and
anti-rabbit IgG (1:450 dilution; cat. no. 4412; both Cell Signaling
Technology, Inc.). The slides were stained with DAPI (Invitrogen;
Thermo Fisher Scientific, Inc.) to visualize the nuclei. Finally, a
fluorescence microscope with ×20 or ×40 objective lens (Leica
microsystem GmbH) was used to view the immunolabeled slides, under
5 fields of view. Immunofluorescence quantification was performed
using ImageJ v5.0 software (National Institutes of Health).
PVA sponge implantation model
The PVA sponge implantation model was generated as
described previously (22). PM2.5
was mixed with quantitative L-polylactic acid powder (PLLA; Jinan
Daigang Biomaterial Co., Ltd.), to create a slow release gel, and
implanted into the back of mice using a sponge. A negative control
gel was created using the same amount of PLLA. Sponges were
harvested after 2 weeks of implantation with either collagenase
digestion for RT-qPCR, or 4% PFA fixation and paraffin embedding
for FITC-labeled lectin, VEGF, and F4/80+
immunofluorescence assays. In vivo experiments the control
groups used the same volume of normal saline as the volume of PM2.5
extract.
Syngeneic growth of LLC cells in
mice
Animals (n=4 mice in each group) were injected
subcutaneously in the right flank with 0.2 ml PBS containing cell
suspension (4×106 LLC cells). Tumor volumes were
measured every other day, beginning 7 days after LLC cell
inoculation, and calculated according to the following formula:
Volume=Length × Width2 ×0.5. Alveolar macrophages of
mice lungs were depleted by intratracheal application of clodronate
liposomes. The liposomes were purchased from Yeasen Biotechnology
Co., Ltd. Liposomally encapsulated clodronate (5 mg/ml; Yeasen
Biotechnology Co., Ltd) was stored at −80°C and thawed immediately
prior to use. Clodronate liposomes (50 µl) were prepared for
intratracheal application as previously described (23). The efficiency of clodronate liposomes
was evaluated using F4/80+ immunofluorescence.
Intratracheal administration of saline containing PM2.5 (320 mg/ml)
began at 7 days of tumor cell inoculation, and administration was
carried out every 3 days. Mice were sacrificed, and tumors were
harvested on the fifteenth day following LLC cell inoculation.
Subsequently, tumors were either extracted for RT-qPCR or fixed
with 4% PFA and paraffin embedded for VEGF, F4/80+,
CD31, and α-SMA immunofluorescence assays. In another tumor-bearing
mouse model, LLC cells were exposed to 500 µg/ml PM2.5-induced
RAW264.7 supernatant for 3 h (37°C). Subsequently the LLC cells
were injected subcutaneously in the right flank of the mice (n=4).
Tumor extraction and the following detection assays were as
aforementioned.
Statistical analysis
All data are presented as the mean ± SD unless
otherwise specified. A total of 3 independent repeat experiments
were performed. Significance was determined by one-way ANOVA
followed by Tukey's post hoc test for multiple group comparisons
and t-test for comparisons between two groups using SPSS v16.0
software (SPSS, Inc.). P≤0.05 was considered to indicate a
statistically significant difference.
Results
Cytotoxic effect of PM2.5 on a variety
of lung cancer cell lines
Cell viability is an important parameter for
evaluating the impact of toxicants on cell growth. The cell
viability of macrophage RAW264.7 and lung cancer H1299, LLC and
A549 cell lines, following long-term exposure to different doses
(100, 200, 400, 600, 800 and 1,200 µg/ml) of PM2.5, was determined
using MTT assays. The results indicated that PM2.5 exhibited an
inhibitory effect on viability in RAW264.7, LLC, H1299 and A549
cell lines, in a dose-dependent manner (Fig. 1A).
 | Figure 1.Role of macrophages in PM2.5-induced
angiogenesis in LLC cells. (A) Cytotoxicity of PM2.5 in RAW264.7
cell, H1299 cells, LLC and A549 cells treated with different
concentrations of PM2.5 for 48 h was measured using MTT assays
(n=4). (B) No significantly elevated expression of several
angiogenic cytokines (VEGF-A, MMP-9, PDGF and bFGF) was identified
in LLC cells following direct exposure to 500 µg/ml PM2.5 for 3 h.
LLC cells treated without PM2.5 were used as a control. (C)
Increased mRNA expression levels of angiogenic cytokines, including
VEGF-A, MMP-9, PDGF and bFGF, in LLC cells was observed following
exposure to 500 µg/ml PM2.5-induced RAW264.7 supernatant for 3 h,
using reverse transcription-quantitative PCR assays. LLC cells
treated without PM2.5-induced RAW264.7 supernatant were used as a
control. Data are presented as the mean ± SD of three independent
experiments and were analyzed using ANOVA. **P<0.01 vs. control;
***P<0.001 vs. control. (D) Following exposure to 500 µg/ml
PM2.5 for 3 h, increased VEGF-A mRNA expression was observed in
RAW264.7 cells. RAW264.7 cells treated without PM2.5 were used as a
control. **P<0.01 vs. control. (E) Increased VEGF protein
expression was detected in RAW264.7 supernatant following exposure
to 500 µg/ml PM2.5 for 12 h. The OD value of VEGF was determined
using ELISA. RAW264.7 cells treated without PM2.5 were used as a
control. **P<0.01 vs. control. (F) CD47 mRNA expression was
increased in LLC cells following exposure to 500 µg/ml
PM2.5-induced RAW264.7 supernatant. LLC cells treated without
PM2.5-induced RAW264.7 supernatant were used as a control.
***P<0.001 vs. RAW264.7 supernatant. (G) High expression levels
of SIRP-α were detected in PM2.5-induced RAW264.7 cells. RAW264.7
cells treated without PM2.5 were used as a control. Data are
presented as the mean ± SD of three independent experiments and
were analyzed using a t-test. *P<0.05 vs. PM2.5 0 µg/ml. PM,
particle matter; LLC, Lewis lung carcinoma; VEGF-A, vascular
endothelial growth factor A; MMP-9, matric metallopeptidase 9;
PDGF, platelet-derived growth factor; bFGF, basic fibroblast growth
factor; OD, optical density; SIRP-α, signal regulatory protein
α. |
Macrophages are crucial for acute
PM2.5 exposure-induced VEGF-A expression in LLC cells
The expression levels of VEGF-A, matrix
metallopeptidase-9 (MMP-9), PDGF and bFGF cytokines in LLC cells
were measured in response to direct exposure to 500 µg/ml PM2.5 for
3 h. No significant differences were observed in the expression
levels of these cytokines in the PM2.5 exposure group compared with
the control LLC cells (Fig. 1B).
After 3 h of treatment with 500 µg/ml PM2.5-induced RAW264.7
supernatant, the results indicated enhanced mRNA expression of
angiogenic cytokines VEGF-A, MMP-9, PDGF and bFGF in LLC cells
compared with cells without PM2.5-induced RAW264.7 supernatant
exposure (Fig. 1C). To explore the
specific mechanisms of this, RAW264.7 cells were exposed to PM2.5
and the results revealed elevated mRNA expression levels of VEGF-A
in RAW264.7 cells and increased VEGF protein expression in RAW264.7
supernatant following exposure to 500 µg/ml PM2.5 for 12 h
(Fig. 1D and E). CD47 mRNA
expression was significantly higher in LLC cells following acute
exposure to 500 µg/ml PM2.5-induced RAW264.7 supernatant compared
with LLC cells treated with normal RAW264.7 supernatant (Fig. 1F). Sirp-α expression was higher in
RAW264.7 cells following exposure to 500 µg/ml PM2.5 (Fig. 1G). To determine the concentration of
PM2.5 to be used in the in vitro experiments, RAW264.7 cells
were exposed to different concentrations of PM2.5 (50, 500, 2,000,
4,000, 6,000, 8,000 µg/ml) for 3 h (37°C). The results revealed
that VEGF-A mRNA expression only increased at 500 µg/ml, which
indicated that this concentration was the most suitable for further
experiments (Fig. S1).
Validation of the angiogenic and
macrophage accumulation effects of PM2.5 in a PVA sponge
implantation mouse model
Little is known regarding PM2.5-regulated
angiogenesis and macrophage accumulation in vivo. Therefore,
a PVA sponge implantation mouse model, which exhibits features of
monocyte/macrophage localization and microvessel formation
(22), was implemented. At 14 days
following subcutaneous implantation, sponges were excised and
evaluated for macrophage localization, VEGF-A mRNA expression and
microvessel formation. Immunofluorescence staining for VEGF,
F4/80+ and lectin in sponges revealed an increase in
VEGF protein release (indicated by VEGF; Fig. 2A), macrophage recruitment (indicated
by F4/80+; Fig. 2A) and
microvessel density (indicated by Lectin; Fig. 2A) following exposure to 10 mg PM2.5
(Fig. 2B). High VEGF-A mRNA
expression was indicated in sponges of the PM2.5 exposure group
(Fig. 2C).
Pro-angiogenesis and macrophage
recruitment effects of PM2.5 in mice bearing LLC cells
A model of C57BL/6 mice bearing LLC cells was
established to determine whether PM2.5 directly promoted tumor
growth in mice. Furthermore, clodronate liposomes were used to
decrease alveolar macrophages in the model. This was confirmed
using measurements of F4/80+ immunofluorescence
(Fig. 3E and G). Tumor size
(Fig. 3A and B) and weight (Fig. 3C) were demonstrated to be
significantly higher in the 38.4 mg PM2.5 exposure group compared
with in the control group. Tumor size and weight (Fig. 3A-C) were also decreased in the
clodronate group, in which macrophage were depleted compared with
that in the 38.4 mg PM2.5 exposure group. Immunofluorescence assays
of tumor tissues demonstrated increased VEGF release (Fig. 3H), macrophage recruitment (indicated
by F4/80+; Fig. 3H) and
microvessel formation (indicated by CD31+ and α-SMA;
Fig. 3H) in the PM2.5-exposed group
compared with in the control group (Fig.
3I). Furthermore, once macrophages were depleted from using
clodronate, the PM2.5-induced pro-angiogenic and macrophage
recruitment effects were attenuated (Fig. 3H-I). Additionally, hematoxylin and
eosin staining of tumor tissues indicated increased vessel
formation in the PM2.5 exposure group, and vessel formation
decreased in the clodronate group compared with that in the 38.4 mg
PM2.5 exposure group (Fig. 3F).
Additionally, the results demonstrated that CD47 mRNA expression
was increased in tumor tissues of the PM2.5 exposure group and
decreased following the exposure of clodronate (Fig. 3D).
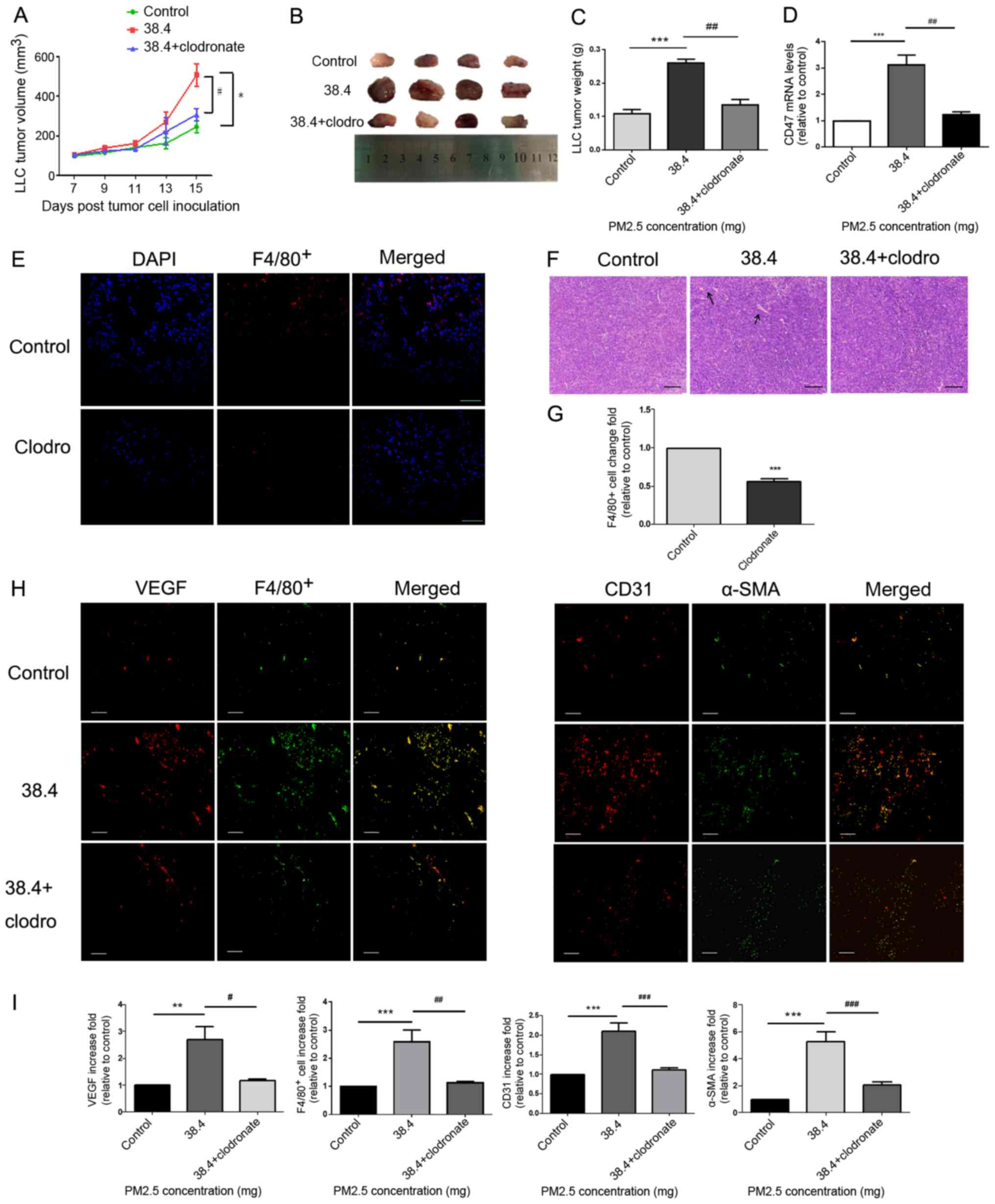 | Figure 3.Pro-angiogenic and macrophage
accumulation effects of PM2.5 in a tumor-bearing model. (A) LLC
cells were inoculated into C57BL/6 mice for 15 days, and
intratracheal instillation normal saline or 38.4 mg PM2.5 was
administered on days 7, 10 and 13. Tumor length and width were
measured every other day beginning 7 days after LLC cell
inoculation. Tumor volume was calculated using the following
formula: Volume=Length × Width2 ×0.5. Mice treated without PM2.5
were used as a control. *P<0.01 vs. control; #P<0.05 vs. 38.4
mg exposure group. (B) Tumor images were obtained after mice were
euthanized on the fifteenth day of cell inoculation (n=4). (C)
Tumor weight was highest in the 38.4 mg PM2.5 exposure group
compared with the control and macrophage depletion groups. (D)
Tumor tissue CD47 mRNA expression was measured using reverse
transcription-quantitative PCR. ***P<0.01 vs. control;
##P<0.01 vs. 38.4 mg exposure group. (E) Clodronate liposomes or
normal saline were intratracheally instilled to mice. F4/80+
immunofluorescence was used to test the efficiency of macrophage
depletion. Scale bar, 100 µm. (F) Hematoxylin and eosin staining
was used to observe angiogenesis in tumor tissues. Scale bar, 50
µm. Arrows indicated vessel. (G) Immunofluorescence quantification
for F4/80+ cells was performed using ImageJ v5.0 software. The
clodronate group exhibited decreased macrophage accumulation in
lung tissues compared with the control group. Data are presented as
the mean ± SD and were analyzed using a t-test. (H) Increased
macrophage recruitment, VEGF protein release and microvessel
formation were detected in response to PM2.5 exposure. VEGF and
F4/80+ staining indicated macrophage recruitment and angiogenic
protein secretion. Microvessel formation in tumor tissues was
examined using CD31 and α-SMA staining, which mark endothelial
cells and vascular smooth muscle cells, respectively. Scale bar, 50
µm. (I) Immunofluorescence quantification for VEGF, F4/80+, CD31
and α-SMA was performed using ImageJ v5.0 software. VEGF, F4/80+,
CD31 and α-SMA immunofluorescence were significantly increased in
the PM2.5 exposure group compared with in the control group. Data
are presented as the mean ± SD and were analyzed by one-way ANOVA.
**P<0.01 vs. control; ***P<0.001 vs. control; #P<0.05 vs.
38.4 mg exposure group; ##P<0.01 vs. 38.4 mg exposure group;
###P<0.001 vs. 38.4 mg exposure group. PM, particle matter; LLC,
Lewis lung carcinoma; VEGF, vascular endothelial growth factor;
α-SMA, α smooth muscle actin; clodro, clodronate. |
Macrophages are important in
PM2.5-induced LLC tumor progression in mice
To test whether macrophages serve a role in
PM2.5-induced lung cancer growth in vivo, a model of C57BL/6
mice bearing LLC cells was created. LLC cells were pre-treated with
PM2.5-induced RAW264.7 supernatant for 3 h prior to inoculation.
Tumor volume (Fig. 4A and B) and
weight (Fig. 4C) were revealed to be
significantly higher in the tumor tissues of mice bearing LLC cells
that were treated with 500 µg/ml PM2.5-induced macrophage
supernatant. Microvessel formation (indicated by CD31+;
Fig. 4E), vascular smooth muscle
cell accumulation (indicated by α-SMA; Fig. 4E), angiogenic-associated protein
release (indicated by VEGF; Fig. 4E)
and macrophage recruitment (indicated by F4/80+;
Fig. 4E) were also indicated in the
PM2.5-induced supernatant group (Fig.
4F). Hematoxylin and eosin staining of tumor tissues indicated
angiogenesis in the PM2.5-induced supernatant group (Fig. 4G). Furthermore, the results indicated
an increase in angiogenesis in the normal macrophage supernatant
group, in which LLC cells were pre-treated with normal RAW264.7
supernatant prior to inoculation, compared with that in the control
group. However, this was lower than that in the PM2.5-induced
supernatant group. Increased CD47 mRNA expression was demonstrated
in the tumor tissues of PM2.5-induced and normal macrophage
supernatant groups, and the former exhibited higher CD47 expression
compared with the normal macrophage supernatant group (Fig. 4D).
 | Figure 4.Macrophages mediated PM2.5-induced
lung cancer angiogenesis in a tumor-bearing model. (A) LLC cells
were exposed to RAW264.7 supernatant with or without PM2.5 for 3 h
and xenografted into C57BL/6 mice for 15 days. Tumor volume was
increased in the 500 µg/ml PM2.5-induced RAW264.7 supernatant
group. ###P<0.001 vs. control; *P<0.05 vs. RAW264.7
supernatant group. (B) Tumor tissue images were obtained after mice
were euthanized on day 15 of cell inoculation (n=4). (C) Tumor
weight was significantly higher in the 500 µg/ml PM2.5-induced
RAW264.7 supernatant group compared with the control and RAW264.7
groups. ###P<0.001 vs. control; **P<0.01 vs. RAW264.7
supernatant group. (D) Increased mRNA expression levels of CD47
were observed in the 500 µg/ml PM2.5-induced RAW264.7 supernatant
group compared with in the control group. *P<0.05 vs. control.
(E) Staining for VEGF, F4/80+, CD31 and α-SMA indicated the
promotion of angiogenesis and macrophage accumulation in mice
bearing LLC cells following exposure to 500 µg/ml PM2.5-induced
RAW264.7 supernatant. Scale bar, 50 µm. (F) Immunofluorescence
quantification was performed using ImageJ v5.0 software.
Immunofluorescence for VEGF, F4/80+, CD31 and α-SMA was
significantly higher in the 500 µg/ml PM2.5-induced RAW264.7
supernatant group compared with the control and RAW264.7 groups.
Data are presented as the mean ± SD and were analyzed by one-way
ANOVA (n=4 mice in each group). ###P<0.001 vs. control;
**P<0.01 vs. RAW264.7 supernatant group; ***P<0.001 vs.
RAW264.7 supernatant group. (G) Hematoxylin and eosin staining
revealed angiogenesis in tumor tissues. Scale bar, 50 µm. Arrows
indicate vessel. PM, particle matter; LLC, Lewis lung carcinoma;
VEGF, vascular endothelial growth factor; α-SMA, α smooth muscle
actin. |
Discussion
Macrophages secrete a variety of inflammatory
cytokines that are released into the tumor stroma and promote
inflammation and angiogenesis in lung cancer (8,24). A
recent study has demonstrated that PM2.5 mediates macrophage
autophagy via activation of PI3K/AKT/mTOR signaling (25). Another study demonstrated that
crosstalk between macrophages and lung cancer cells promotes the
progression of lung cancer via C-C motif chemokine receptor 2 and
C-X3-C motif chemokine receptor 1 (26). However, to the best of our knowledge,
the mechanisms associated with macrophage regulation of
PM2.5-induced lung cancer promotion have not yet been determined.
To assess the pro-angiogenic concentration of PM2.5 in
vitro, VEGF-A mRNA expression was examined in RAW264.7 cells
following short-term exposure to a variety of PM2.5 concentrations.
The results revealed that VEGF-A expression was increased in
RAW264.7 cells at a PM2.5 concentration of 500 µg/ml (Fig. S1). Only the effects of PM2.5 under
acute exposure conditions were examined. Therefore, the results may
change following chronic PM2.5 exposure. In the present study, no
significant differences were observed between angiogenic cytokines
in PM2.5-induced LLC cells. Therefore, LLC cells were exposed to
PM2.5-induced RAW264.7 supernatant for 3 h and high expression of
angiogenic cytokines was measured. These findings suggested that
macrophages are important in PM2.5-mediated angiogenesis in lung
cancer.
To test if this theory could be applied to in
vivo models, a mouse sponge implantation model and an LLC cell
xenograft mouse model were created. After implanting sponges
containing PM2.5 for 2 weeks to create a model in normal mice,
macrophage accumulation, microvessel formation and mRNA expression
levels of VEGF were revealed to increase in PM2.5-induced sponges.
Following tumor establishment in mice bearing LLC cells, it was
observed that PM2.5-induced lung cancer angiogenesis and growth
were closely associated with macrophage activation, since mice
bearing LLC cells exposed to PM2.5 or PM2.5-induced RAW264.7
supernatant exhibited increased VEGF release, macrophage
recruitment and angiogenesis in tumors. Interestingly, the present
study also demonstrated that LLC cells treated with normal
macrophage supernatant promoted tumor progression, and this may be
associated with the production of pro-angiogenic and
pro-inflammatory cytokines by macrophages in normal media. These
results revealed that PM2.5 promoted angiogenesis in normal and
lung cancer tissues, and macrophages were demonstrated to serve an
important role in this process. In the present study, the control
group in the in vitro experiments received the same volume
of PBS as the volume of PM2.5 extract. The same volume of normal
saline as the volume of PM2.5 extract was used as the control in
the in vivo experiments.
Furthermore, the present study examined
macrophage-mediated mechanisms, and the results revealed that the
mRNA and protein expression levels of macrophage-secreted VEGF were
increased following PM2.5 exposure. The present study only
demonstrated that CD47/Sirp-α expression increased in in
vivo and in vitro models. The specific role of the
CD47/Sirp-α signaling pathway in PM2.5-induced lung cancer invasion
requires further exploration, such as a CD47 knockout mice model.
Therefore, the present study may provide a basis for studying the
association between lung cancer progression and the CD47/Sirp-α
signaling pathway. Clodronate liposomes were used to deplete
macrophages in mice bearing LLC cells, which may further indicate
the mediation of macrophages. Therefore, the results demonstrated
that macrophages may regulate PM2.5-induced lung cancer progression
by releasing VEGF and triggering the CD47/Sirp-α signaling
pathway.
Previous studies have demonstrated that macrophages
function as significant regulators in the disease-associated
microenvironment and particularly in cancer (27,28). The
present study connected PM2.5 exposure to macrophage-mediated tumor
progression, and the results demonstrated that PM2.5 may promote
lung cancer in a macrophage-dependent manner.
In summary, the present study demonstrated that
PM2.5 promoted lung cancer progression in vivo and in
vitro, and macrophages were crucial in this process. As shown
in previous studies, VEGF can induce cancer growth by promoting
angiogenesis, whereas the CD47/SIRP-α signaling pathway is able to
increase tumor progression by affecting the phagocytosis of
macrophages and inflammatory response in the tumor microenvironment
(10,18). The possible mechanisms by which this
occurs may include macrophage-associated cytokine release and
CD47/Sirp-α signaling pathway activation, but further evidence is
required to support this hypothesis (Fig. 5). The specific association between
VEGF release and CD47/SIRP-α signaling pathway activation also
requires further exploration. The present study may provide novel
insights into the mechanisms of PM2.5-induced lung cancer
progression and provides potentially novel targets for lung cancer
treatment.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81573579) and the
Funding Program for New Interdisciplinary Subjects of Traditional
Chinese Medicine in Shanghai (grant no. E2-F18003).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding authors on reasonable
request.
Authors' contributions
RZ and JZ contributed to the design and analysis of
data. RL and LY contributed to the design of the study and wrote
the manuscript. NJ, FW and PZ revised the draft manuscript and
performed the experiments. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal studies were approved by the
Institutional Animal Care and Use Committee of Zhengzhou University
(Zhengzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rimkunas VM, Crosby KE, Li D, Hu Y, Kelly
ME, Gu TL, Mack JS, Silver MR, Zhou X and Haack H: Analysis of
receptor tyrosine kinase ROS1-Positive tumors in non-small cell
lung cancer: Identification of a FIG-ROS1 fusion. Clin Cancer Res.
18:4449–4457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ren YP, Tang AG, Zhou QX and Xiang ZY:
Clinical significance of simultaneous determination of serum
tryptophan and tyrosine in patients with lung cancer. J Clin Lab
Anal. 25:246–250. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang H, Luo J, Liu Z, Zhou R and Luo H:
MicroRNA-138 regulates DNA damage response in small cell lung
cancer cells by directly targeting H2AX. Cancer Invest. 33:126–136.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vinikoor-Imler LC, Allen DJ and Luben TJ:
An ecologic analysis of county-level PM2.5 concentrations and lung
cancer incidence and mortality. Int J Environ Res Public Health.
8:1865–1871. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao YB, Gao PP, Yang WD and Ni HG:
Vehicle exhaust: An overstated cause of haze in china. Sci Total
Environ. 612:490–491. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu CL, Guo H, Cheng X, Shao M, Wu C, Wang
S, Li H, Wei L, Gao Y, Tan W, et al: Exposure to airborne PM2.5
suppresses microRNA expression and deregulates target oncogenes
that cause neoplastic transformation in NIH3T3 cells. Oncotarget.
6:29428–29439. 2015.PubMed/NCBI
|
|
7
|
Zhou W, Tian DD, He J, Wang Y, Zhang L,
Cui L, Jia L, Zhang L, Li L, Shu Y, et al: Repeated PM2.5 exposure
inhibits BEAS-2B cell P53 expression through ROS-Akt-DNMT3B
pathway-mediated promoter hypermethylation. Oncotarget.
7:20691–20703. 2016.PubMed/NCBI
|
|
8
|
Chen JJ, Yao PL, Yuan A, Hong TM, Shun CT,
Kuo ML, Lee YC and Yang PC: Up-regulation of tumor interleukin-8
expression by infiltrating macrophages: Its correlation with tumor
angiogenesis and patient survival in non-small cell lung cancer.
Clin Cancer Res. 9:729–737. 2003.PubMed/NCBI
|
|
9
|
Koch S, Tugues S, Li X, Gualandi L and
Claesson-Welsh L: Signal transduction by vascular endothelial
growth factor receptors. Biochem J. 437:169–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Potente M and Carmeliet P: The link
between angiogenesis and endothelial metabolism. Ann Rev Physiol.
79:43–66. 2017. View Article : Google Scholar
|
|
11
|
Eichmann A and Simons M: VEGF signaling
inside vascular endothelial cells and beyond. Curr Opin Cell Biol.
24:188–193. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Siveen KS, Prabhu K, Krishnankutty R,
Kuttikrishnan S, Tsakou M, Alali FQ, Dermime S, Mohammad RM and
Uddin S: Vascular endothelial growth factor (VEGF) signaling in
tumour vascularization: Potential and challenges. Curr Vas
Pharmacol. 15:339–351. 2017.
|
|
13
|
Bekki K, Ito T, Yoshida Y, He C,
Arashidani K, He M, Sun G, Zeng Y, Sone H, Kunugita N and Ichinose
T: PM2.5 collected in china causes inflammatory and oxidative
stress responses in macrophages through the multiple pathways.
Environ Toxicol Pharmacol. 45:362–369. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao Q, Chen H, Yang T, Rui W, Liu F,
Zhang F, Zhao Y and Ding W: Direct effects of airborne PM 2.5
exposure on macrophage polarizations. Biochim Biophys Acta.
1860:2835–2843. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lindberg FP, Gresham HD, Schwarz E and
Brown EJ: Molecular cloning of integrin-associated protein: An
immunoglobulin family member with multiple membrane-spanning
domains implicated in alpha v beta 3-dependent ligand binding. J
Cell Biol. 123:485–496. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chao MP, Alizadeh AA, Tang C, Jan M,
Weissman-Tsukamoto R, Zhao F, Park CY, Weissman IL and Majeti R:
Therapeutic antibody targeting of CD47 eliminates human acute
lymphoblastic leukemia. Cancer Res. 71:1374–1384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murata Y, Kotani T, Ohnishi H and Matozaki
T: The CD47-SIRPα signalling system: Its physiological roles and
therapeutic application. J Biochem. 155:335–344. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oldenborg PA, Zheleznyak A, Fang YF,
Lagenaur CF, Gresham HD and Lindberg FP: Role of CD47 as a marker
of self on red blood cells. Science. 288:2051–2054. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jaiswal S, Chao MP, Majeti R and Weissman
IL: Macrophages as mediators of tumor immunosurveillance. Trends
Immunol. 31:212–219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seiffert M, Brossart P, Cant C, Cella M,
Colonna M, Brugger W, Kanz L, Ullrich A and Bühring HJ:
Signal-regulatory protein alpha (SIRPalpha) but not SIRPbeta is
involved in T-cell activation, binds to CD47 with high affinity,
and is expressed on immature CD34(+)CD38(−) hematopoietic cells.
Blood. 97:2741–2749. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang J, Modi Y, Yarovinsky T, Yu J,
Collinge M, Kyriakides T, Zhu Y, Sessa WC, Pardi R and Bender JR:
Macrophage β2 integrin-mediated, HuR-Dependent stabilization of
angiogenic factor-encoding mRNAs in inflammatory angiogenesis. Am J
Pathol. 180:1751–1760. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsushima Y, Jang JH, Yamada Y, Schwendener
R, Suzuki K, Weder W and Jungraithmayr W: The depletion of donor
macrophages reduces ischaemia-reperfusion injury after mouse lung
transplantation. Eur J Cardiothorac Surg. 45:703–709. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Carrillo de Santa Pau E, Arias FC, Caso
Peláez E, Muñoz Molina GM, Sánchez Hernández I, Muguruza Trueba I,
Moreno Balsalobre R, Sacristán López S, Gómez Pinillos A and del
Val Toledo Lobo M: Prognostic significance of the expression of
vascular endothelial growth factors A, B, C, and D and their
receptors R1, R2, and R3 in patients with nonsmall cell lung
cancer. Cancer. 115:1701–1712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Su R, Jin X, Zhang W, Li Z, Liu X and Ren
J: Particulate matter exposure induces the autophagy of macrophages
via oxidative stress-mediated PI3K/AKT/mTOR pathway. Chemosphere.
167:444–453. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schmall A, Altamari HM, Herold S,
Kampschulte M, Weigert A, Wietelmann A, Vipotnik N, Grimminger F,
Seeger W, Pullamsetti SS and Savai R: Macrophage and cancer cell
cross-talk via CCR2 and CX3CR1 is a fundamental mechanism driving
lung cancer. Am J Respir Crit Care Med. 191:437–447. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen M, Zhang J, Hu F, Liu S and Zhou Z:
Metformin affects the features of a human hepatocellular cell line
(HepG2) by regulating macrophage polarization in a co-culture
microenvironment. Diabetes Metab Res Rev. 31:781–789. 2016.
View Article : Google Scholar
|
|
28
|
Wang Q, Shu C, Su J and Li X: A crosstalk
triggered by hypoxia and maintained by MCP-1/miR-98/IL-6/p38
regulatory loop between human aortic smooth muscle cells and
macrophages leads to aortic smooth muscle cells apoptosis via Stat1
activation. Int J Clin Exp Pathol. 8:2670–2679. 2015.PubMed/NCBI
|
















