Introduction
Wilms' tumor (WT), also known as nephroblastoma, is
a malignant form of renal cancer that originates from the
metanephric blastema and is one of the most common types of
pediatric cancer (1). The incidence
rate of nephroblastoma makes it the fifth most common type of
pediatric malignant neoplasm, and ~90% of childhood renal tumors
are of the WT in 2001 (2,3). With advancements in diagnosis and
therapy, the survival rate of children with nephroblastoma has
greatly improved and the 5-year survival rate is >85% in
European children (1978–1997) (4,5).
However, some children with WT still have a much poorer prognosis,
such as those with bilateral WT and focal anaplastic WT, and cannot
be treated completely; thus having a poor clinical outcome due to
tumor recurrence and metastasis (6,7).
Therefore, there is an urgent requirement to identify potential
biomarkers or therapeutic targets for WT.
Recent evidence suggests that microRNAs (miRNAs) and
long non-coding RNAs (lncRNAs) could play an important role in the
occurrence and development of WT (8–10).
lncRNAs are non-coding RNA transcripts of >200 nucleotides in
length (11). In human cells,
lncRNAs are widely expressed and play a key regulatory role in
cellular activity (12). miRNAs are
a class of non-coding small RNA molecules that can inhibit the
expression of target genes by interacting with miRNA response
elements (13). Cao et al
(14) demonstrated that Stat3 can
promote cell proliferation through the upregulation of miRNA-370,
which directly inhibits the expression of Wilms’ tumor gene on X
chromosome (WTX), a tumor repressor of WT. Jiang and Li (15) found that p73 was the target mRNA of
miR-1180 and that the inhibition of miR-1180 could promote
apoptosis in the SK-NEP-1 cell line and inhibit tumor growth in
mice. Low expression levels of miR-21 in SK-NEP-1 cells were found
to promote cellular proliferation and migration by targeting PTEN,
which is a tumor suppressor (16).
Liu et al (17) demonstrated
that the knockdown of miR-19b can inhibit the proliferation,
invasion and migration of SK-NEP-1 WT cells. However, up to now,
only 4 lncRNAs (8,14,18,19) and
10 miRNAs (15–17,20,21) have
been intensively studied in WT, and the function and mechanism of
most of these remain unknown.
In 2011, Salmena et al (22) proposed the competitive endogenous
(ceRNA) regulatory network hypothesis. This hypothesis suggests
that lncRNAs could not only directly participate in the expression
of the target regulatory gene, but also may contain the seed
sequence of the core of the miRNAs. The target gene can be further
regulated by adsorbing the corresponding miRNA, thereby affecting
the number and abundance of miRNAs, ultimately affecting gene
expression. With the development of high-throughput sequencing
technology, an lncRNA-miRNA-mRNA ceRNA regulatory network has been
constructed for various disease types, such as gastric cancer,
colorectal cancer, breast cancer, liver cancer and periodontitis
(23–27). However, the construction of a ceRNA
network based on high-throughput sequencing has not yet been
generated for WT. The present study aimed to construct a ceRNA
regulatory network by investigating the associations between
lncRNA, miRNAs and mRNAs, and to identify candidate prognostic
biomarkers based on this network.
The Cancer Genome Atlas (TCGA) project was
constructed to understand the causes and pathogenesis of cancer
from a molecular perspective, thereby further improving early
diagnosis, treatment and, ultimately, cancer prevention (28). In the present study, RNAsequencing
(seq) and miRNAseq datasets were downloaded from TCGA database. The
data obtained were processed using the edgeR software, in order to
identify differentially expressed (DE)RNAs. Subsequently, DERNAs
were predicted and integrated to construct a lncRNA-miRNA-mRNA
ceRNA network for WT. Survival analysis was further conducted to
identify prognostic biomarkers in the ceRNA network. The
χ2 test was used to assess the association between the
expression of prognostic RNA and histology classification/clinical
staging. The present study aids our further understanding of the
molecular basis of WT, as well as the discovery of potential
prognostic markers for diagnosis and treatment.
Materials and methods
Data download and pre-processing
The RNA-sequencing (RNA-seq) data and the miRNA
sequencing data were downloaded from the TCGA data portal
[https://tcga-data.nci.nih.gov/tcga/;
Data Release v.15.0; release time: Feb 20, 2019; DbGaP (The
database of Genotypes and Phenotypes) study accession, phs000218).
The mRNA sequencing data included 120 WT malignant and 6 adjacent
normal tissues, the miRNA sequencing data included 126 WT malignant
tissues and 6 adjacent normal tissues. The GENCODE database is
currently the main genome annotation database (29). The GENCODE (22nd edition) GTF file
was used to annotate the expression profile of RNAseq files and
quantify lncRNAs and mRNAs, RNA not included in the GENCODE
database was excluded. Next, mRNA and lncRNA expression profiles
were extracted from the RNAseq expression matrix. Thus, three
expression profiles were obtained for mRNA, lncRNA and miRNA. The
clinical follow-up datasets from 126 patients with WT, including
clinical characteristics such as sex, age, ethnicity, pathological
stage, histology classification and survival status, were also
obtained from the TCGA database. The clinical characteristics of
patients with WT are summarized in Table
I.
 | Table I.Clinical characteristics of all
patients (n=128) in The Cancer Genome Atlas cohort. |
Table I.
Clinical characteristics of all
patients (n=128) in The Cancer Genome Atlas cohort.
|
Characteristics | Value |
|---|
| Age at diagnosis,
days |
|
Mean | 1,688 |
|
Range | 156-5,698 |
| Sex, n (%) |
|
Male | 54 (42.2) |
|
Female | 74 (57.8) |
| Ethnicity, n
(%) |
|
White | 95 (74.2) |
| Black
or African American | 19 (14.8) |
| Other
(Asian, Native Hawaiian, American Indian) | 5 (3.9) |
| Not
reported | 9 (7.0) |
| Clinical stage, n
(%) |
| I | 17 (13.3) |
| II | 49 (38.3) |
|
III | 47 (36.7) |
| IV | 14 (10.9) |
|
IV/V | 1 (0.8) |
| Histology
classification of primary tumor, n (%) |
|
FAWT | 84 (65.6) |
|
DAWT | 44 (34.4) |
| Adverse event |
|
Relapse | 94 (73.4) |
|
None | 27 (21.1) |
|
Progression | 7 (5.5) |
| Overall survival
time, days |
|
Mean | 1,728 |
|
Range | 180-4,795 |
| Vital status, n
(%) |
|
Alive | 76 (59.4) |
|
Deaths | 52 (40.6) |
Screening of DERNAs
Expression profiles from tumor and normal samples
were processed, and all data representing unexpressed RNA were
firstly filtered out. The remaining data were further analyzed
using the edgeR software to obtain Differentially expressed lncRNA
(DElncRNA), Differentially Expressed mRNA (DEmRNA) and
Differentially Expressed miRNA (DEmiRNA). EdgeR (v.3.28.0) is an R
package specifically designed to analyze DE genes (30). By following the steps outlined in the
edgeR operating guidelines, all P-values were corrected for
multiple tests using the false discovery rate (FDR). Subsequently,
the DERNAs were screened out using a specific cut-off value
[FDR<0.01 and log|fold change (FC)|>2]. Hierarchical
Clustering method was used to cluster DERNAs and samples. A volcano
plot and heat map plot were generated using the gplots package
(https://cran.r-project.org/web/packages/gplots/index.html;
v.3.0.12).
Construction of the ceRNA regulatory
network
The miRcode database (http://www.mircode.org/mircode/) is a web search
platform, which is dedicated to the prediction of target miRNAs by
uploading relevant lncRNA and miRNA (31). By uploading the DElncRNAs, miRNAs
that interact with DElncRNAs were screened out, and then overlapped
with DEmiRNAs in order to derive common miRNAs; the common miRNAs
were paired with the corresponding DElncRNAs to derive lncRNA-miRNA
pairing files. The miRDB (http://www.mirdb.org/; v.6.0) (32) is an online database for miRNA target
prediction using the MirTarget bioinformatics tool (v.4.0), which
was developed by analyzing thousands of miRNA-target interactions
from high-throughput sequencing experiments. The miRTarBase
(http://mirtarbase.mbc.nctu.edu.tw/php/index.php;
version 7.0) (33) is also a miRNA
target gene database, which is validated in in vivo
experiments. TargetScan (http://www.targetscan.org/vert_72/; v.7.2) predicts
miRNA targets by determining mRNAs with conserved sequence
complementarity to the seed (nucleotides 2–7) of the input miRNA
(34). The target mRNAs of the
miRNAs in the lncRNA-miRNA pairs were predicted using these three
databases (miRDB, miRTarBase and TargetScan), and defined target
mRNAs that were predicted by all three databases, as the final
screened-out target genes. Subsequently, the intersection elements
were obtained between the target mRNAs and DEmRNAs and these
intersection mRNAs were selected to build a miRNA-mRNA pairing
file. Cytoscape (v.3.3.2) is an open source bioinformatics software
platform for visualizing and analyzing molecular interaction
networks (35). The NetWorkAnalyzer
toolkit (v.3.3.2) from Cytoscape was used to analyze the
characteristics of the ceRNA topology network, including network
connections, the path length, the closest centrality of nodes, and
the degree of connectivity. Based on the node degree, the hub
lncRNAs (degree >5) and its associated mRNAs and miRNAs in the
network were identified. It was reported that lncRNAs can
positively regulate mRNAs by competitive combination with miRNAs.
Therefore, correlation analysis was performed for each candidate
ceRNA pair, and only pairs with correlation coefficients >0.4
and P<0.05 were selected as the final ceRNA pairs. Finally, a
ceRNA topological network was constructed.
Functional and enrichment analyses of
mRNAs in the ceRNA network
Gene Ontology (GO) is a database established by the
Gene Ontology Consortium and aims to annotate genes and gene
products from different organisms using three ontologies, including
cellular component (CC), molecular function (MF) and biological
process (BP) (36). The Kyoto
Encyclopedia of Genes and Genomes (KEGG) is a database that
systematically analyzes the metabolic pathways of gene products and
their functions (37). The
clusterProfiler (v.3.14.3) is a bioconductor package, which can
perform statistical analyses and visualization of functional
clustering of gene sets or gene clusters (38). In order to explore the function of
mRNAs in the ceRNA network, a GO and KEGG pathway analysis was
conducted.
Protein-protein interaction (PPI)
network analysis
To determine the interactive associations between
DEmRNAs in the ceRNA network, a PPI network analysis was performed
with the online software Search Tool for the Retrieval of
Interacting Genes/Proteins (STRING (version 11.0; http://string-db.org); confidence score >0.4)
(39).
Survival analysis of the ceRNA
module
Clinical information was downloaded from TCGA
database, and the survival data of patients were extracted and
combined with the expression matrix of RNAs in the ceRNA
topological network. A Kaplan-Meier (KM) survival curve was
analyzed for each node in the ceRNA topological network using the
survival package (v.3.1–8) in R. The median value was used as the
cut-off for the gene expression value and the log-rank test was
performed to determine the differences between the high and the low
expression groups (40). P<0.05
was considered to indicate a statistically significant
difference.
Screening of important
prognosis-associated DERNAs
Following expression and survival analyses, four
types of prognosis-associated DERNAs were identified: i) High
expression of DERNAs were associated with a poor prognosis in
patients with WT, ii) low expression of DERNAs were associated with
a poor prognosis in patients with WT, iii) high expression of
DERNAs were associated with a good prognosis in patients with WT
and iv) low expression of DERNAs were associated with a good
prognosis in patients with WT. The abnormal expression of DERNAs,
which were associated with a poor prognosis in patients with WT,
were selected as the prognosis-associated DERNAs for further
experimentation. χ2 tests were performed to analyze the
associations between the important prognosis-associated DERNAs and
clinical characteristics.
Validation of the expression of
prognosis-related DERNAs
In order to further confirm the expression levels of
the candidate prognosis-associated DERNAs, validation datasets were
downloaded from the Gene Expression Omnibus (GEO) database
(https://www.ncbi.nlm.nih.gov/geo/). A
total of 28 samples of WT tissues and 4 adjacent non-tumor tissues
were included in the GSE66405 dataset. The expression profile of
GSE50505 contained 28 samples of WT tissues and 6 adjacent
non-tumor tissues. The miRNA expression profiles were also obtained
from the GEO database, including GSE50505 (26 WT samples and 12
adjacent non-tumor tissues) and GSE57370 (62 WT samples and 4
normal controls (41,42). All validation datasets were
downloaded from the Gene Expression Omnibus (GEO) database.
Unpaired t-tests were used to compare the differences in expression
levels. P<0.05 was considered to indicate a statistically
significant difference.
Results
Identification of Significantly
DERNAs
The clinical data, RNAseq data and miRNAseq data
regarding WT were downloaded from TCGA database. Subsequently, the
edgeR package was used to analyze the original expression profiles
between WT and normal tissues. Based on the cut-off conditions
(FDR<0.01 and log|FC|>2), a total of 3,337 DEmRNAs were
screened, including 1,577 upregulated mRNAs and 1,760 downregulated
mRNAs (Table SI). For lncRNAs,
1,784 DElncRNAs were identified, including 833 that were
upregulated and 951 that were downregulated (Table SII). The differential expression
profiles of miRNAs were also compared in tumor and normal tissues;
114 DEmiRNAs were identified, of which 49 were upregulated and 65
were downregulated (Table SIII).
The volcano plot and heat maps are shown in Fig. 1.
Construction of the ceRNA network
To further the understanding of the role of these
DERNAs in WT, a ceRNA network was constructed to understand the
interaction between them. Firstly, 23 DEmiRNAs that interacted with
DElncRNAs were predicted, based on the results from the miRcode
database. The mRNAs targeted by these 23 DEmiRNAs were retrieved
from the miRTarBase, TargetScan and miRDB databases. Subsequently,
these targeted mRNAs were compared with the 3,337 DEmRNAs obtained
from the aforementioned differential analyses. The mRNAs that were
not included in the 3,337 DEmRNAs were excluded, resulting in 133
DEmRNAs and 19 DEmiRNAs in the ceRNA networks. Following this, the
19 DEmiRNAs were compared with miRNAs in the lncRNA-miRNA pairing
file, resulting in 189 DElncRNAs. Finally, 735 DElncRNA-DEmiRNA
pairs and 188 DEmiRNA-DEmRNA interaction pairs were identified from
189 DElncRNAs, 19 DEmiRNAs and 133 DEmRNAs. In order to verify the
reliability of the network, network analyses were performed in
order to understand the characteristics of the ceRNA network. With
an increase in node degree, the number of nodes decreased (Fig. S1A). The closeness centrality is a
measure centrality which describes how fast information spreads
from a given node to other reachable nodes in the network. A number
of nodes displayed the numbers of closeness centrality ~0.4, which
indicated these nodes were relatively centralized within the
network. Only a few nodes have closeness centrality ~0.5, which
indicated those nodes were relatively sparsely distributed
(Fig. S1B). The distribution of
shared neighbors is shown in Fig.
S1C; most nodes in the network had few shared neighbors.
Shortest paths is a measure of a network's overall navigability
(43). Fig. S1D demonstrates the distribution of
the shortest path and the path length was relatively shorter
(<4), which means the network has better navigability. As the
hub genes with higher degree in biological networks were more
likely to be important, a hub lncRNA (degree >5) and its linked
mRNAs and miRNAs in the ceRNA network were screened. According to
the correlation analyses (Cor >0.4; P<0.05; Table SIV), candidate ceRNA pairs were
chosen as the final ceRNA pairs. Finally, a ceRNA topological
network was reconstructed, which included 218 DElncRNA-DEmiRNA
pairs and 138 DEmiRNA-DEmRNA interaction pairs from 38 DElncRNAs,
18 DEmiRNAs and 99 DEmRNAs (Table
SV). Subsequently, the Cytoscape software was used to visualize
this information, and the constructed ceRNA regulatory network from
WT is shown in Fig. 2.
Functional enrichment of the
DEmRNAs
In order to investigate the function of the 99
DEmRNAs in the ceRNA network and the signaling pathways involved,
functional and pathway enrichment analyses were performed using the
clusterProfiler package. From GO analyses, 73 GO entries (FDR
<0.01) were obtained. The results showed that these pathways
were mainly enriched in the ‘positive regulation of smooth muscle
cell proliferation’, ‘muscle cell proliferation’ and ‘regulation of
transforming growth factor β production’. MF was mainly directed to
‘transcription factor activity’. The CC mainly included ‘RNA
polymerase II transcription factor complex’, ‘nuclear chromatin’,
‘nuclear transcription factor complex’ and ‘transcription factor
complex’ (Fig. 3A). From KEGG
pathway analyses, it was found that these DEmRNAs were
significantly enriched in the pathways of the ‘cell cycle’, ‘small
cell lung cancer’, ‘miRNAs in cancer’, ‘human papilloma virus
infection’ and ‘bladder cancer’ (Fig.
3B). To better understand the role of DEmRNAs, a PPI network
was established using the STRING online software, including 96
nodes and 298 edges (Fig. 4A), of
which the main hub nodes were CHEK1 (checkpoint kinase 1), CDC25A
(cell division cycle 25A), SKP2 (S-phase kinase associated protein
2), STAT3 (signal transducer and activator of transcription 3) and
E2F1 (E2F transcription factor 1) (Fig.
4B).
Identification of prognosis-associated
genes in WT
KM analysis was performed to investigate the overall
survival of patients with WT for the DERNAs (38 DElncRNAs, 18
DEmiRNAs and 99 DEmRNAs) in the ceRNA network. As a result, the
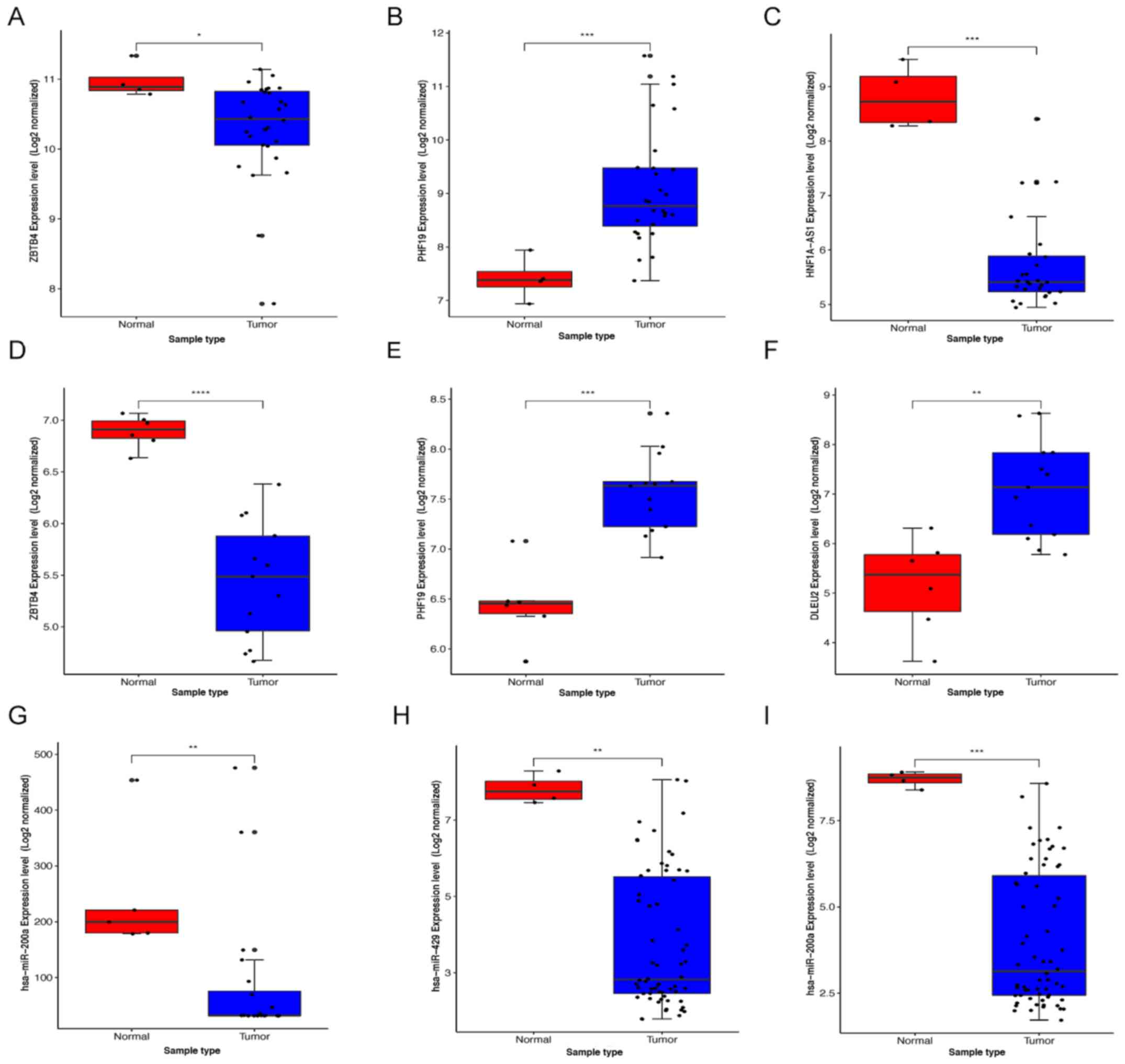
expression levels of 2 DEmRNAs [zinc finger and BTB domain
containing 4 (ZBTB4) and PHD finger protein 19 (PHF19)] (Figs. 5C and D), 5 DElncRNAs [maternally
expressed 3 (MEG3), rhabdomyosarcoma 2 associated transcript
(RMST), ZNF503 antisense RNA 1 (ZNF503-AS1), HNF1A antisense RNA 1
(HNF1A-AS1), deleted in lymphocytic leukemia 2 (DLEU2)] (Figs. S2A-C and 5E and F) and 4 DEmiRNAs (hsa-miR-132,
hsa-miR-200a, hsa-miR-429 and hsa-miR-506) (Figs. 5G-I and S2D) were associated with the
overall survival time of patients with WT (P<0.05). High
expression of PHF19, DLEU2, ZNF503-AS1 and hsa-miR-506 was
associated with low survival time. High expression of ZBTB4, MEG3,
RMST, HNF1A-AS1, hsa-miR-132, hsa-miR-200a and hsa-miR-429 was
associated with high survival time.
 | Figure 5.Screening of the key RNAs in WT.
Identification of key (A) upregulated competitive endogenous
RNA-associated DERNAs and (B) downregulated DERNAs by combining
expression and prognosis analyses. Expression and prognostic value
of (C) ZBTB4, (D) PHF19, (E) DLEU2, (F) HNF1A-AS1, (G) hsa-miR-132,
(H) hsa-miR-200a, and (I) hsa-miR-429 in *P<0.05, **P<0.01,
***P<0.001. WT. WT, Wilms' tumor; DE, differentially expressed;
ZBTB4, zinc finger and BTB domain containing 4; PHF19, PHD finger
protein 19; DLEU2, deleted in lymphocytic leukemia 2; HNF1A
antisense RNA 1; miR, microRNA. |
By combining the results of expression
and survival analysis, DERNAs whose abnormal expression resulted in
a trend for decreased survival times in WT patients were screened
out
Among the 11 prognosis-related DERNAs, 2 RNAs (PHF19
and DLEU2) that were not only significantly upregulated in WT but
also for which increased expression indicated a poor prognosis
(Fig. 5A, D and E), 5 RNAs (ZBTB4,
HNF1A-AS1, hsa-miR429, hsa-miR-132 and hsa-miR-200a) had low
expression in tumor samples and the low expression of these
indicated a poor prognosis (Fig. 5B, C
and F-I). High expression levels of MEG3 and RMSET were
associated with a good prognosis in patients with WT (Fig. S2A and B), while low expression
levels of ZNF503-AS1 and hsa-miR-506 were associated with a good
prognosis in patients with WT (Fig. S2C
and D). A total of 7 RNAs (PHF19, DLEU2, ZBTB4, HNF1A-AS1,
hsa-miR-429, hsa-miR-132 and hsa-miR-200a) which were associated
with a poor prognosis in patients with WT were selected for further
analysis. The association between the expression of these seven
RNAs and histology classification and clinical staging were
subsequently analyzed. For mRNAs, the results demonstrated that the
expression of ZBTB4 was associated with clinical staging and
histology classification (P<0.05; Fig. 6A and C). For lncRNAs, DLEU2 was also
associated with both histology classification and clinical staging
(P<0.05; Fig. 6B and D). For
miRNAs, hsa-miR-132 was only significantly associated with
histology classification (P<0.05; Fig. 6E).
GEO dataset verification
A total of seven aforementioned prognosis-associated
RNAs (PHF19, DLEU2, ZBTB4, HNF1A-AS1, hsa-miR-429, hsa-miR-132 and
hsa-miR-200a) were selected for validation. Consistent with the
aforementioned results, downregulation of ZBTB4 (Fig. 7A and D) and upregulation of PHF19
(Fig. 7B and E) were confirmed in
GSE66405 and GSE110696 datasets, respectively. The mean expression
levels of HNF1A-AS1 were significantly lower in WT tissues compared
with that in normal tissues in the GSE66405 dataset (Fig. 7C). DLEU2 was demonstrated to be
significantly higher in WT tissues compared with that in normal
tissues in the GSE110696 dataset (Fig.
7F). DLEU was not obtained from the GSE66405 dataset, as well
as HNF1A-AS1 in the GSE110696 dataset; this may be as the two genes
were not considered when designing probe sequences. For miRNA, in
the GSE50505 dataset, the expression levels of hsa-miR-200a were
significantly lower in WT tissues compared with that in normal
tissue (Fig. 7G). In the GSE57370
dataset, hsa-miR-200a and hsa-miR-429 were significantly lower in
WT tissues (Fig. 7H and I). No
significant differences were found in hsa-miR-132 or hsa-miR-429
expression in the GSE50505 (Fig. S3A
and B). hsa-miR-132 was not found to be DE in the GSE57370
dataset (Fig. S3C). This may be due
to the small number of patients studied.
Flow diagram representing construction
and analysis of the ceRNA regulatory network
A flow diagram representing the construction of the
lncRNA-miRNA-mRNA regulatory network and the screening of
prognostic RNAs in WT is shown in Fig.
8.
 | Figure 8.Flow diagram of the construction and
analysis of the competitive endogenous RNA regulatory network in
Wilms' tumor. Pale yellow denotes RNA, which was associated with
clinical staging and histology classification. Light orange
represents RNA, which was associated with histology classification.
TCGA, The Cancer Genome Atlas; lncRNA, long non-coding RNA;
miRNA/miR, microRNA; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of
Genes and Genomes; PPI, protein-protein interaction; KM,
Kaplan-Meier; ZBTB4, zinc finger and BTB domain containing 4;
DLEU2, deleted in lymphocytic leukemia 2; DE, differentially
expressed; cor, correlation. |
Discussion
WT is the most common renal tumor in children and
its carcinogenesis and progression is driven by multiple
interacting mechanisms (44). The
ceRNA hypothesis provides important clues and directions for the
study of tumor pathogenesis, and provides a novel theoretical basis
for the diagnosis and treatment of tumor (45). Studies have shown that lncRNA, miRNA
and mRNA in the ceRNA network are in a certain equilibrium state,
and that disease occurs once the balance is broken (46,47). The
ceRNA hypothesis integrates the relationship between lncRNAs, mRNAs
and miRNAs and can better explain the interaction among a variety
of types of RNAs (22). In order to
lay a useful foundation for studying the regulatory function of
ceRNA in WT, a ceRNA network was constructed at the
transcriptome-wide level and screened for prognosis-associated
biomarkers for diagnosis and treatment purposes.
In the present study, a ceRNA regulatory network for
WT was successfully constructed. The network was composed of 38
DElncRNAs, 18 DEmiRNAs and 99 DEmRNAs. Among these RNAs, combined
with expression and the survival analysis, 7 DERNAs (PHF19, DLEU2,
ZBTB4, HNF1A-AS1, hsa-miR-429, hsa-miR-132 and hsa-miR-200a) were
significantly associated with prognosis. These RNAs may be
potential biomarkers for predicting prognosis in WT. The expression
of DLEU2 was associated with the histology classification and
clinical staging, indicating that DLEU2 is an important lncRNA and
the over expression of DLEU2 may play a role in the pathogenesis
and progression of WT. Similar studies have found that the
inactivation of DLEU2 can promote cell proliferation and tumor
progression through functional loss of miR-15a/miR-16-1 (48–50). The
present study demonstrated that DLEU2 was associated with two key
DEmiRNAs (hsa-miR-21 and hsa-miR-506) and competed to regulate the
mRNA expression in WT (Table SV).
Therefore, an in-depth study of the mechanism of action of DLEU2
and its associated miRNAs maybe beneficial, as a potential
treatment target for WT.
Previous studies have revealed that miRNAs have been
extensively investigated in cancer (51) and may play important biological
functions by regulating target genes in WT. For example, miR-100-5p
and miR-130b-3p could be potential biomarkers for WT and the
expression levels of these miRNAs in serum was stable over time
with different serum storage conditions (52). The present study demonstrated that
the expression level of hsa-miR-132 was lower in WT tissues
compared with that in normal tissue and could be a significant
indicator for poor prognosis in patients with WT. In addition, it
was found that the expression level of hsa-miR-132 was associated
with histology classification. hsa-miR-132, as a member of the
miR-212/132 family, which is highly conserved in vertebrates, has
been reported as a tumor-associated miRNA in a variety of cancer
types, including liver, colorectal, pancreatic and ovarian cancer
(53–56). From the aforementioned studies, it
has been suggested that miR-132 may have a potential involvement in
the occurrence and development of WT.
In addition, among the 99 target mRNAs in the ceRNA
regulatory network, functional enrichment results indicated that
these genes are involved in pathways associated with cancer
development. Survival analyses demonstrated that two target mRNAs
(ZBTB4 and PHF19) were significantly associated with the prognosis
of patients with WT. Patients with low expression of ZBTB4 have
poor prognoses, whereas low expression of PHF19 contributes to
prolongation of survival time in patients with WT. It was also
found that the expression level of ZBTB4 was associated with
histology classification and clinical staging (P<0.05), which
indicated that ZBTB4 may have an important role in the
tumorigenesis of WT. ZBTB4, is a mammalian DNA-binding protein, and
contains C2H2 zinc fingers and a POZ/BTB domain, and functions as a
transcriptional repressor protein (57). Loss of ZBTB4 expression has been
observed in several types of cancer, including breast cancer,
prostate cancer and Ewing sarcoma, and was associated with shorter
relapse-free patient survival (58–60).
Roussel-Gervais et al (61)
reported that ZBTB4 had the capacity for methyl-CpG-binding and had
a conserved role in the preservation of genomic stability in 8
tumor types: breast-invasive carcinoma, prostate adenocarcinoma,
uterine endometrioid carcinoma, kidney renal cell carcinoma,
cervical squamous cell carcinoma, lung adenocarcinoma, stomach
adenocarcinoma and head and neck squamous cell cancer. Furthermore,
the loss of ZBTB4 induced transcriptional alterations indicative of
aneuploidy and mitotic checkpoint deregulation (61). A recent study showed that high PHF19
expression was associated with a shorter survival time in patients
with ovarian carcinoma, and that the silencing of PHF19 could
reduce cell proliferation (62).
Thus, further study on the mechanism of action of these two mRNAs
is required, which may prove beneficial as possible targets for the
treatment of WT.
The present study successfully identified numerous
DElncRNAs, DEmiRNAs and DEmRNAs in WT. Moreover, a WT-specific
lncRNA-associated ceRNA regulatory network was constructed,
providing a potential target for the diagnosis, treatment and
prognosis evaluation of WT. Importantly, novel candidate biomarkers
consisting of two DElncRNAs (HNF1A-AS1 and DELU2), three DEmiRNAs
(hsa-miR-429, hsa-miR-132 and hsa-miR-200a) and two DEmRNAs (ZBTB4
and PHF19) were identified. These biomarkers were significantly
associated with overall survival time in patients with WT. Notably,
ZBTB4 and DLEU2 were significantly associated both with clinical
staging and histology classification. ZBTB4 and DLEU2 may therefore
be considered as promising targets for therapy in WT. Moreover,
further studies are required in order to validate the role of newly
discovered genes in the mechanism of WT progression.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Ms Lingyan Dai, Mrs
Mianli Xiao, Ms Jieyu Feng, Ms Yue Wu and Mr Sixia You from
Tianpeng Technology Co., Ltd. (Guangzhou, China) for their
assistance and contributions in processing and analyzing the data.
The results published here are in whole or part based upon data
generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Funding
This study was supported by the Natural Science
Foundation of Guangdong Province (grant no. 2016A030310187).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XL, JX and ZL contributed to the study design. CL
searched for and downloaded the gene expression profiles from the
Gene Expression Omnibus database. YR, WC and CL performed the
analysis and interpretation of the data. ZL, WZ, CL and YR
critically revised the manuscript for important intellectual
content. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Breslow N, Olshan A, Beckwith JB and Green
DM: Epidemiology of Wilms tumor. Med Pediatr Oncol. 21:172–181.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brok J, Treger TD, Gooskens SL, van den
Heuvel-Eibrink MM and Pritchard-Jones K: Biology and treatment of
renal tumours in childhood. Eur J Cancer. 68:179–195. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chu A, Heck JE, Ribeiro KB, Brennan P,
Boffetta P, Buffler P and Hung RJ: Wilms' tumour: A systematic
review of risk factors and meta-analysis. Paediatr Perinat
Epidemiol. 24:449–469. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pastore G, Znaor A, Spreafico F, Graf N,
Pritchard-Jones K and Steliarova-Foucher E: Malignant renal tumours
incidence and survival in European children (1978–1997): Report
from the Automated Childhood Cancer Information System project. Eur
J Cancer. 42:2103–2114. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Magnani C, Pastore G, Coebergh JW, Viscomi
S, Spix C and Steliarova-Foucher E: Trends in survival after
childhood cancer in Europe, 1978-1997: Report from the Automated
Childhood Cancer Information System project (ACCIS). Eur J Cancer.
42:1981–2005. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Geller JI: Current standards of care and
future directions for ‘high-risk’ pediatric renal tumors:
Anaplastic Wilms tumor and Rhabdoid tumor. Urol Oncol. 34:50–56.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Spreafico F, Pritchard Jones K,
Malogolowkin MH, Bergeron C, Hale J, de Kraker J, Dallorso S, Acha
T, de Camargo B, Dome JS, et al: Treatment of relapsed Wilms
tumors: Lessons learned. Expert Rev Anticancer Ther. 9:1807–1815.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu S, Fu W, Zhang L, Fu K, Hu J, Jia W
and Liu G: LINC00473 antagonizes the tumour suppressor miR-195 to
mediate the pathogenesis of Wilms tumour via IKKα. Cell Prolif. Nov
20–2017.(Epub ahead of print). doi:
https://doi.org/10.1111/cpr.12416.
|
|
9
|
Yu X, Li Z, Chan MT and Wu WK: The roles
of microRNAs in Wilms' tumors. Tumour Biol. 37:1445–1450. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Su L, Wu A, Zhang W and Kong X: Silencing
long non-coding RNA SNHG6 restrains proliferation, migration and
invasion of Wilms' tumour cell lines by regulating miR-15a. Artif
Cells Nanomed Biotechnol. 47:2670–2677. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Muers M: RNA: Genome-wide views of long
non-coding RNAs. Nat Rev Genet. 12:742–743. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ye W, Lv Q, Wong CK, Hu S, Fu C, Hua Z,
Cai G, Li G, Yang BB and Zhang Y: The effect of central loops in
miRNA:MRE duplexes on the efficiency of miRNA-mediated gene
regulation. PLoS One. 3:e17192008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao X, Liu D, Yan X, Zhang Y, Yuan L,
Zhang T, Fu M, Zhou Y and Wang J: Stat3 inhibits WTX expression
through up-regulation of microRNA-370 in Wilms tumor. FEBS Lett.
587:639–644. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang X and Li H: MiR-1180-5p regulates
apoptosis of Wilms' tumor by targeting p73. OncoTargets Ther.
11:823–831. 2018. View Article : Google Scholar
|
|
16
|
Cui M, Liu W, Zhang L, Guo F, Liu Y, Chen
F, Liu T, Ma R and Wu R: Over-expression of miR-21 and lower PTEN
levels in Wilms' tumor with aggressive behavior. Tohoku J Exp Med.
242:43–52. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu GL, Yang HJ, Liu B and Liu T: Effects
of MicroRNA-19b on the proliferation, apoptosis, and migration of
Wilms' tumor cells via the PTEN/PI3K/AKT signaling pathway. J Cell
Biochem. 118:3424–3434. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Okamoto K, Morison IM, Taniguchi T and
Reeve AE: Epigenetic changes at the insulin-like growth factor
II/H19 locus in developing kidney is an early event in Wilms
tumorigenesis. Proc Natl Acad Sci USA. 94:5367–5371. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Hou T, Qi X, Wang J and Sun X:
SOX21-AS1 is associated with clinical stage and regulates cell
proliferation in nephroblastoma. Biosci Rep. May 17–2019.(Epub
ahead of print). doi: 10.1042/BSR20190602.
|
|
20
|
Peng Y and Croce CM: The role of MicroRNAs
in human cancer. Signal Transduct Target Ther. 1:150042016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Su H, Wang X, Song J, Wang Y, Zhao Y and
Meng J: MicroRNA-539 inhibits the progression of Wilms' tumor
through downregulation of JAG1 and Notch1/3. Cancer Biomark.
24:125–133. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu H, Zhang Z, Wu N, Guo H, Zhang H, Fan
D, Nie Y and Liu Y: Integrative analysis of dysregulated
lncRNA-associated ceRNA network reveals functional lncRNAs in
gastric cancer. Genes (Basel). Jun 18–2018.(Epub ahead of print).
doi: 10.3390/genes9060303.
|
|
24
|
Arun K, Arunkumar G, Bennet D,
Chandramohan SM, Murugan AK and Munirajan AK: Comprehensive
analysis of aberrantly expressed lncRNAs and construction of ceRNA
network in gastric cancer. Oncotarget. 9:18386–18399. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao C, Li H, Zhuang J, Zhang H, Wang K,
Yang J, Liu C, Liu L, Zhou C and Sun C: The construction and
analysis of ceRNA networks in invasive breast cancer: A study based
on The Cancer Genome Atlas. Cancer Manag Res. 11:1–11. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yan Y, Yu J, Liu H, Guo S, Zhang Y, Ye Y,
Xu L and Ming L: Construction of a long non-coding RNA-associated
ceRNA network reveals potential prognostic lncRNA biomarkers in
hepatocellular carcinoma. Pathol Res Pract. 214:2031–2038. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yuan W, Li X, Liu L, Wei C, Sun D, Peng S
and Jiang L: Comprehensive analysis of lncRNA-associated ceRNA
network in colorectal cancer. Biochem Biophys Res Commun.
508:374–379. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Weinstein JN, Collisson EA, Mills GB, Shaw
KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C and Stuart JM;
Cancer Genome Atlas Research Network, : The Cancer Genome Atlas
Pan-Cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aken BL, Ayling S, Barrell D, Clarke L,
Curwen V, Fairley S, Fernandez Banet J, Billis K, García Girón C,
Hourlier T, et al: The Ensembl gene annotation system. Database
(Oxford). Jun 23–2016.(Epub ahead of print). doi:
10.1093/database/baw093. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jeggari A, Marks DS and Larsson E:
miRcode: A map of putative microRNA target sites in the long
non-coding transcriptome. Bioinformatics. 28:2062–2063. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wong N and Wang X: miRDB: An online
resource for microRNA target prediction and functional annotations.
Nucleic Acids Res. 43:D146–D152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hsu SD, Lin FM, Wu WY, Liang C, Huang WC,
Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al: miRTarBase: A
database curates experimentally validated microRNA-target
interactions. Nucleic Acids Res. 39 (Suppl 1):D163–D169. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Blake JA and Harris MA: The Gene Ontology
(GO) project: Structured vocabularies for molecular biology and
their application to genome and expression analysis. Curr Protoc
Bioinformatics. Sep 1–2008.(Epub ahead of print). doi:
https://doi.org/10.1002/0471250953.bi0702s23. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47:D607–D613. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bland JM and Altman DG: The logrank test.
BMJ. 328:10732004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ludwig N, Werner TV, Backes C, Trampert P,
Gessler M, Keller A, Lenhof HP, Graf N and Meese E: Combining miRNA
and mRNA Expression Profiles in Wilms Tumor Subtypes. Int J Mol
Sci. 17:4752016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu M, Roth A, Yu M, Morris R, Bersani F,
Rivera MN, Lu J, Shioda T, Vasudevan S, Ramaswamy S, et al: The
IGF2 intronic miR-483 selectively enhances transcription from IGF2
fetal promoters and enhances tumorigenesis. Genes Dev.
27:2543–2548. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Barabási AL and Oltvai ZN: Network
biology: Understanding the cell's functional organization. Nat Rev
Genet. 5:101–113. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tian F, Yourek G, Shi X and Yang Y: The
development of Wilms tumor: From WT1 and microRNA to animal models.
Biochim Biophys Acta. 1846:180–187. 2014.PubMed/NCBI
|
|
45
|
Qi X, Zhang DH, Wu N, Xiao JH, Wang X and
Ma W: ceRNA in cancer: Possible functions and clinical
implications. J Med Genet. 52:710–718. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chan JJ and Tay Y: Noncoding RNA:RNA
Regulatory Networks in Cancer. Int J Mol Sci. 19:E13102018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Klein U, Lia M, Crespo M, Siegel R, Shen
Q, Mo T, Ambesi-Impiombato A, Califano A, Migliazza A, Bhagat G, et
al: The DLEU2/miR-15a/16-1 cluster controls B cell proliferation
and its deletion leads to chronic lymphocytic leukemia. Cancer
Cell. 17:28–40. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lerner M, Harada M, Lovén J, Castro J,
Davis Z, Oscier D, Henriksson M, Sangfelt O, Grandér D and Corcoran
MM: DLEU2, frequently deleted in malignancy, functions as a
critical host gene of the cell cycle inhibitory microRNAs miR-15a
and miR-16-1. Exp Cell Res. 315:2941–2952. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xie ZZ, Xiao ZC, Song YX, Li W and Tan GL:
Long non-coding RNA Dleu2 affects proliferation, migration and
invasion ability of laryngeal carcinoma cells through triggering
miR-16-1 pathway. Eur Rev Med Pharmacol Sci. 22:1963–1970.
2018.PubMed/NCBI
|
|
51
|
Tan W, Liu B, Qu S, Liang G, Luo W and
Gong C: MicroRNAs and cancer: Key paradigms in molecular therapy.
Oncol Lett. 15:2735–2742. 2018.PubMed/NCBI
|
|
52
|
Ludwig N, Nourkami-Tutdibi N, Backes C,
Lenhof HP, Graf N, Keller A and Meese E: Circulating serum miRNAs
as potential biomarkers for nephroblastoma. Pediatr Blood Cancer.
62:1360–1367. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mokutani Y, Uemura M, Munakata K, Okuzaki
D, Haraguchi N, Takahashi H, Nishimura J, Hata T, Murata K,
Takemasa I, et al: Down-Regulation of microRNA-132 is Associated
with Poor Prognosis of Colorectal Cancer. Ann Surg Oncol. 23 (Suppl
5):599–608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Park JK, Henry JC, Jiang J, Esau C, Gusev
Y, Lerner MR, Postier RG, Brackett DJ and Schmittgen TD: miR-132
and miR-212 are increased in pancreatic cancer and target the
retinoblastoma tumor suppressor. Biochem Biophys Res Commun.
406:518–523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tian H, Hou L, Xiong YM, Huang JX, Zhang
WH, Pan YY and Song XR: miR-132 targeting E2F5 suppresses cell
proliferation, invasion, migration in ovarian cancer cells. Am J
Transl Res. 8:1492–1501. 2016.PubMed/NCBI
|
|
56
|
Zhang X, Tang W, Li R, He R, Gan T, Luo Y,
Chen G and Rong M: Downregulation of microRNA-132 indicates
progression in hepatocellular carcinoma. Exp Ther Med.
12:2095–2101. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Filion GJ, Zhenilo S, Salozhin S, Yamada
D, Prokhortchouk E and Defossez PA: A family of human zinc finger
proteins that bind methylated DNA and repress transcription. Mol
Cell Biol. 26:169–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kim K, Chadalapaka G, Lee SO, Yamada D,
Sastre-Garau X, Defossez PA, Park YY, Lee JS and Safe S:
Identification of oncogenic microRNA-17-92/ZBTB4/specificity
protein axis in breast cancer. Oncogene. 31:1034–1044. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ross-Adams H, Lamb AD, Dunning MJ, Halim
S, Lindberg J, Massie CM, Egevad LA, Russell R, Ramos-Montoya A,
Vowler SL, et al CamCaP Study Group, : Integration of copy number
and transcriptomics provides risk stratification in prostate
cancer: A discovery and validation cohort study. EBioMedicine.
2:1133–1144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yu Y, Shang R, Chen Y, Li J, Liang Z, Hu
J, Liu K and Chen C: Tumor suppressive ZBTB4 inhibits cell growth
by regulating cell cycle progression and apoptosis in Ewing
sarcoma. Biomed Pharmacother. 100:108–115. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Roussel-Gervais A, Naciri I, Kirsh O,
Kasprzyk L, Velasco G, Grillo G, Dubus P and Defossez PA: Loss of
the Methyl-CpG-Binding Protein ZBTB4 Alters Mitotic Checkpoint,
Increases Aneuploidy, and Promotes Tumorigenesis. Cancer Res.
77:62–73. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tao F, Tian X, Ruan S, Shen M and Zhang Z:
miR-211 sponges lncRNA MALAT1 to suppress tumor growth and
progression through inhibiting PHF19 in ovarian carcinoma. FASEB J.
Jun 6–2018.(Epub ahead of print). doi: 10.1096/fj.201800495RR.
View Article : Google Scholar
|