Introduction
Among the numerous types of primary liver
malignancy, intrahepatic cholangiocarcinoma accounts for ~15% of
cases globally. In previous years, the incidence of intrahepatic
cholangiocarcinoma has risen, and this rise is anticipated to
increase further (1). However,
>50% of patients who undergo excision exhibit disease
recurrence, which most commonly occurs in the remnant liver
(2). The molecular mechanisms of the
progression of intrahepatic cholangiocarcinoma have yet to be
determined. Therefore, it is necessary to identify the molecular
mechanisms associated with the development of intrahepatic
cholangiocarcinoma.
High-throughput microarray and RNA sequencing
technology, which are able to identify changes in gene expression,
have been widely used in studying the etiology of numerous cancer
types, including hepatocellular carcinoma (3), breast cancer (4) and pancreatic cancer (5). Screening novel biomarkers for tumor
diagnosis and the pathways associated with tumor development,
recurrence and drug resistance, are also critical. Using
bioinformatics analysis, Xiao et al (6) revealed five potential biomarkers that
serve a key function in the progression of adrenocortical carcinoma
and are associated with a poor outcome. Similarly, through
integrated analysis, Huang et al (7) investigated five genes that were
indicated to contribute to multidrug resistance in patients with
Hodgkin's lymphoma. However, the molecular mechanisms associated
with the progression of intrahepatic cholangiocarcinoma are yet to
be determined.
In the present study, GSE107943 was used as a
discovery cohort to identify differentially expressed genes (DEGs)
and perform weighted gene co-expression network analysis (WGCNA),
while GSE26566 was used to identify tissue-specific genes.
Furthermore, GSE119336 was used as a validation cohort. Subsequent
to the removal of tissue-specific genes, real DEGs were used to
construct the WGCNA. Followed by Gene Ontology (GO) enrichment
analysis, Kyoto Encyclopaedia of Genes and Genomes (KEGG) analysis,
protein-protein network (PPI) interactions, receiver operating
characteristic curve (ROC) analysis and immumohistochemical
staining on intrahepatic cholangiocarcinoma tissues, the hub genes
were identified. These hub genes may be used for future
developments for the diagnosis and treatment of intrahepatic
cholangiocarcinoma.
Materials and methods
Data processing
Gene expression profile data GSE107943, GSE119336
and GSE26566 were obtained from the Gene Expression Omnibus
(http://www.ncbi.nlm.nih.gov/geo)
(8). A total of 27 adjacent tissues
and 30 intrahepatic cholangiocarcinoma tissues were included in the
GSE107943 profile. A total of 15 adjacent tissues and 15
intrahepatic cholangiocarcinoma tissues were included in the
GSE119336 profile. A total of 59 normal liver tissues and 6 normal
bile duct tissues were included in the GSE26556 profile. The
GSE107943 array data were acquired from Illumina NextSeq 500 (Homo
sapiens; GPL18573). The GSE119336 array data were acquired from
Illumina HiSeq 2000 (Homo sapiens; GPL11154). The GSE26566 array
data were acquired from Illumina humanRef-8 v2.0 expression
beadchip (GPL6104). The gene expression profile data were
normalized using R software (version: 3.5.2) prior to DEG
analysis.
DEG analysis
The R software was used to identify DEGs in 27
adjacent non-tumor tissues and 30 intrahepatic cholangiocarcinoma
tissues. The tissue-specific genes between 59 normal liver tissues
and 6 normal bile duct tissues were also analyzed. A |log2 fold
change (FC)|≥2 and an adjusted value of P<0.05 were considered
to indicate a statistically significant difference. The expressions
of all genes are presented in a volcano plot, while the expression
of DEGs in each sample is presented in a heatmap. Following the
removal of tissues-specific genes, real DEGs were obtained and
included in the WGCNA analysis.
Construction of WGCNA
The R package ‘WGCNA’ was used in the present study
to construct a co-expression network for the real DEGs identified
in the 30 intrahepatic cholangiocarcinoma samples. To construct the
WGCNA, the expression profile of DEGs and their clinical trait
information were imported into R software. A Pearson's correlation
analysis was subsequently performed to cluster samples and detect
outliers. The threshold for identifying outlier samples was 80
cut-height, and the results identified no outlier samples (Fig. S1). All gene pairs were then analyzed
using Pearson's correlation analysis, and a matrix of similarity
was constructed based on this analysis. Subsequently, to achieve a
scale-free co-expression network, the matrix of similarity was
constructed using a soft power of β=5. The adjacency matrix was
then translated into a topological overlap matrix (TOM).
Furthermore, the median linkage hierarchical clustering was
analyzed using the TOM-based dissimilarity measure with the
mini-size set as 30.
Identification of clinically
significant modules and verification of module hub genes
Following the construction of the WGCNA, different
module eigengenes and their corresponding clinical traits were
correlated. These traits included the recurrence, disease free
survival in months, tumor associated mortality, overall survival in
months, carcinoembryonic antigen expression, carbohydrate antigen
19-9 expression, tumor differentiation, tumor class (Child-Pugh
score) (9), American Joint Committee
on Cancer stage (10), tumor size,
vascular invasion and hepatitis B infection. Modules were
considered relevant to the clinical phenotype if their correlation
score with clinical traits was >0.3 and if they exhibited a
P<0.05 statistically significant difference. Modules associated
with multiple clinical traits (the number of clinical traits >3)
were set as key modules. These modules were then selected and
enrolled in further analysis. The gene significance (GS), which was
quantified using associations between individual genes and the
clinical trait of interest, and the module membership (MM), which
was the correlation between the module eigengenes and the gene
expression profiles, were calculated. MM was defined as >0.8 and
GS was defined as >0.2 for screening module core genes. If
modules were associated with numerous clinical traits, the
Bioinformatics & Evolutionary Genomics Venn Diagram tool
(http://bioinformatics.psb.ugent.be/webtools/Venn/) was
used to locate intersected module core genes.
GO and pathway enrichment
analysis
DAVID 6.8 (http://david-d.ncifcrf.gov/) (11,12) was
used in the present study to perform functional enriched analysis
and pathway enrichment analysis for the module core genes,
including in GO (biological process, molecular function and
cellular component) and KEGG pathway analysis. P<0.05 was
considered to indicate a statistically significant result. The
results of GO and KEGG analysis were visualized using R
software.
Construction of the PPI network
STRING (available online: http://string-db.org) (13) was used in the present study to
construct the PPI network, and the node and edge information was
exported into a text file. Cytoscape software (version: 6.1;
http://cytoscape.org/) was used to visualize the
PPI network and analyze the combined degree of module hub genes
(14). Finally, degree analysis was
performed and genes with the highest degree score were identified
as hub genes in the PPI network.
Diagnostic value of hub genes
The sensitivity and specificity of the hub genes
identified in the present study were evaluated through the ROC
curve analysis based on the gene expression profile GSE119336. The
ROC curve was created using SPSS 20.0 (IBM Corp., Armonk, NY, USA).
Genes exhibiting an area under the curve (AUC) value of >0.7
were considered to exhibit a good diagnostic value.
Tissues ethics
The procedures of the present study were ethically
approved by the Clinical Ethics Management Committee of Renmin
Hospital of Wuhan University. A total of 23 intrahepatic
cholangiocarcinoma tissues and corresponding adjacent tissues were
obtained between April 2019 and August 2019. None of the patients
enrolled in the present study received neoadjuvant chemotherapy,
radiotherapy or immunotherapy prior to surgery. Similarly, these
patients had no other systemic diseases, such as diabetes mellitus
and rheumatic disease. All patients (mean age, 54.3±9.2, range,
41–69 years; 10 males and 13 females) who provided tissue samples
provided written informed consent.
Immumohistochemical staining
All tissues were fixed in 4% paraformaldehyde for 30
min under room temperature and sectioned into 4-µm thick
paraffin-embedded sections. After heating at 60°C for 1 h, the
specimens were deparaffinized using xylene under room temperature
and rehydrated with graded ethanol (100, 80, 60 and 40%).
Subsequent to antigen retrieval with sodium citrate (100 mM), the
sections were blocked using 3% H2O2 for 20
min and 5% bovine serum albumin (Wuhan Servicebio Technology Co.,
Ltd., Wuhan, China) for 30 min under room temperature. The
specimens were then incubated with the primary antibodies,
including CCNB1 (1:400; cat. no. A16800; ABconal Biotech Co.,
Ltd.), CDC20 (1:400; cat. no. A15656; ABconal Biotech Co., Ltd.),
CDCA8 (1:400; cat. no. A12594; ABconal Biotech Co., Ltd.), CDK1
(1:400; cat. no. A2861; ABconal Biotech Co., Ltd.), CEP55 (1:500;
cat. no. 23891-1-AP; ProteinTech Group, Inc.), KIF2C (1:400; cat.
no. A5449; ABconal Biotech Co., Ltd.), TOP2A (1:200; cat. no.
A0726; ABconal Biotech Co., Ltd.) and TPX2 (1:200; cat. no. A4522;
ABconal Biotech Co., Ltd.) for 12 h at 4°C. Subsequently, the
sections were immunohistochemically stained with horseradish
peroxidase (HRP)-conjugated secondary goat anti-mouse and -rabbit
antibodies (cat. no. G1210-2-A-100; dilution: 1:200; Wuhan
Servicebio Technology Co., Ltd.) for 2 h at room temperature.
Subsequent to incubation with the Cell and Tissue Staining
HRP-3,3′-diaminobenzidine kit (Wuhan Servicebio Technology Co.,
Ltd.), an orthophotomicroscope was used to collect images. Finally,
the protein levels of the target genes were evaluated according to
the sum of the intensity and percentage scores. Stain intensity
score: 0 (no staining), 1 (+), 2 (++) and 3 (+++); the proportion
of positive cells score: 0 (0-1%), 1 (1-33%), 2 (34-66%) and 3
(67-100%). The total staining scores were defined as follows: 0–2
(low expression); 3–4 (moderate expression); 5–6 (high expression).
The intensity and percentage scores were determined using Image pro
plus software (version number: 6.0; Media Cybernetics, Inc.).
Results
Identification of real DEGs
A GSE107943 profile including 30 intrahepatic
cholangiocarcinoma tissues and 27 adjacent non-tumor tissues was
used to identify DEGs. DEGs were identified using R software and
the cutoff was set as |log2FC|≥2 and an adjusted P-value <0.05.
A total of 1,675 DEGs were identified between intrahepatic
cholangiocarcinoma and the adjacent non-tumor tissues, including
1,129 upregulated DEGs and 546 downregulated DEGs (Fig. 1A and B). The adjacent tissues
originated from liver tissues, therefore, genes known to be
differentially expressed between liver and bile duct tissues needed
to be removed. The gene expression profile of GSE26566 was used to
identify specific genes that differed between liver and bile duct
tissues. The results indicated that 153 genes were upregulated in
normal bile duct tissues and 3 genes were upregulated in normal
liver tissues (Fig. 1C). Subsequent
to removing tissues-specific genes which were contained in the DEGs
indicated by the analysis of GSE107943, the 1,643 real DEGs, which
included 1,098 real upregulated DEGs and 545 real downregulated
DEGs, were identified (Fig. 1D). All
real DEGs were subsequently included in the WGCNA analysis.
WCGNA construction
To construct the WGCNA, a soft power of β=5
(scale-free R2=0.88) was selected as the soft threshold
to ensure a scale-free network (Fig.
2A-D). A total of seven co-expressed modules were identified
(black, brown, blue, yellow, green, red and turquoise), while genes
with no cluster were distributed in the grey module (Fig. 2E). The seven co-expressed networks
were then used in further analysis.
Identification of significant modules
and module core genes
The results revealed five modules that were
significantly associated with clinical traits in patients with
intrahepatic cholangiocarcinoma, including black, brown, blue,
yellow and red modules. Genes in blue and black modules were
significantly associated with tumor class (R=0.41, P=0.02; R=0.71,
P=1×10−05; respectively), while genes in red modules
were significantly associated with tumor class (R=−0.74,
P=3×10−06) and HBV infection (R=0.41, P=0.02). Genes in
brown modules were significantly associated with tumor recurrence
(R=0.52, P=0.003), tumor-associated mortality (R=0.47, P=0.009),
serum carbohydrate antigen 19-9 (CA19-9) expression (R=0.45,
P=0.01) and tumor class (R=0.49, P=0.006). Furthermore, genes in
yellow modules were significantly associated with recurrence
(R=0.49, P=0.006), disease-free survival in months (R=−0.41,
P=0.02), tumor-associated mortality (R=0.52, P=0.003) and tumor
class (R=0.83, P=2e-08; Fig. 3). Due
to genes in brown and yellow modules being significantly associated
with the number of clinical traits (>3), these modules were
regarded as key modules and used in the subsequent analysis. The MM
in the brown module and the gene significance for tumor recurrence,
mortality, serum CA199 expression and tumor class exhibited a high
correlation (cor=0.5 for recurrence; cor=0.31 for tumor-associated
mortality; cor=0.45 for CA199 expression; cor=0.38 for tumor class;
Fig. 4A-D). Furthermore, a total of
38 genes were demonstrated to meet the threshold in the brown
module for the four clinical traits, and these 38 genes were
identified as core genes (Fig. 4E).
Additionally, the MM in the yellow module and the gene significance
for tumor recurrence, disease-free survival in months,
tumor-associated mortality and tumor class also exhibited a high
correlation (cor=0.59 for recurrence; cor=0.29 for disease-free
survival in months; cor=0.55 for tumor-associated mortality;
cor=0.85 for tumor class; Fig.
4F-I). Furthermore, a total of 25 genes in the yellow module
met the threshold for the four clinical traits, and these 25 genes
were identified as core genes (Fig.
4J). Therefore, a total of 63 core genes in the brown and
yellow modules were used in further analysis.
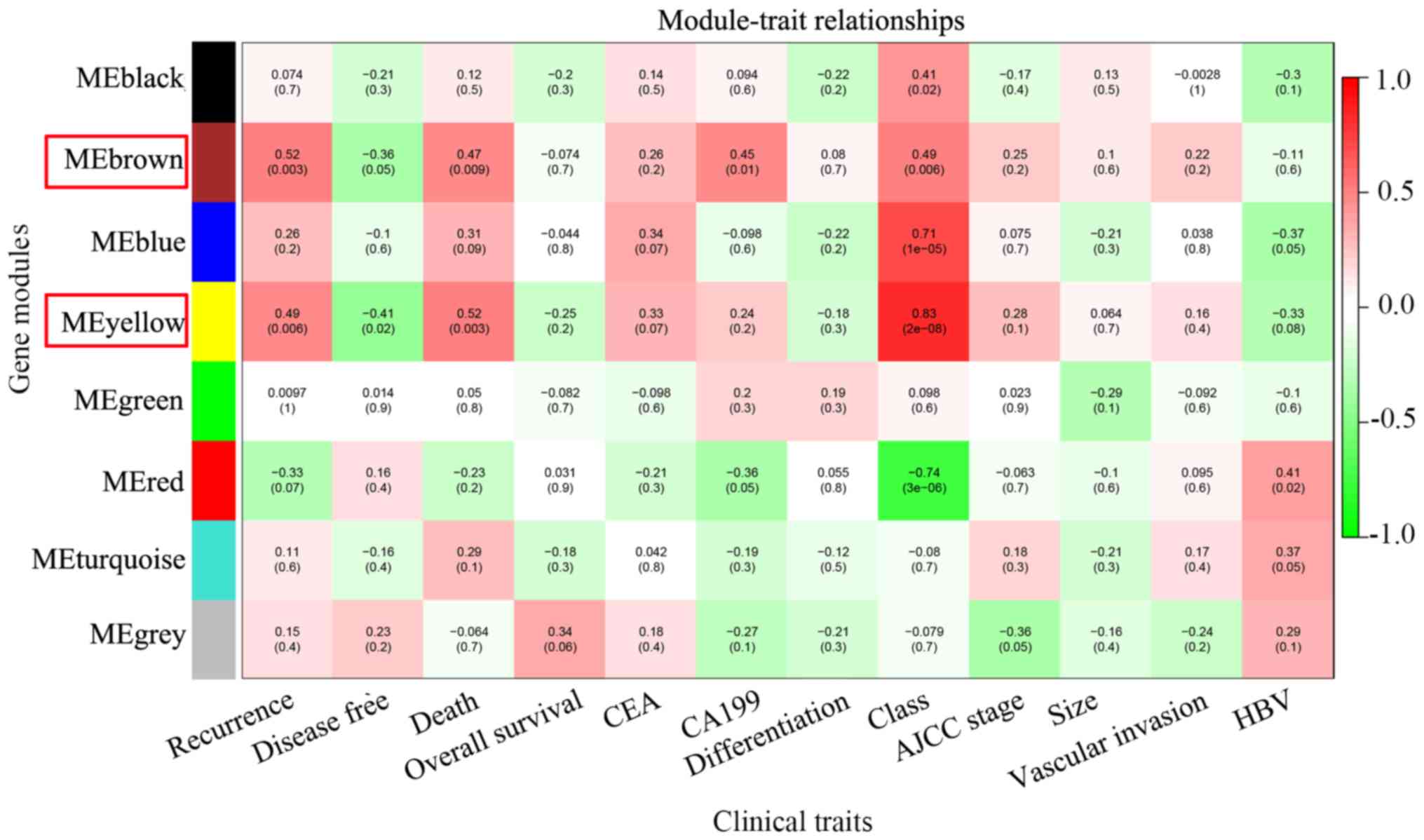 | Figure 3.Identification of key modules.
Heatmap of the correlation between module eigengenes and clinical
traits of intrahepatic cholangiocarcinoma. A total of 12 clinical
traits were included: Recurrence, disease-free survival months,
tumor-associated mortality, overall survival months, CEA
expression, CA199 expression, tumor differentiation, tumor class
(Child-Pugh score), AJCC stage, tumor size, vascular invasion and
HBV infection. Brown and yellow modules were associated with
multiple clinical traits (the number of clinical traits >3), and
they were regarded as key modules and highlighted with a red frame.
HBV, hepatitis B virus; AJCC, American Joint Committee on Cancer;
CEA, carcinoembryonic antigen; CA199, carbohydrate antigen
19-9. |
Functional and pathway analysis of
core genes
To understand the enriched function and the KEGG
pathway in the 63 core genes, the DAVID online tool was used. The
results demonstrated that the top five biological processes that
were associated with these 63 core genes were mitotic spindle
assembly, mitotic metaphase plate congression, positive regulation
of mitosis, mitotic sister chromatid segregation and the spindle
checkpoint (Fig. 5A). Only three
molecular function terms were indicated, and these were genes that
were enriched in ATP binding, chromatin binding and DNA binding
(Fig. 5B). Furthermore, the top five
cellular components which were associated with these genes were the
nucleus, cytoplasm, nucleoplasm, midbody and cell membrane
(Fig. 5C). Furthermore, the results
revealed that the core genes were enriched in five pathways: The
cell cycle pathway, oocyte meiosis pathway, p53 signaling pathway,
insulin secretion pathway and progesterone-mediated oocyte
maturation pathway (Fig. 5D).
Construction of PPI network and
verification of hub genes
To identify hub genes in intrahepatic
cholangiocarcinoma, a total of 63 module core genes were used to
construct the PPI network. A total of 56 nodes and 512 edges were
indicated in the network (Fig. 6A).
Furthermore, the results revealed that CCNB1, CDC20, CDCA8, CDK1,
CEP55, KIF2C, TOP2A and TPX2 exhibited the highest degree score in
the PPI network and these genes were subsequently identified as hub
genes (Fig. 6B).
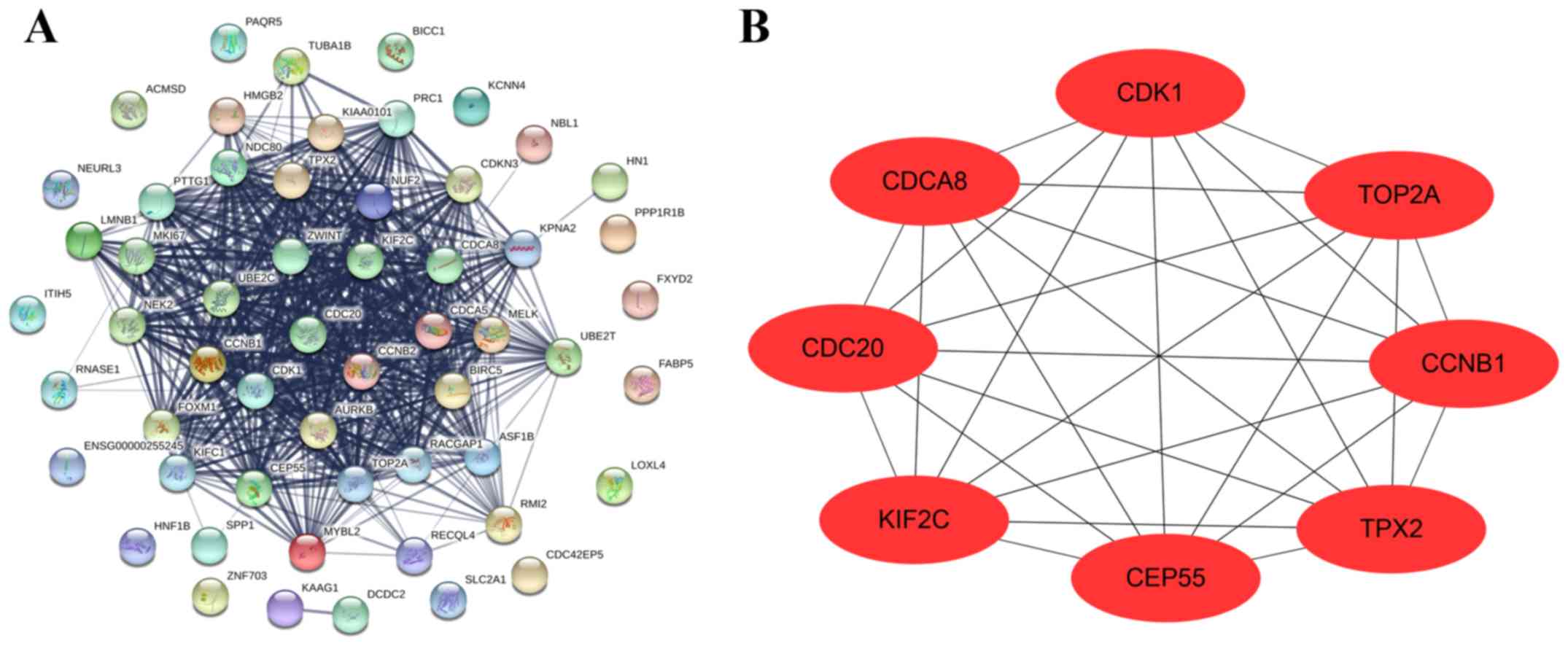 | Figure 6.Construction of PPI and selection of
hub genes. (A) Core genes were imported into STRING to construct a
PPI network. Nodes indicate core genes, while edges indicate the
correlation between core genes. (B) Cytoscape was used to analyze
the PPI network, while hub genes were indicated by degree score and
used to construct the PPI sub-network. PPI, protein-protein
interactions; CCNB1, cyclin B1; CDC20, cell division cycle 20;
CDCA8, cell division cycle associated 8; CDK1, cyclin dependent
kinase 1; CEP55, centrosomal protein 55; KIF2C, kinesin family
member 2C; TOP2A, DNA topoisomerase IIα; TPX2, TPX2 microtubule
nucleation factor. |
Diagnosis value of hub genes
In order to detect the diagnostic value of these
eight hub genes, the gene expressed profile GSE119336, including 15
adjacent tissues and 15 intrahepatic cholangiocarcinoma tissues,
was used and a ROC analysis was performed (Fig. 7). The results revealed that CCNB1,
CDC20, CDCA8, CDK1, CEP55, KIF2C, TOP2A and TPX2 all exhibited good
diagnostic values for intrahepatic cholangiocarcinoma, using a
cut-off AUC value of 0.7. The results of the AUC were as follows:
CCNB1 (AUC=0.969), CDC20 (AUC=0.964), CDCA8 (AUC=0.960), CDK1
(AUC=0.929), CEP55 (AUC=0.964), KIF2C (AUC=0.973), TOP2A
(AUC=0.942) and TPX2 (AUC=0.911).
 | Figure 7.Verification of the diagnostic value
of hub genes. Gene expression profile GSE119336 was obtained and
receiver operating characteristic curve analysis was performed to
identify the diagnostic value of hub genes CCNB1, CDC20, CDCA8,
CDK1, CEP55, KIF2C, TOP2A and TPX2. CCNB1, cyclin B1; CDC20, cell
division cycle 20; CDCA8, cell division cycle associated 8; CDK1,
cyclin dependent kinase 1; CEP55, centrosomal protein 55; KIF2C,
kinesin family member 2C; TOP2A, DNA topoisomerase IIα; TPX2, TPX2
microtubule nucleation factor. |
Verification of the expression of hub
genes
In order to verify the results obtained from
prediction, immumohistochemical staining was performed in 23
intrahepatic cholangiocarcinoma tissues and corresponding adjacent
tissues. The present study observed the expression of hub genes in
intrahepatic cholangiocarcinoma tissues, adjacent normal liver
tissues and intrahepatic bile duct tissues, and revealed that the
expression of all hub genes was increased in most intrahepatic
cholangiocarcinoma tissues compared with adjacent normal liver
tissues and intrahepatic bile duct tissues (Table I; Fig.
8).
 | Figure 8.Representative figure of
immumohistochemical staining for CCNB1, CDC20, CDCA8, CDK1, CEP55,
KIF2C, TOP2A and TPX2 in intrahepatic cholangiocarcinoma tissues,
adjacent liver tissues and intrahepatic bile duct tissues. CCNB1,
cyclin B1; CDC20, cell division cycle 20; CDCA8, cell division
cycle associated 8; CDK1, cyclin dependent kinase 1; CEP55,
centrosomal protein 55; KIF2C, kinesin family member 2C; TOP2A, DNA
topoisomerase IIα; TPX2, TPX2 microtubule nucleation factor. |
 | Table I.Analysis of the expression of
targeted genes in intrahepatic cholangiocarcinoma tissues, adjacent
liver tissues and intrahepatic bile duct tissues. |
Table I.
Analysis of the expression of
targeted genes in intrahepatic cholangiocarcinoma tissues, adjacent
liver tissues and intrahepatic bile duct tissues.
| Targets | Tissues | Low expression
(0–2) | Medium expression
(3–4) | High expression
(5–6) |
|---|
| CCNB1 | Intrahepatic
cholangiocarcinoma tissues | 2 | 5 | 16 |
|
| Adjacent liver
tissues | 5 | 16 | 2 |
|
| Intrahepatic bile
duct tissues | 20 | 2 | 1 |
| CDC20 | Intrahepatic
cholangiocarcinoma tissues | 1 | 4 | 18 |
|
| Adjacent liver
tissues | 5 | 11 | 7 |
|
| Intrahepatic bile
duct tissues | 17 | 4 | 2 |
| CDCA8 | Intrahepatic
cholangiocarcinoma tissues | 3 | 6 | 14 |
|
| Adjacent liver
tissues | 9 | 11 | 3 |
|
| Intrahepatic bile
duct tissues | 16 | 7 | 0 |
| CDK1 | Intrahepatic
cholangiocarcinoma tissues | 3 | 5 | 15 |
|
| Adjacent liver
tissues | 4 | 16 | 3 |
|
| Intrahepatic bile
duct tissues | 18 | 4 | 1 |
| CEP55 | Intrahepatic
cholangiocarcinoma tissues | 2 | 3 | 18 |
|
| Adjacent liver
tissues | 7 | 15 | 1 |
|
| Intrahepatic bile
duct tissues | 13 | 7 | 3 |
| KIF2C | Intrahepatic
cholangiocarcinoma tissues | 1 | 1 | 21 |
|
| Adjacent liver
tissues | 8 | 12 | 3 |
|
| Intrahepatic bile
duct tissues | 16 | 5 | 2 |
| TOP2A | Intrahepatic
cholangiocarcinoma tissues | 2 | 4 | 17 |
|
| Adjacent liver
tissues | 6 | 13 | 4 |
|
| Intrahepatic bile
duct tissues | 16 | 5 | 2 |
| TPX2 | Intrahepatic
cholangiocarcinoma tissues | 2 | 4 | 17 |
|
| Adjacent liver
tissues | 3 | 16 | 4 |
|
| Intrahepatic bile
duct tissues | 17 | 3 | 3 |
Discussion
Intrahepatic cholangiocarcinoma is the second most
common primary liver malignant tumor globally, and cases of this
disease are increasing annually. Unfortunately, the majority of
patients are diagnosed at a nonsurgical stage and only ~1 in 5
cases are surgically resectable (15). However, a majority of patients are
still at risk of tumor recurrence and drug resistance (16). Therefore, there is an urgent
requirement to investigate the mechanisms of intrahepatic
cholangiocarcinoma development, and the knowledge gained maybe
useful in the development of novel clinical treatment
strategies.
In the present study, a total of 1,643 real DEGs
were identified and used to construct a WGCNA. Subsequent to
analyzing the association between gene modules and clinical traits,
brown and yellow modules were identified as key modules. A total of
63 core genes in the brown and yellow gene module were identified.
The 63 core genes were highly enriched in mitotic spindle assembly
(GO term), in ATP binding (GO term) and in the nucleus (GO term),
and the pathways these genes were enriched in were the cell cycle
pathway, oocyte meiosis pathway, p53 signaling pathway, insulin
secretion pathway and progesterone-mediated oocyte maturation
pathway. Following the construction of the PPI network and the
results of the ROC curve, a total of eight genes, including CCNB1,
CDC20, CDCA8, CDK1, CEP55, KIF2C, TOP2A and TPX2 were identified as
hub genes for intrahepatic cholangiocarcinoma. Furthermore, the
protein expression levels of all these genes were increased in most
intrahepatic cholangiocarcinoma tissues.
CCNB1 and CDK1 were indicated to be two genes that
are able to directly regulate the cell cycle. CDK1 and CCNB1
proteins are located in the matrix of mitochondria. Here, they
increase mitochondrial respiration, enhance oxygen consumption and
promote ATP generation, which provides cells with enough bioenergy
to complete the G2/M transition, and shortens the overall time to
complete the cell cycle (17).
Additionally, CDK1 and CCNB1 exhibit the potential to phosphorylate
a number of pro-apoptotic and anti-apoptotic proteins, and are able
to induce the inhibition of apoptosis in tumor cells (18). Furthermore, CDK1 and CCNB1 crosstalk
with the p53 pathway, and activated CDK1 and CCNB1 are able to
inhibit the effect of p53, and may result in cell proliferation and
escape from apoptosis (19,20). CDC20 is a downstream factor of the
spindle assembly checkpoint and is a key co-factor of the
anaphase-promoting complex or cyclosome E3 ubiquitin ligase
(21). The potential of CDC20 as a
therapeutic target for human cancer treatment has been reported in
previous studies (22). Wu et
al (23) investigated the
effects of CDC20 overexpression in docetaxel- and
castration-resistant prostate cancer cell lines, and demonstrated
that CDC20 mediated resistance in a Bim-dependent manner. Paul
et al (24) indicated that
CDC20 regulated the proteasome-mediated degradation of the tumor
suppressor BTG3 associated nuclear protein, and inhibited apoptosis
in breast cancer cells. Furthermore, Zhang et al (25) demonstrated that the inhibition of
CDC20 using curcumin is a useful method in the treatment of
patients with pancreatic cancer. CDCA8 is a component of the
chromosomal passenger complex 52, which serves a vital function in
mitosis and cell division (26).
Previous studies have demonstrated that high CDCA8 expression is
positively associated with a poor prognosis in a variety of cancer
types, including in cutaneous melanoma (27) and bladder cancer (28). In breast cancer, the expression of
CDCA8 has been demonstrated to be associated with disease-free
survival rate (29). Additionally,
CDCA8 exhibits the potential to promote breast cancer cell
proliferation following stimulation with estrogen (30). However, the exact function of CDCA8
in intrahepatic cholangiocarcinoma has, to the best of our
knowledge, not yet been determined.
CEP55 is located on chromosome 10q23.33, which is
associated with cytokinesis. A variety of studies have demonstrated
that CEP55 serves a key function in numerous biological processes
within cancer cells, including in proliferation, migration and
differentiation (31). The knockdown
of CEP55 expression has been indicated to inhibit the proliferation
of breast cancer cells (32). In
lung adenocarcinoma, the high expression of CEP55 is negatively
associated with patient overall survival rate (33). KIF2C is the most important member of
the motor proteins family and serves a crucial function in spindle
assembly and ensuring proper chromosome segregation during cell
mitosis (34). The effect of KIF2C
on the progression of cancer has been previously reported, and
KIF2C is an identified biomarker for the prognosis of human glioma
(35). KIF2C has also been
demonstrated to be overexpressed in primary breast cancer tissues
and cell lines, and this may be suppressed by the ectopic
introduction of p53 (36,37). TOP2A encodes for a DNA topoisomerase,
which exhibits the capacity to regulate the topologic states of DNA
during transcription (38). TOP2A is
overexpressed in nasopharyngeal carcinoma and is associated with a
poor outcome (39). Additionally,
high TOP2A expression in breast cancer is able to predict poor
prognosis (40). Furthermore, TOP2A
has been indicated to be highly expressed in 33% cases of patients
with intrahepatic cholangiocarcinoma (41). TPX2 serves a key function in spindle
formation and microtubule nucleation (42). High TPX2 expression in hepatocellular
carcinoma has been associated with a poor prognosis and has been
indicated to contribute to cell proliferation and metastasis
(43). The inhibition of TPX2 may
activate the p53 pathway and promote apoptosis in breast cancer
(44).
Through WGCNA, ROC analysis and immumohistochemical
staining, the present study identified eight hub genes (CCNB1,
CDC20, CDCA8, CDK1, CEP55, KIF2C, TOP2A and TPX2), which may
contribute to the development of intrahepatic cholangiocarcinoma,
and may also be novel biomarkers for the diagnosis of this disease.
However, further research into the hub genes screened in the
present study is urgently required to determine the underlying
mechanisms associated with their function in intrahepatic
cholangiocarcinoma.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Program For
Tackling Key Problems in Science and Technology (grant. no.
2007AA301B35-2), the Natural Science Foundation of Hubei Province
of China (grant. no. 2010CDB06807) and the Important Project of
Wuhan Administration of Science and Technology (grant. no.
2.008E+11).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZY, ZZ and DW were responsible for the data
collection and analysis. ZC designed the study. SL and YS collected
the clinical samples. ZY, ZZ, DW, SL and YS performed the
experiments. ZC wrote the manuscript. All authors read and approved
the final version of the article.
Ethics approval and consent to
participate
All patients who had provided samples provided
informed consent. The present study was approved by the Ethics
Committee of the Renmin Hospital of Wuhan University (approval nr.
WDRY2019-K075) and was performed in accordance with the Declaration
of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chun YS and Javle M: Systemic and adjuvant
therapies for intrahepatic cholangiocarcinoma. Cancer Control.
24:10732748177292412017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nakano M, Ariizumi SI and Yamamoto M:
Intrahepatic cholangiocarcinoma. Semin Diagn Pathol. 34:160–166.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu Q, Sun Y, Zhou Q, He Q and Qian H:
Identification of key genes and pathways by bioinformatics analysis
with TCGA RNA sequencing data in hepatocellular carcinoma. Mol Clin
Oncol. 9:597–606. 2018.PubMed/NCBI
|
|
4
|
Tang D, Zhao X, Zhang L, Wang Z and Wang
C: Identification of hub genes to regulate breast cancer metastasis
to brain by bioinformatics analyses. J Cell Biochem. 120:9522–9531.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma X, Tao R, Li L, Chen H, Liu Z, Bai J,
Shuai X, Wu C and Tao K: Identification of a 5microRNA signature
and hub miRNAmRNA interactions associated with pancreatic cancer.
Oncol Rep. 41:292–300. 2019.PubMed/NCBI
|
|
6
|
Xiao H, Xu D, Chen P, Zeng G, Wang X and
Zhang X: Identification of five genes as a potential biomarker for
predicting progress and prognosis in adrenocortical carcinoma. J
Cancer. 9:4484–4495. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang Y, Huang Y, Zhang L, Chang A, Zhao
P, Chai X and Wang J: Identification of crucial genes and
prediction of small molecules for multidrug resistance of Hodgkin's
lymphomas. Cancer Biomark. 23:495–503. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Edgar R, Domrachev M and Lash AE: Gene
Expression Omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang K, Yu J, Yu X, Han Z, Cheng Z, Liu F
and Liang P: Clinical and survival outcomes of percutaneous
microwave ablation for intrahepatic cholangiocarcinoma. Int J
Hyperthermia. 34:292–297. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meng ZW, Pan W, Hong HJ, Chen JZ and Chen
YL: Modified staging classification for intrahepatic
cholangiocarcinoma based on the sixth and seventh editions of the
AJCC/UICC TNM staging systems. Medicine (Baltimore). 96:e78912017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic acids Res. 47:D607–D613. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu L, Tsilimigras DI, Farooq A, Hyer JM,
Merath K, Paredes AZ, Mehta R, Sahara K, Shen F and Pawlik TM:
Potential survival benefit of radiofrequency ablation for small
solitary intrahepatic cholangiocarcinoma in nonsurgically managed
patients: A population-based analysis. J Surg Oncol. 120:1358–1364.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laurent S, Verhelst X, Geerts A, Geboes K,
De Man M, Troisi R, Vanlander A, Rogiers X, Berrevoet F and Van
Vlierberghe H: Update on liver transplantation for
cholangiocarcinoma: A review of the recent literature. Acta
Gastroenterol Belg. 82:417–420. 2019.PubMed/NCBI
|
|
17
|
Wang Z, Fan M, Candas D, Zhang TQ, Qin L,
Eldridge A, Wachsmann-Hogiu S, Ahmed KM, Chromy BA, Nantajit D, et
al: Cyclin B1/Cdk1 coordinates mitochondrial respiration for
cell-cycle G2/M progression. Dev Cell. 29:217–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie B, Wang S, Jiang N and Li JJ: Cyclin
B1/CDK1-regulated mitochondrial bioenergetics in cell cycle
progression and tumor resistance. Cancer Lett. 443:56–66. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huskey NE, Guo T, Evason KJ, Momcilovic O,
Pardo D, Creasman KJ, Judson RL, Blelloch R, Oakes SA, Hebrok M and
Goga A: CDK1 inhibition targets the p53-NOXA-MCL1 axis, selectively
kills embryonic stem cells, and prevents teratoma formation. Stem
Cell Rep. 4:374–389. 2015. View Article : Google Scholar
|
|
20
|
Lu M, Breyssens H, Salter V, Zhong S, Hu
Y, Baer C, Ratnayaka I, Sullivan A, Brown NR, Endicott J, et al:
Restoring p53 function in human melanoma cells by inhibiting MDM2
and Cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell.
30:822–823. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kapanidou M, Curtis NL and Bolanos-Garcia
VM: Cdc20: At the crossroads between chromosome segregation and
mitotic exit. Trends Biochem Sci. 42:193–205. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang L, Zhang J, Wan L, Zhou X, Wang Z and
Wei W: Targeting Cdc20 as a novel cancer therapeutic strategy.
Pharmacol Ther. 151:141–151. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu F, Lin Y, Cui P, Li H, Zhang L, Sun Z,
Huang S, Li S, Huang S, Zhao Q and Liu Q: Cdc20/p55 mediates the
resistance to docetaxel in castration-resistant prostate cancer in
a Bim-dependent manner. Cancer Chemother Pharmacol. 81:999–1006.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Paul D, Ghorai S, Dinesh US, Shetty P,
Chattopadhyay S and Santra MK: Cdc20 directs proteasome-mediated
degradation of the tumor suppressor SMAR1 in higher grades of
cancer through the anaphase promoting complex. Cell Death Dis.
8:e28822017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y, Xue YB, Li H, Qiu D, Wang ZW and
Tan SS: Inhibition of cell survival by curcumin is associated with
downregulation of cell division cycle 20 (Cdc20) in pancreatic
cancer cells. Nutrients. 9:E1092017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hindriksen S, Meppelink A and Lens SM:
Functionality of the chromosomal passenger complex in cancer.
Biochem Soc Trans. 43:23–32. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ci C, Tang B, Lyu D, Liu W, Qiang D, Ji X,
Qiu X, Chen L and Ding W: Overexpression of CDCA8 promotes the
malignant progression of cutaneous melanoma and leads to poor
prognosis. Int J Mol Med. 43:404–412. 2019.PubMed/NCBI
|
|
28
|
Bi Y, Chen S, Jiang J, Yao J, Wang G, Zhou
Q and Li S: CDCA8 expression and its clinical relevance in patients
with bladder cancer. Medicine (Baltimore). 97:e118992018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Phan NN, Wang CY, Li KL, Chen CF, Chiao
CC, Yu HG, Huang PL and Lin YC: Distinct expression of CDCA3,
CDCA5, and CDCA8 leads to shorter relapse free survival in breast
cancer patient. Oncotarget. 9:6977–6992. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bu Y, Shi L, Yu D, Liang Z and Li W: CDCA8
is a key mediator of estrogen-stimulated cell proliferation in
breast cancer cells. Gene. 703:1–6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jeffery J, Sinha D, Srihari S, Kalimutho M
and Khanna KK: Beyond cytokinesis: The emerging roles of CEP55 in
tumorigenesis. Oncogene. 35:683–690. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Y, Jin T, Dai X and Xu J:
Lentivirus-mediated knockdown of CEP55 suppresses cell
proliferation of breast cancer cells. Biosci Trends. 10:67–73.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu S, Wu D, Pan Y, Liu H, Shao Z and Wang
M: Correlation between EZH2 and CEP55 and lung adenocarcinoma
prognosis. Pathol Res Pract. 215:292–301. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ritter A, Kreis NN, Louwen F, Wordeman L
and Yuan J: Molecular insight into the regulation and function of
MCAK. Crit Rev Biochem Mol Biol. 51:228–245. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bie L, Zhao G, Wang YP and Zhang B:
Kinesin family member 2C (KIF2C/MCAK) is a novel marker for
prognosis in human gliomas. Clin Neurol Neurosurg. 114:356–360.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dai X, Hua T and Hong T: Integrated
diagnostic network construction reveals a 4-gene panel and 5 cancer
hallmarks driving breast cancer heterogeneity. Sci Rep. 7:68272017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shimo A, Tanikawa C, Nishidate T, Lin ML,
Matsuda K, Park JH, Ueki T, Ohta T, Hirata K, Fukuda M, et al:
Involvement of kinesin family member 2C/mitotic
centromere-associated kinesin overexpression in mammary
carcinogenesis. Cancer Sci. 99:62–70. 2008.PubMed/NCBI
|
|
38
|
Nelson WG, Haffner MC and Yegnasubramanian
S: The structure of the nucleus in normal and neoplastic prostate
cells: Untangling the role of type 2 DNA topoisomerases. Am J Clin
Exp Urol. 6:107–113. 2018.PubMed/NCBI
|
|
39
|
Lan J, Huang HY, Lee SW, Chen TJ, Tai HC,
Hsu HP, Chang KY and Li CF: TOP2A overexpression as a poor
prognostic factor in patients with nasopharyngeal carcinoma. Tumour
Biol. 35:179–187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
An X, Xu F, Luo R, Zheng Q, Lu J, Yang Y,
Qin T, Yuan Z, Shi Y, Jiang W and Wang S: The prognostic
significance of topoisomerase II alpha protein in early stage
luminal breast cancer. BMC Cancer. 18:3312018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Potkonjak M, Miura JT, Turaga KK, Johnston
FM, Tsai S, Christians KK and Gamblin TC: Intrahepatic
cholangiocarcinoma and gallbladder cancer: Distinguishing molecular
profiles to guide potential therapy. HPB (Oxford). 17:1119–1123.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Neumayer G, Belzil C, Gruss OJ and Nguyen
MD: TPX2: Of spindle assembly, DNA damage response, and cancer.
Cell Mol Life Sci. 71:3027–3047. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hsu CW, Chen YC, Su HH, Huang GJ, Shu CW,
Wu TT and Pan HW: Targeting TPX2 suppresses the tumorigenesis of
hepatocellular carcinoma cells resulting in arrested mitotic phase
progression and increased genomic instability. J Cancer.
8:1378–1394. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen M, Zhang H, Zhang G, Zhong A, Ma Q,
Kai J, Tong Y, Xie S, Wang Y, Zheng H, et al: Targeting TPX2
suppresses proliferation and promotes apoptosis via repression of
the PI3k/AKT/P21 signaling pathway and activation of p53 pathway in
breast cancer. Biochem Biophys Res Commun. 507:74–82. 2018.
View Article : Google Scholar : PubMed/NCBI
|


















