Introduction
Ovarian cancer (OC) is the most lethal gynecological
malignancy and one of the leading causes of cancer-related deaths
in women (1–3). The high mortality rate is due to the
late diagnosis or advanced stage of at the time of diagnosis, with
the majority of patients possessing stage III–IV cancer (2). The incidence of OC is still increasing.
Currently, the most commonly used therapeutic strategies are
surgery and chemotherapy, with radiotherapy occasionally being used
(4–6). However, the 5-year survival rate is
only ~50% due to the development of recurrent disease that is often
resistant to chemotherapy (7). The
treatment of recurrent OC is limited and recurrence of OC is still
considered as incurable. Thus, the development of new and efficient
therapeutic strategies is urgently needed.
MicroRNA (miRNA/miR) is a small non-coding RNA, 22
nt in length, that is able to regulate gene expression by binding
to the complementary sequence at the 3′-untranslated region
(3′-UTR) of its target mRNAs (8–12). Each
miRNA can have multiple targets, inducing either upregulation or
downregulation of the expression of each target (13). It has been reported that the
dysregulation of miRNAs is related to a variety of human diseases,
including cancer (14–16). miRNAs act as either tumor suppressors
or oncogenes in different types of cancer and their role is
dependent on their expression pattern and function (16,17). In
OC, a number of miRNAs have been identified to have altered
expression leading to tumorigenesis. Among these miRNAs, miR-16,
miR-20a, miR-27a, miR-26, miR-182, miR-146, miR-221 and miR-508
have been reported to be upregulated in OC, while miR-145,
miR-125b, miR-377, miR-210, miR-493 and miR-106b have been reported
to be downregulated in OC (18–20).
Investigating the role of these miRNAs in the development and
progression of OC may provide new insights for the detection,
diagnosis and treatment of OC.
miR-32 (miR-32-5p) has been reported to be
overexpressed in several types of cancer, including breast,
prostate, endometrial, colorectal and hepatocellular cancer, and
has been shown to promote cancer cell proliferation and development
(21–26). By contrast, miR-32 acts as a tumor
suppressor in human oral squamous cell carcinoma (27). However, its expression and biological
role in OC is still largely unknown. Therefore, the present study
aimed to investigate the expression levels and functional roles of
miR-32 in OC tissues and cell lines. The findings of the present
study might highlight the potential of miR-32 as a therapeutic
target for treatment of OC in the future.
Patients and methods
Patients and clinical specimens
A total of 38 paired malignant OC tissues and
adjacent normal ovarian tissues were collected in Tianjin Medical
University General Hospital (Tianjin, China) from female patients
aged of 24–73 who underwent surgical resection between December
2015 and December 2016. Written informed consent was obtained from
each patient, and the study was approved by The Ethics Committee of
the Tianjin Medical University. All patient information is listed
in Table I. The collected tissues
were immediately frozen in liquid nitrogen and stored at −80°C
prior to RNA isolation.
 | Table I.Associations between miR-32 expression
and clinicopathological characteristics of 38 ovarian cancer
patients. |
Table I.
Associations between miR-32 expression
and clinicopathological characteristics of 38 ovarian cancer
patients.
|
|
| miR-32 expression,
n |
|
|---|
|
|
|
|
|
|---|
| Characteristics | Patients, n | High (n=23) | Low (n=15) | P-value |
|---|
| Age, years |
|
|
| 0.646 |
|
<50 | 16 | 9 | 7 |
|
|
≥50 | 22 | 14 | 8 |
|
| Clinical stage |
|
|
| 0.021a |
|
I–II | 8 | 2 | 6 |
|
|
III–IV | 30 | 21 | 9 |
|
| Pathological
grade |
|
|
| 0.225 |
|
1-2 | 11 | 5 | 6 |
|
| 3 | 27 | 18 | 9 |
|
| Histological
type |
|
|
| 0.475 |
|
Serous | 28 | 16 | 12 |
|
|
Non-serous | 10 | 7 | 3 |
|
| Residual tumors
after surgery, cm |
|
|
| 0.254 |
|
<1 | 21 | 11 | 10 |
|
| ≥1 | 17 | 12 | 5 |
|
Cell culture and transfection
Three human OC cell lines: OVCAR3 (cat. no.
HTB-161), SKOV3 (cat. no. HTB-77) and ES-2 (cat. no. CRL-1978) were
purchased from the American Type Culture Collection and one human
ovarian surface epithelial (HOSE) cell line (IOSE80; cat. no.
CVCL_5546) was obtained from the Canadian Ovarian Tissue Bank
(University of British Columbia). Cells were cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS) and 100 U/ml
penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.), at
37°C in a humidified chamber with 5% CO2 atmosphere.
Around 5–6×105 cells were seeded into 6-well plates at
24 h prior to transfection. 50 nM of miR-32 inhibitor, inhibitor
negative control (NC), miR-32 mimic, mimic NC, SMG1 small hairpin
(sh)RNAoligo and SMG1 NC were purchased from Shanghai GenePharma
Co., Ltd., and transfected into ES-2 cells using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) following the manufacturer's instructions. A
total of 50 nM RNA was used for each transfection. At 48 h after
transfection, the functional experiments were performed. The
inhibitor NC and the miR-32 mimic NC were synthesized with
non-specific sequences of the same length as the miR-32 inhibitor
and mimic, which could eliminate non-sequence-specific effects in
the experiments. The sequences of genes mentioned above were listed
as following. miR-32 inhibitor, 5′-UGCAACUUAGUAAUGUGCAAUA-3′;
inhibitor NC, 5′-CAGUACUUUUGUGUAGUACAA-3′; miR-32 mimic, sense:
5′-UAUUGCACAUUACUAAGUUGCA-3′, antisense:
5′-CAACUUAGUAAUGUGCAAUAUU-3′; mimic NC,
5′-UUCUCCGAACGUGUCACUGUU-3′; SMG1 (nonsense mediated mRNA decay
associated PI3K related kinase) shRNA,
5′-GCCAUGACUAACACUGAAAdTdT-3′.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the indicated cell
lines, including OVCAR3, SKOV3, ES-2 and IOSE80, and the patient
samples using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
cDNA was reverse transcribed from RNA using the PrimeScript RT
reagent kit (Promega Corp.) (50-55°C for 10 min, 80°C for 10 min).
U6 snRNA was used as normalization control gene for the detection
of miR-32. RT-qPCR analyses for SMG1 and the normalization control
gene GAPDH were performed using SYBR Premix Ex Taq (Takara
Biotechnology Co., Ltd.). The condition for qPCR was: 95°C 30 sec;
95°C 5 sec, 60°C 30 sec for 40 cycles. The relative expression of
each gene was calculated and normalized to U6 snRNA or GAPDH using
the 2−ΔΔCq method (28).
The correlation between miR-32 and SMG1 mRNA expression in OC
tissues was analyzed by the Spearman's correlation analysis. The
following primers were used for RT-qPCR: miR-32, Forward:
5′-GCGGCGTATTGCACATTACT-3′, reverse: 5′-TCGTATCCAGTGCAGGGTC-3′;
SMG1, forward: 5′-GTGCATTAGCCACCAAAGAC-3′ and reverse:
5′-CTCAGAGAAGCACAGAGAAG-3′.
Cell proliferation assay
Transfected cells were placed into 96-well plates at
a density of 2×103 cells/well and were cultured for 24,
48, 72 and 96 h. Next, 10 µl Cell Counting Kit-8 (CCK-8) reagent
(Beyotime Institute of Biotechnology) was added into each well. The
96-well plates were placed in a 5% CO2 incubator at 37°C
and the cells were incubated for 2 h. The absorbance was measured
at 450 nm using a microplate reader.
Cell migration and invasion
assays
Following transfection for 24 h, the cells were
cultured in serum-free medium for another 12 h. The cells were then
collected and their density was adjusted to 4–5×105/ml.
A Transwell chamber with 8-µm pores (Corning, Inc.) was used for
the migration assay. Complete DMEM (500 µl) (Hyclone; GE Healthcare
Life Sciences), containing 10% FBS, was added in the lower layer,
and 200 µl of the cell suspension in serum-free media was added in
the upper chamber. After a 10-h incubation, the cells on the lower
surface of the chamber were fixed with glacial acetic acid for
15–30 min at room temperature and stained with 0.2% crystal violet
for 30 min at room temperature. A total of 10 fields from each
chamber were selected randomly for counting. The cells
(4-5×105/ml) were plated into the upper layer of the
chamber covered with Matrigel (BD Biosciences), and the same
culture method was used to perform cell invasion assays. After
staining with 0.2% crystal violet, at least 10 fields from each
chamber were selected and the invasive cells were counted and
quantified under an inverted light microscope (Olympus) with ×20
magnification.
Bioinformatics analysis
TargetScan version 6.2 (targetscan.org/vert_72/) was used to predict the
potential targets of miR-32. Several potential targets, including
ANP32E, ARRDC3, FXR1, SMG1, EVI5, GRAMD1B, KIF1B, BMP7 and SPHK2,
were selected to analyze the target-miR-32 association and the role
of miR-32 in the regulation of their expression. The primers of
ANP32E, ARRDC3, FXR1, SMG1, EVI5, GRAMD1B, KIF1B, BMP7 and SPHK2
are listed in Table SI.
Dual-luciferase reporter assay
The 3′-UTR sequence of wild-type (WT) SMG1 and
target-site mutant-type (MT) PCR products were cloned into a
dual-luciferase reporter vector plasmid (Promega Corp.), and the
products were termed as pGL3-SMG1-3′-UTR-WT (WT vector) and
pGL3-SMG1-3′-UTR-MT (MT vector). Logarithmic growth-phase ES-2
cells were seeded into 96-well plates at a density of
1.5×104 cells/well prior to transfection. ES-2 cells
were then co-transfected with the WT or MT vector and miR-32 mimic
or mimic NC using the Attractene Transfection Reagent (Qiagen,
Inc.), because miR-32 was most significantly expressed in ES-2
cells. After 48 h of transfection, the Firefly to Renilla
luciferase activity ratio was detected using a dual-luciferase
reporter system (Promega Corp.). Also, subsequent experiments were
conducted in ES-2 cells.
Western blot analysis
Lysis buffer [150 M NaCl, 20 mM Tris-HCl (pH 7.4), 1
mM EDTA, 1 mM DTT, 1 mM PMSF and 10% glycerol)] was used to digest
the sample tissues and cells. The protein concentration of each
sample was measured using a BCA protein assay kit (cat. no. 23225;
Thermo Fisher Scientific, Inc.). Total proteins (30 µg) from each
sample were separated by polyacrylamide gel electrophoresis with
10% SDS and then transferred to polyvinylidene fluoride membranes
at 100 V for 1.5 h. The membranes were blocked with 5% skimmed milk
in TBST (1 ml/l Tween-20, 100 mM Tris-Cl, 9 g/l NaCl, pH 7.5) for 1
h at room temperature, and were incubated with primary antibodies
(anti-SMG1; ab30916; 1:500; Abcam,) at 4°C overnight. After
washing, secondary antibodies (horseradish peroxidase-conjugated
goat anti-rabbit immunoglobulin G; 1;1,000; cat. no. ab6721; Abcam)
were added and the membranes were incubated at room temperature for
2 h. Protein bands were visualized using enhanced chemiluminescence
(ECL) reagents (EMD Millipore). ImageJ version 1.46 software
(National Institutes of Health) was used to quantify the protein
expression levels. GAPDH (cat. no. ab181602; 1:1,000; Abcam) was
used as the loading control.
Statistical analysis
Statistical analysis was performed using the SPSS
17.0 software (SPSS, Inc.). Data are expressed as the mean ± SD.
The independent-samples or paired t-test was used for comparisons
between two groups. One-way ANOVA, followed by Bonferroni's
post-hoc test, was performed to analyze the differences among more
than two groups. The correlation between miR-32 and SMG1 mRNA
expression in OC tissues was analyzed by Spearman's correlation
analysis. The Pearson's χ2 test was used to analyze the
association between miR-32 expression and clinicopathological
parameters. Each experiment was repeated at least three times with
triplicates in each experiment. P<0.05 was considered to
indicate a statistically significant difference.
Results
miR-32 is upregulated in OC tissues
and cell lines
To determine the expression profile of miR-32 in OC,
RT-qPCR was performed to detect the mRNA level of miR-32 in 38
paired human OC tissues and normal ovarian tissues. The expression
of miR-32 was elevated in OC tissues compared with that in the
normal ovarian tissues (P<0.01; Fig.
1A). Furthermore, the expression of miR-32 was significantly
higher in the three OC cell lines (OVCAR3, SKOV3, ES-2) compared
with that in the HOSE cell line (IOSE80) (P<0.01; Fig. 1B). These results indicate that the
expression of miR-32 is upregulated in OC.
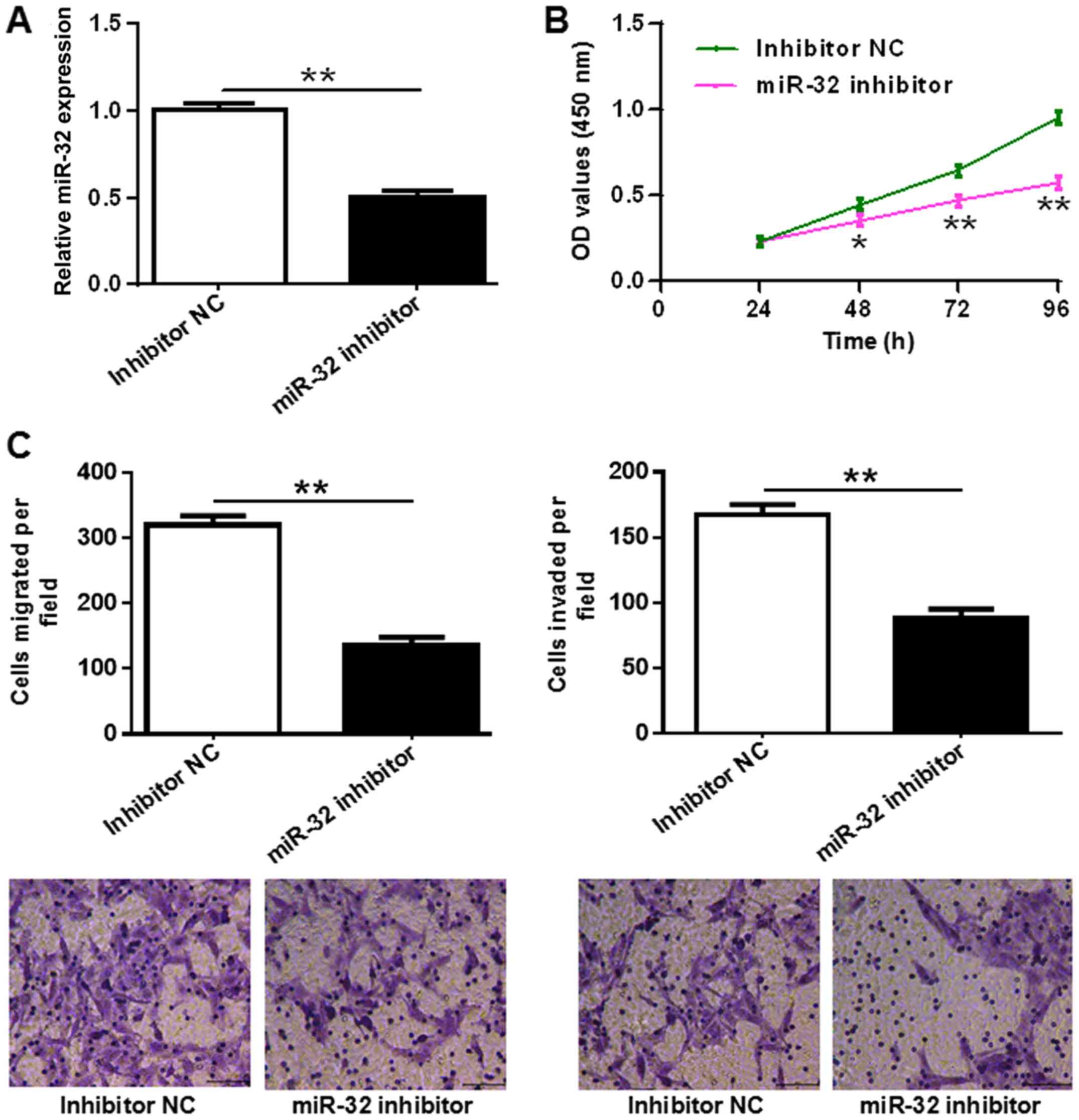
Inhibition of miR-32 suppresses OC
cell proliferation and motility
To investigate the effect of miR-32 on OC cell
proliferation and motility, ES-2 cells were transfected with miR-32
inhibitor or inhibitor NC. Transfection efficiency of miR-32
expression in ES-2 cells was confirmed by RT-qPCR (P<0.01;
Fig. 2A). A CCK-8 assay was
performed to detect the proliferation of the transfected ES-2
cells. The results showed that miR-32 inhibitor significantly
suppressed OC cell proliferation compared with the inhibitor NC
(P<0.05 at 48 h, and P<0.01 at 72 and 96 h; Fig. 2B). Next, cell motility was measured
by Transwell migration and Matrigel invasion assays, and it was
shown that the inhibition of miR-32 effectively suppressed OC cell
migration and invasion (both P<0.01; Fig. 2C). These results reveal that the
inhibition of miR-32 suppresses OC cell proliferation and
motility.
SMG1 is a direct target of miR-32
TargetScan 6.2 was used to explore the potential
targets of miR-32 in OC. SMG1, a tumor suppressor in human
tumorigenesis, was predicted and selected as the target of miR-32
in the present study (Fig. 3A). To
confirm this, the cells were co-transfected with WT or MT SMG1
3′-UTR vectors, with miR-32 mimic or mimic NC (Fig. S1), and a luciferase activity assay
was conducted. The results showed that the overexpression of miR-32
significantly suppressed the WT, but not the MT 3′-UTR of SMG1,
while inhibition of miR-32 significantly promoted the WT, but not
the MT 3′-UTR of SMG1 (P<0.01; Fig.
3B). In addition, RT-qPCR and western blot analysis revealed
that the inhibition of miR-32 significantly increased the mRNA and
protein levels of SMG1, while overexpression of miR-32
significantly decreased the mRNA and protein levels of SMG1 (both
P<0.01; Fig. 3C and D). The
decrease in SMG1 protein expression induced by miR-32 mimic is
lower than the decrease in SMG1 mRNA expression, because the
post-transcriptional, translational and degradation regulation
determines the concentration of the protein, thus, the protein and
mRNA levels may not be well correlated. Taken together, these
results demonstrate that SMG1 is a direct target of miR-32.
SMG1 expression is decreased in both
OC tumor tissues and tumor cells, and negatively correlated with
the expression of miR-32
Next, the expression of SMG1 in both OC tumors and
tumor cell lines was analyzed. RT-qPCR results demonstrated that
the expression of SMG1,compared with other potential targets,
including ANP32E, ARRDC3, FXR1, SMG1, EVI5, GRAMD1B, KIF1B, BMP7
and SPHK2, was significantly decreased in both OC tumor tissues and
tumor cell lines compared with that in normal adjacent tissues and
the HOSE cell line (IOSE80), respectively (all P<0.01; Figs. 4A, B and S1). Hence, SMG1 was the most relevant
target to be used in this study. Moreover, the correlation between
the SMG1 and miR-32 expression levels was also analyzed. The
results revealed that SMG1 was negatively correlated with miR-32
(r2=0.3460, P<0.0001; Fig.
4C). Taken together, these results suggest that SMG1 is
downregulated in OC and negatively correlated with miR-32.
Interference of SMG1 restores
miR-32-mediated OC cell proliferation and motility
Since SMG1 is a direct target of miR-32,
downregulation of SMG1 was performed to determine its effect on the
inhibition of miR-32-induced cell proliferation and motility
regression. To this end, the SMG1-targeting shRNA oligo was
employed to deplete endogenous SMG1 in OC cells. The downregulation
effect was confirmed by western blot analysis (Fig. 5A). miR-32 inhibitor or inhibitor NC
and SMG1 shRNA oligo or SMG1 NC were co-transfected into ES-2
cells. According to the results, SMG1 shRNA attenuated miR-32
inhibitor-triggered SMG1 protein elevation in ES-2 cells
(P<0.01; Fig. 5A). In addition,
downregulation of SMG1 by shRNA also attenuated the miR-32
inhibitor-induced OC cell proliferation regression (*P<0.05 and
**P<0.01 vs. control group; Fig.
5B). Moreover, downregulation of SMG1 by shRNA also attenuated
miR-32 inhibitor-induced ES-2 cell migration and invasion
regression (P<0.01; Fig. 5C).
These findings suggest that miR-32 promotes OC cell proliferation
and motility by the regulation of SMG1.
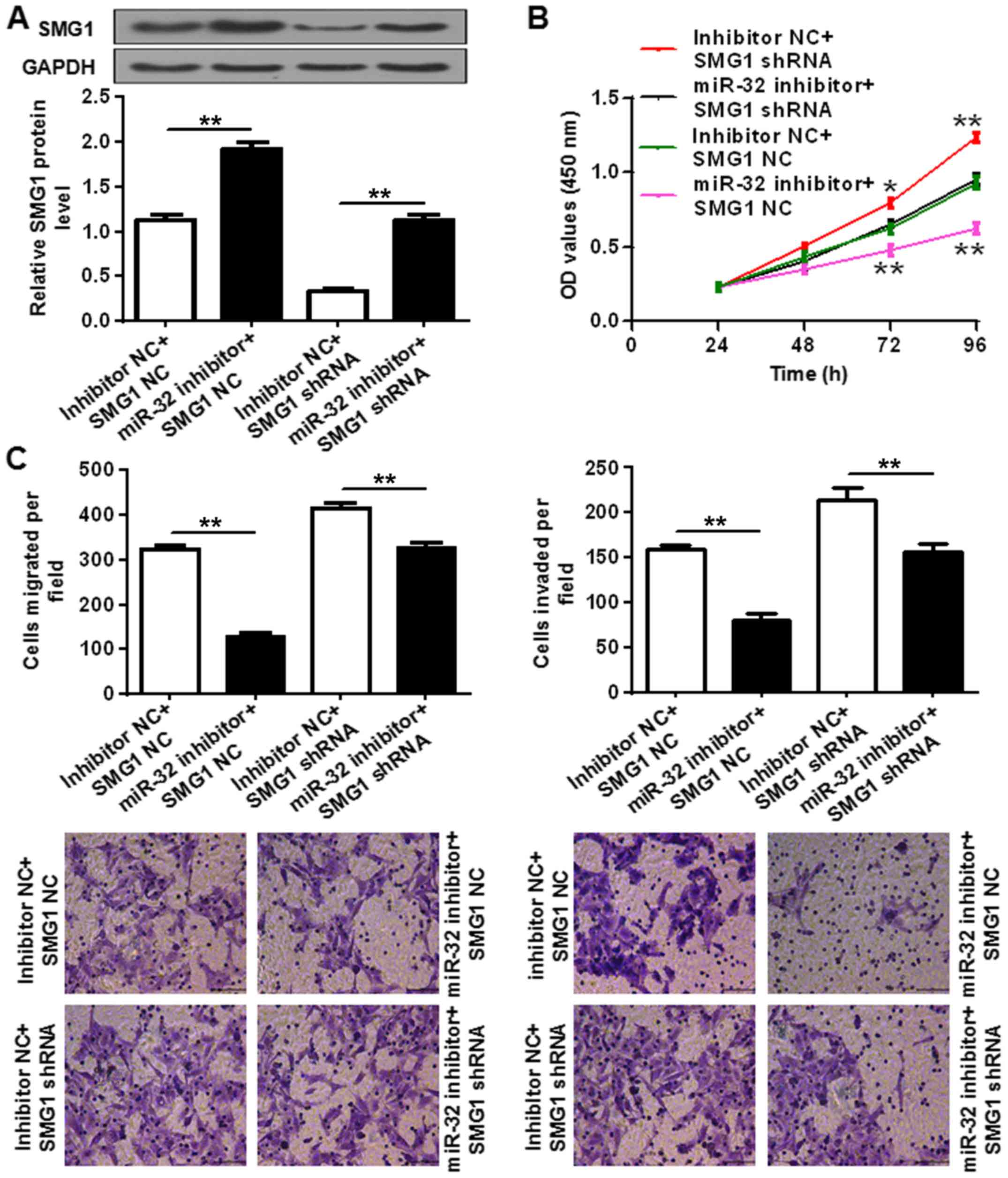 | Figure 5.Effect of SMG1 depletion on
miR-32-mediated OC cell proliferation and motility. (A) SMG1
protein expression was determined in ES-2 cells co-transfected with
miR-32 inhibitor, inhibitor NC, SMG1 shRNA oligo or SMG1 NC. (B)
Cell proliferation and (C) motility (migration and invasion) were
determined in ES-2 cells co-transfected with miR-32 inhibitor,
inhibitor NC, SMG1 shRNA oligo or SMG1 NC by Transwell assay and
staining with 0.2% crystal violet respectively. The experiments
were repeated at least three times and similar results were
obtained. *P<0.05 and **P<0.01 vs. control group. SMG1,
suppressor of morphogenesis in genitalia 1; OC, ovarian cancer; NC,
negative control; OD, optical density; miR, microRNA. Scale bar,
200 µm. |
Discussion
miRNAs have been studied for decades, and the
dysregulation of miRNAs has been reported in tumor tissues and
serums (29,30). miRNAs act as tumor suppressors or
oncogenes in the development and progression of different types of
cancer, depending on their proliferation, biological function and
targets. Therefore, the investigation of specific miRNAs, their
role in cancer and their targets would be valuable for cancer
diagnosis and therapy. miR-32, located at chromosome band Xq26.2,
has been reported to serve as an oncogene in several types of
cancer, including breast, prostate, endometrial, colorectal and
hepatocellular cancer (21–26), while acting as a tumor suppressor in
human oral squamous cell carcinoma (27). However, its expression profile and
biological function in OC is still under investigation. In this
study, the expression and biological function of miR-32 in OC was
explored. There are two types of miR-32, miR-32-5p and its
complementary strand miR-32-3p, derived from the miR-32-5p/-3p
duplex, which is processed from intron 14 of the C9orf5 gene. Since
miR-32-5p has been widely explored and identified as an important
regulator in tumorigenesis in different types of cancer, its role
in OC was explored in the present study to characterize its target
genes and physiological functions.
In the present study, different types of OC patients
were enrolled and the expression of miR-32 was analyzed in tumor
tissues and paired adjacent normal tissues. The data indicated that
miR-32 was significantly upregulated in both OC tumor tissues and
cell lines, when compared with normal adjacent tissues and normal
ovarian cells, respectively. These results suggest that miR-32 may
play an oncogenic role in OC development and progression. However,
there are still limitations to our understanding of the expression
profile of miR-32 due to the lack of a large number of
participants. In future studies, more OC patients will be included
to confirm the oncogenic role in the development and progression of
OC. Furthermore, a CCK8 assay showed that downregulation of miR-32
markedly inhibited OC cell proliferation, and a Transwell assay
proved that the downregulation of miR-32 profoundly inhibited cell
motility by decreasing cell migration and invasion. Together, these
findings confirm the oncogenic role of miR-32 in OC cells.
Each miRNA can have multiple targets and can
regulate its targets to either promote or inhibit tumor cell
proliferation, growth and motility. In breast cancer, miR-32 was
reported to promote cell proliferation and motility, and suppress
apoptosis by targeting FBXW7 (22).
In hepatocellular carcinoma, miR-32 was proven to induce cell
proliferation and motility by targeting PTEN (24). Also, in human squamous cell
carcinoma, miR-32 was shown to act as tumor suppressor and directly
target EZH2 (27). Thus, exploring
the targets of miR-32 could advance our understanding of the
mechanism of miR-32 in OC development and progression. In the
present study, several potential targets, including ANP32E, ARRDC3,
FXR1, SMG1, EVI5, GRAMD1B, KIF1B, BMP7 and SPHK2, were detected and
RT-qPCR was performed to check the mRNA expression level of these
targets in both OC cell lines and related normal cells (Figs. 4B and S2). Among these potential targets, the
expression decrease of SMG1 was most significant. Hence, SMG1 was
selected for further research. SMG1 is an enzyme encoded by the
SMG1 gene that belongs to the phosphatidylinositol 3-kinase-related
kinase protein family, which is involved in nonsense-mediated mRNA
decay (31,32). Recent studies have shown that SMG1 is
a potential tumor suppressor gene in acute myeloid leukemia and in
planarians (33,34). In the present study, SMG1 was
predicted and proven to be a direct target of miR-32.
Downregulation of SMG1 restored miR-32-mediated OC cell
proliferation, migration and invasion. Therefore, miR-32 may
promote OC cell growth and motility by targeting SMG1. Thus,
inhibition of miR-32 by its inhibitor has the potential to be
considered as a therapeutic strategy for the treatment of OC.
In conclusion, the present results revealed that
miR-32 was upregulated in OC tissue samples and cells, and that
downregulation of miR-32 inhibited OC cell proliferation and
motility. To the best of our knowledge, this is the first time that
the oncogenic role of miR-32 in the development and progression of
OC has been demonstrated. Inhibition of miR-32 may be a therapeutic
strategy in the treatment of OC. Furthermore, SMG1 was shown to be
a direct target of miR-32. Downregulation of SMG1 was found to
attenuate the inhibition of miR-32-triggered OC cell proliferation
and motility. Hence, miR-32 promotes OC cell proliferation and
motility via regulation of SMG1. Together, these results uncover
the mechanism through which miR-32 serves as an oncogene in OC to
promote cancer development and progression, and miR-32 can be
explored as a therapeutic target for the clinical treatment of OC.
However, there are still limitations to this study. Since ES-2
cells have the highest expression of miR-32, only ES-2 was selected
to be extensively studied. In the future, more OC cell lines will
be used to confirm the role of miR-32 in OC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SZ and FX designed and performed the study. SL, JF
and JG participated in conducting the experiments and in the
statistical analysis. SZ and FX wrote the manuscript. FX revised
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The study was approved by The Ethics Committee of
Tianjin Medical University (Tianjin, China). Written informed
consent was obtained from each patient.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hennessy BT, Coleman RL and Markman M:
Ovarian cancer. Lancet. 374:1371–1382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ozols RF, Bundy BN, Greer BE, Fowler JM,
Clarke-Pearson D, Burger RA, Mannel RS, DeGeest K, Hartenbach EM
and Baergen R; Gynecologic Oncology Group, : Phase III trial of
carboplatin and paclitaxel compared with cisplatin and paclitaxel
in patients with optimally resected stage III ovarian cancer: A
Gynecologic Oncology Group study. J Clin Oncol. 21:3194–3200. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bristow RE, Santillan A, Salani R,
Diaz-Montes TP, Giuntoli RL II, Meisner BC, Armstrong DK and Frick
KD: Intraperitoneal cisplatin and paclitaxel versus intravenous
carboplatin and paclitaxel chemotherapy for Stage III ovarian
cancer: A cost-effectiveness analysis. Gynecol Oncol. 106:476–481.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ozols RF, Bookman MA, du Bois A, Pfisterer
J, Reuss A and Young RC: Intraperitoneal cisplatin therapy in
ovarian cancer: Comparison with standard intravenous carboplatin
and paclitaxel. Gynecol Oncol. 103:1–6. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Greenlee RT, Hill-Harmon MB, Murray T and
Thun M: Cancer statistics, 2001. CA Cancer J Clin. 51:15–36. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Filipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: Are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lagos-Quintana M, Rauhut R, Lendeckel W
and Tuschl T: Identification of novel genes coding for small
expressed RNAs. Science. 294:853–858. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mukherji S, Ebert MS, Zheng GX, Tsang JS,
Sharp PA and van Oudenaarden A: MicroRNAs can generate thresholds
in target gene expression. Nat Genet. 43:854–859. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Selbach M, Schwanhäusser B, Thierfelder N,
Fang Z, Khanin R and Rajewsky N: Widespread changes in protein
synthesis induced by microRNAs. Nature. 455:58–63. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Iorio MV and Croce CM: MicroRNA
dysregulation in cancer: Diagnostics, monitoring and therapeutics.
A comprehensive review. Embo Molecular Medicine. 4:143–159. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Heneghan HM, Miller N and Kerin MJ: MiRNAs
as biomarkers and therapeutic targets in cancer. Curr Opin
Pharmacol. 10:543–550. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nam EJ, Yoon H, Kim SW, Kim H, Kim YT, Kim
JH, Kim JW and Kim S: MicroRNA expression profiles in serous
ovarian carcinoma. Clin Cancer Res. 14:2690–2695. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang L, Volinia S, Bonome T, Calin GA,
Greshock J, Yang N, Liu CG, Giannakakis A, Alexiou P, Hasegawa K,
et al: Genomic and epigenetic alterations deregulate microRNA
expression in human epithelial ovarian cancer. Proc Natl Acad Sci
USA. 105:7004–7009. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dahiya N and Morin PJ: MicroRNAs in
ovarian carcinomas. Endocr Relat Cancer. 17:F77–F89. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jayaraman M, Radhakrishnan R, Mathews CA,
Yan M, Husain S, Moxley KM, Song YS and Dhanasekaran DN:
Identification of novel diagnostic and prognostic miRNA signatures
in endometrial cancer. Genes Cancer. 8:566–576. 2017.PubMed/NCBI
|
|
22
|
Xia W, Zhou J, Luo H, Liu Y, Peng C, Zheng
W and Ma W: MicroRNA-32 promotes cell proliferation, migration and
suppresses apoptosis in breast cancer cells by targeting FBXW7.
Cancer Cell Int. 17:142017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang H, Li Y, Zhong X, Luo P, Luo P, Sun
R, Xie R, Fu D, Ma Y, Cong X and Li W: Upregulation of microRNA-32
is associated with tumorigenesis and poor prognosis in patients
with hepatocellular carcinoma. Oncol Lett. 15:4097–4104.
2018.PubMed/NCBI
|
|
24
|
Yan SY, Chen MM, Li GM, Wang YQ and Fan
JG: MiR-32 induces cell proliferation, migration, and invasion in
hepatocellular carcinoma by targeting PTEN. Tumor Biol.
36:4747–4755. 2015. View Article : Google Scholar
|
|
25
|
Latonen L, Scaravilli M, Gillen A,
Hartikainen S, Zhang FP, Ruusuvuori P, Kujala P, Poutanen M and
Visakorpi T: In vivo expression of miR-32 induces proliferation in
prostate epithelium. Am J Pathol. 187:2546–2557. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu W, Yang J, Feng X, Wang H, Ye S, Yang
P, Tan W, Wei G and Zhou Y: MicroRNA-32 (miR-32) regulates
phosphatase and tensin homologue (PTEN) expression and promotes
growth, migration, and invasion in colorectal carcinoma cells. Mol
Cancer. 12:302013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang D, Ni Z, Xu X and Xiao J: MiR-32
functions as a tumor suppressor and directly targets EZH2 in human
oral squamous cell carcinoma. Med Sci Monit. 20:2527–2535. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Croce CM: Causes and consequences of
microRNA dysregulation in cancer. Nat Rev Genet. 10:704–744. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K,
Guo J, Zhang Y, Chen J, Guo X, et al: Characterization of microRNAs
in serum: A novel class of biomarkers for diagnosis of cancer and
other diseases. Cell Res. 18:997–1006. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McIlwain DR, Pan Q, Reilly PT, Elia AJ,
McCracken S, Wakeham AC, Itie-Youten A, Blencowe BJ and Mak TW:
Smg1 is required for embryogenesis and regulates diverse genes via
alternative splicing coupled to nonsense-mediated mRNA decay. Proc
Natl Acad Sci USA. 107:12186–12191. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nickless A, Bailis JM and You ZS: Control
of gene expression through the nonsense-mediated RNA decay pathway.
Cell Biosci. 7:262017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
González-Estévez C, Felix DA, Smith MD,
Paps J, Morley SJ, James V, Sharp TV and Aboobaker AA: SMG-1 and
mTORC1 act antagonistically to regulate response to injury and
growth in planarians. PLoS Genet. 8:e10026192012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Du Y, Lu F, Li P, Ye J, Ji M, Ma D and Ji
C: SMG1 acts as a novel potential tumor suppressor with epigenetic
inactivation in acute myeloid leukemia. Int J Mol Sci.
15:17065–17076. 2014. View Article : Google Scholar : PubMed/NCBI
|