Introduction
Lung adenocarcinoma (LUAD) is a subtype of non-small
cell lung cancer (NSCLC) and represents the most common
histological type of NSCLC (1).
Although significant progress has been made in the treatment of
LUAD with the use of molecular targeted therapies, the 5-year
survival rate of patients with LUAD is still <20% (2). Therefore, novel biomarkers are urgently
required for significant improvements in prognosis. Progress has
been made in the application of immunotherapy in the treatment of
LUAD. For example, the application of programmed cell death-1
(PD-1) inhibitors, PD-ligand 1 inhibitors and cytotoxic
T-lymphocyte-associated protein 4 (CTLA-4) inhibitors have been
shown to enhance intratumoral immune responses and improve
prognosis in patients with LUAD in numerous preclinical or clinical
studies (3–6). However, some studies have also
identified the limitations of immunotherapy, including severe
adverse effects and low response in some patients (7). One of the factors which diminishes the
efficacy of tumor immunotherapy is the tumor microenvironment (TME)
(8,9), which is heterogeneous in composition
and contains cellular components, growth factors, proteases and
extracellular matrix (10). As a
result, it is particularly important to identify TME-related
biomarkers in order to identify patients who will have an improved
prognosis after receiving immunotherapy.
The TME regulates major hallmarks of cancer,
including angiogenesis, inflammation, immune suppression,
epithelial-mesenchymal transition and metastasis (11). Previous studies have demonstrated
that immune-inflamed TMEs express high levels of cytotoxic
lymphocytes as well as immune activation markers, and tumor purity
is decreased (12,13). Tumors with this type of TMEs are
often associated with a favorable prognosis (14), however, patients with immune-excluded
TMEs or a high tumor purity have a poorer survival instead. In
addition, Givechian et al (15) reported that patients with
immune-inflamed LUAD were associated with improved overall survival
(OS) compared with patients with immune-excluded LUAD. Behind this
phenomenon, genes such as CD8 and PRF1 (12,15–17) or
signaling pathways such as ribosomal, metabolic and chemokine
signaling pathways (15,18) may serve an important role. Therefore,
recognizing these genes and utilizing them provides a deep
understanding of TME in patients with LUAD, which could guide
immunotherapy. With the development of bioinformatics, some
algorithms have been applied to evaluate the tumor purity of TME
according to the specific gene expression signature of immune
or/and stromal cells (19,20). In 2013, Yoshihara et al
(20) invented an algorithm termed
Estimation of STromal and Immune cells in MAlignant Tumors using
Expression data (ESTIMATE) to analyze stromal and immune cells that
form the major non-tumor constituents of tumor samples. This
algorithm calculates the stromal and immune scores to predict the
tumor purity of tumor tissues. In the present study, The Cancer
Genome Atlas (TCGA) database and ESTIMATE algorithm were utilized
to identify TME-related genes to predict outcomes in patients with
LUAD.
Materials and methods
Database
Level 3 gene expression data for 517 patients with
LUAD was downloaded from TCGA data portal (https://tcga-data.nci.nih.gov/tcga/) and was analyzed
using the Illumine Hiseq 2000 RNA Sequencing v.2 platform
[University of North Carolina (UCSC) TCGA genome characterization
center; October 10, 2017] and RNA sequencing data was downloaded
from UCSC Xena browser (https://xena.ucsc.edu/). Clinical data, including age,
sex, histological type, tumor metastasis conditions, epidermal
growth factor receptor (EGFR) mutation status, overall survival
time and outcome were downloaded from TCGA data portal. Immune and
stromal scores of 517 patients with LUAD were calculated by
applying the ESTIMATE algorithm to the downloaded dataset. For
validation, the Gene Expression Omnibus (GEO) database was used to
compare gene expression profiling of patients with LUAD with
clinical data of survival and outcome. Finally, two independent
datasets, GES37745 (n=106) (21) and
GES29013 (n =31) (22), were used to
validate the identified genes.
Identification of differentially
expressed genes (DEGs)
Based on the ESTIMATE results, all samples were
divided into high/low immune-score groups and high/low
stromal-score groups to select intersection genes. The cut-off
value of identifying high immune score group or low immune score
group was 980.35. The cut-off value for identifying high stromal
score group or low stromal score group was 36.85. DEG data analysis
was performed using limma package (23). The cut-off values for screening DEGs
were set as fold change (FC) >2 or <-2 and P<0.05. Volcano
plots were generated using the ggplot2 package in R software v.3.5
(24).
Construction of protein-protein
interaction (PPI) network
The Search Tool for the Retrieval of Interacting
Genes (STRING) online database was used to analyze the PPI network
of DEGs (25). The DEGs were
uploaded to the STRING online website and the interactive
relationships were determined. The cut-off value of the minimum
required interaction score was set as 0.700. Subsequently, the
Cytoscape software v.3.6 (26) was
used to construct the PPI network and Molecular Complex Detection
(MCODE) was used to identify the top three complete module clusters
(26).
Functional analysis of DEGs
The Database for Annotation, Visualization and
Integrated Discovery (DAVID) website was used to perform Gene
Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes
(KEGG) analysis and the false discovery rate <0.05 was defined
as the cut-off value (27).
Statistical analysis
All data are presented expressed as the mean ±
standard deviation. Student's t-test (two groups) and one-way ANOVA
(multiple groups) were used to compare the immune and stromal
scores in different groups using Graph-Pad Prism v7.0 software.
(GraphPad Software, Inc.). The post hoc test used following ANOVA
was Tukey's multiple comparisons test. OS curves were created using
the Kaplan-Meier survival analysis and estimated using two-sided
log rank test. Differential analysis of expressed genes and volcano
plots was performed using R software v.3.5. P<0.05 was
considered to indicate a statistically significant difference.
Results
Stromal and immune scores are
significantly associated with TNM stage, epidermal growth factor
receptor mutation status, distant metastasis and clinical outcome
in patients with LUAD
To determine the relationship between ESTIMATE
scores and clinical characteristics, the gene expression data as
well as clinical information of 517 patients with LUAD were
downloaded from the TCGA database. The dataset included patients in
which initial pathological diagnosis was made between 1991 and
2013. Among these patients, 277 cases (54%) were male and 240 cases
(46%) were female. The immune and stromal scores of each patient
were determined, the stromal scores ranged from −1,959.31 to
2,098.77, and the immune scores ranged from −1,355.85 to 3,286.67.
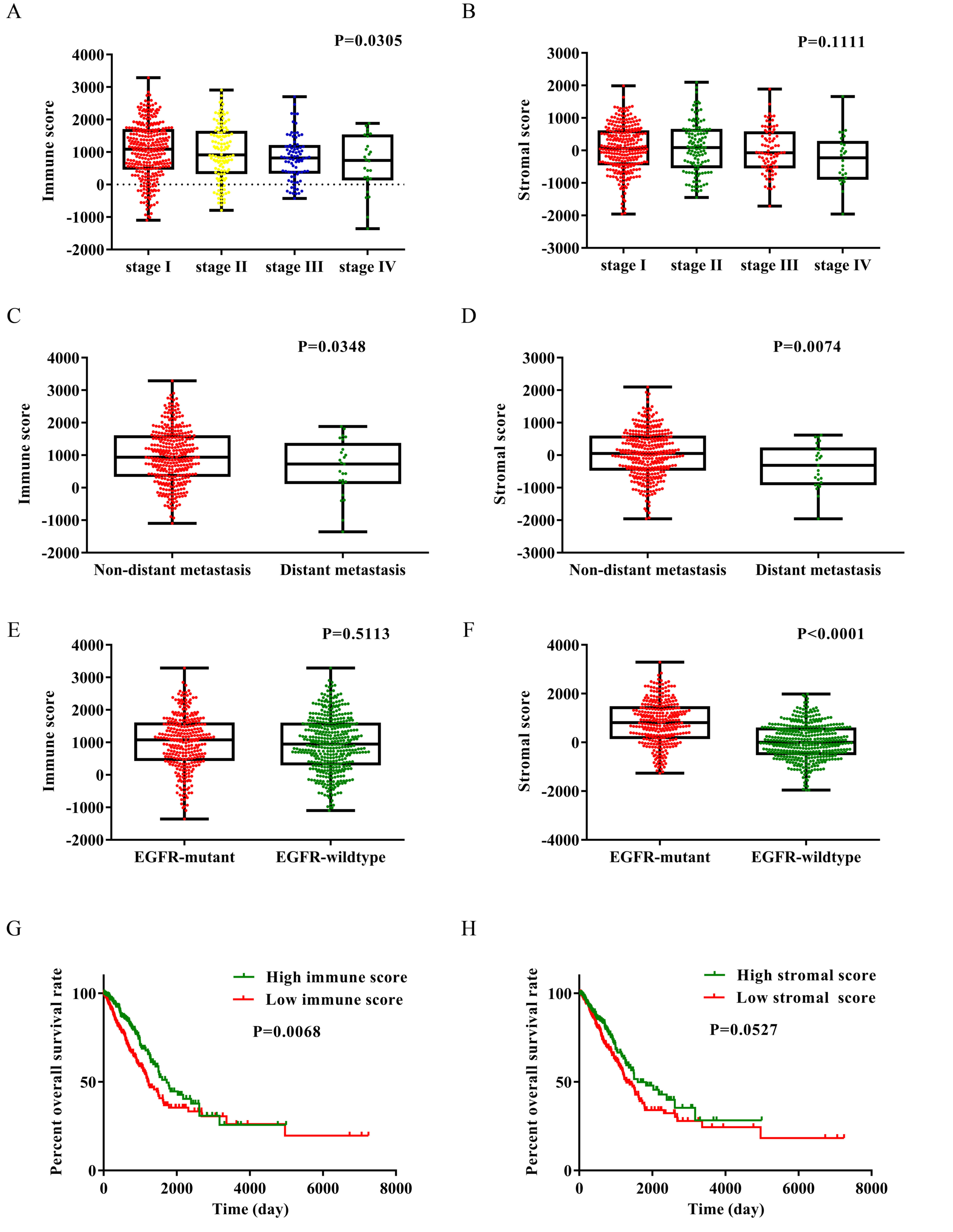
As presented in Fig. 1A, the immune
scores of each TNM stage were compared. The average immune scores
of patients with stage I were the highest amongst all of the TNM
stages, followed by that of stage II, and stage III. Patients with
stage IV had the lowest immune scores (Fig. 1A; P=0.0305, one-way ANOVA). Fig. 1B revealed the stromal scores of each
TNM stage. The stromal scores were highest in patients with stage
II LUAD, followed by stage I, stage III and stage IV (Fig. 1B; P=0.1111).
Subsequently, the immune and stromal scores were
compared between the distant metastasis group and the non-distant
metastasis group. The average immune and stromal scores of the
distant metastasis group were significantly lower compared with
that in the non-distant metastasis group (Fig. 1C and D). The immune and stromal
scores between lymph node metastasis group and non-lymph node
metastasis were also compared, however, the results revealed there
was no statistically significant difference between the two groups
(Fig. S1A and B).
To the best of our knowledge, the frequency of EGFR
gene mutation among Asian populations with NSCLC is ~30% (28). In Fig. 1E
and F, the average immune and stromal scores in the EGFR mutant
group and the EGFR wild-type group were shown. The stromal score of
the EGFR mutant group was significantly higher compared with that
of the EGFR wild-type group (P<0.0001), while the immune score
of the EGFR mutant group was also higher compared with that of the
EGFR wild-type group, although the result was not statistically
significant (P=0.5113).
To determine the association between OS and immune
and stromal scores, patients were divided into two groups based on
high or low scores using median value and Kaplan-Meier survival
curves were subsequently plotted. As indicated by Fig. 1G, the median OS time of the low
immune score group was significantly shorter compared with that of
the high immune score group (1,229 vs. 1,725 days; P=0.0068 using
the log-rank test). As shown in Fig.
1H, the median OS time of the low stromal score group was
shorter compared with that of the high stromal score group (1,293
vs. 1,600 days; P=0.0527; log-rank test), although there were no
statistically significant differences between the two groups.
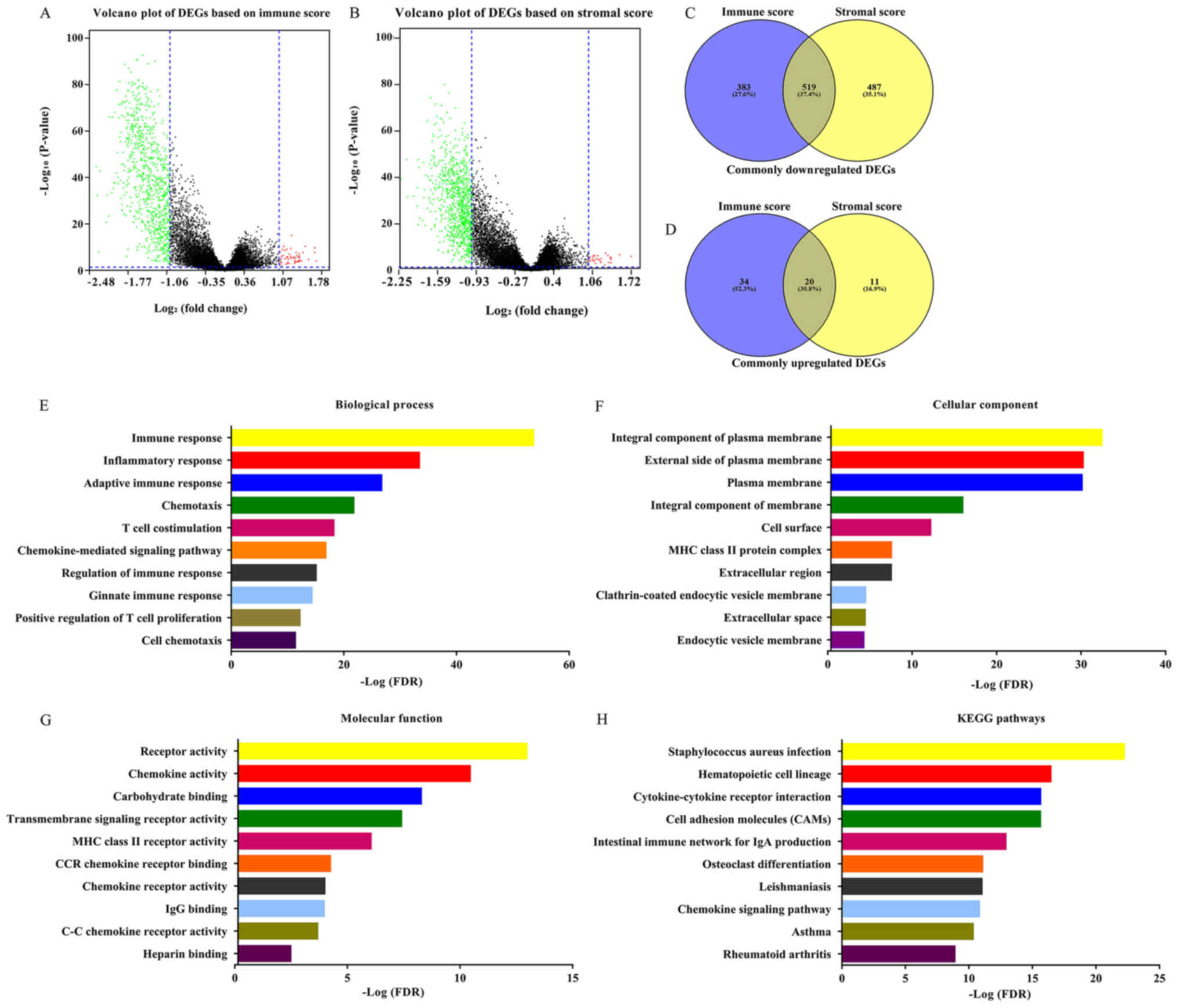
Identification of DEGs of immune and
stromal score groups
The gene expression matrix file of 517 patients with
LUAD downloaded from TCGA database was used to assess the gene
expression profile differences between high immune/stromal scores
and low immune/stromal scores. As shown in Fig. 2A and B, volcano plots revealed the
DEG profile of low immune/stromal score groups compared with high
immune/stromal score groups. A total of 902 downregulated genes and
54 upregulated genes were identified in the immune score group (low
vs. high, FC >2 or <-2; P<0.05). Similarly, a total of
1,006 downregulated genes and 31 upregulated genes were identified
in the stromal score group (low vs. high; FC >2 or <-2;
P<0.05). Furthermore, 519 downregulated DEGs as well as 20
upregulated DEGs were found in both the immune and stromal groups
(Fig. 2C and D; Table SI). Only the 519 intersection
downregulated genes were included in the further analysis. The 20
intersection upregulated DEGs were not included due to being the
minority of common DEGs. Furthermore, the current analysis was
primary focused on downregulated genes, the overexpression of which
might become a target for future gene therapy. The prognostic value
of the 20 upregulated genes was performed (Table SII) and the results revealed that
the hazard ratio (HR) of these genes was ~1, and the P-value of
most of these genes was >0.05. The 519 DEGs were used for all
subsequent analysis. The DAVID online tool was used to determine
the potential functions of these genes. Fig. 2E-G revealed the top GO terms of these
genes. These genes were involved in biological processes (BPs),
including ‘immune response’, ‘inflammatory response’ and
‘chemotaxis’ (Fig. 2E). The
molecular functions (MFs) of these genes included ‘receptor
activity’, ‘chemokine activity’ and ‘transmembrane signaling
receptor activity’ (Fig. 2G).
Cellular components (CCs) of these genes included integral
component of plasma membrane, ‘external side of plasma membrane’
and ‘plasma membrane’ (Fig. 2F). The
potential pathways these genes may be associated with were also
investigated using DAVID. The top 10 KEGG pathways these genes are
involved in included ‘Staphylococcus aureus infection’,
‘cytokine-cytokine receptor interaction’, ‘cell adhesion molecules
(CAMs)’ and ‘chemokine signaling pathway’.
Prognostic value analysis of 519
common downregulated DEGs
To determine if the 519 common downregulated genes
were associated with OS, Kaplan-Meier survival curves were plotted.
A total of 281 genes were identified to be associated with
favorable OS using log-rank test (P<0.05; HR<1; Table SIII). Kaplan-Meier survival curves
for some of the genes are presented in Fig. 3. Kaplan-Meier survival curves for 10
representative genes were selected and these 10 genes were found to
have multiple connections with other genes (Fig. 4).
PPI network construction of 281 DEGs
of prognostic value
The PPI network using STRING online tool was used to
identify the interaction among the identified DEGs. This network
was comprised of 273 nodes and 580 edges and Cytoscape software was
used to visualize the network (Fig.
4A). The top three modules of this PPI network were selected
using the MCODE plugin. The first module (Fig. 4B) contained 26 nodes and 162 edges,
and protein tyrosine phosphatase receptor type C, lymphocyte
cytosolic protein 2, HLA class II histocompatibility antigen DR
beta 5 chain, HLA class II histocompatibility antigen DQ alpha 1
chain, HLA class II histocompatibility antigen DQ beta 1 chain and
C-C chemokine receptor type 5 were of interest as they were found
to have multiple connections with other genes. The second module
(Fig. 4C) contained seven nodes and
21 edges, identifying CTLA-4 and B-lymphocyte antigen CD19 as the
most connected genes. The third module (Fig. 4D) was formed by 11 nodes and 32
edges, and integrin alpha-L (ITGAL) and P2X purinoceptor 1 had the
highest connectivity degree values.
Functional analysis of genes of
prognostic value
GO and KEGG pathway analysis of genes of prognostic
value was subsequently performed. Fig.
5A shows the BPs in which these genes are involved, and the top
10 terms were identified including ‘immune response’, ‘T cell
receptor signaling pathway’, ‘adaptive immune response’, and
‘regulation of immune response’. Fig.
5B shows the top 10 CC terms, including ‘external side of
membrane’, ‘MHC class II protein complex’ and plasma membrane.
Fig. 5C shows the top 10 significant
MF terms of these genes, which were ‘transmembrane signaling
receptor activity’, ‘MHC class II receptor activity’, ‘C-C
chemokine receptor activity’ and ‘peptide antigen binding
activity’. A total of 10 significant KEGG pathway terms were also
identified including ‘cell adhesion molecules (CAMs)’ and
‘cytokine-cytokine receptor interaction’.
Validation in the GEO database
To confirm that the prognostic genes identified
using TCGA analysis were also important in other LUAD cases, two
independent eligible cohorts of LUAD cases from the GEO database
(accession numbers GES37745 and GES29013) were selected for
validation, and the calculated OS rate of genes was performed the
GEO database (P<0.05; HR<1; Table
SIV). Only 4 genes were validated as positively associated with
prognosis. They were GTPase IMAP family member 1 (GIMAP1), T-cell
surface glycoprotein CD1b (CD1B), ITGAL and leukocyte surface
antigen CD53 (CD53). The HR value of these 4 genes in the GSE37745
dataset were as follows: GIMAP1, 0.51 (P=0.01); CD1B, 0.51
(P=0.01); ITGAL, 0.58 (P=0.01) and CD53, 0.63 (P=0.02). The HR
value of the 4 aforementioned genes in the GSE29013 dataset were as
follows: GIMAP1, 0.50 (P=0.02); CD1B, 0.51 (P=0.04); ITGAL, 0.71
(P=0.01) and CD53, 0.54 (P=0.04). Although other genes were
associated with prognosis in the TCGA database, they were not
associated with prognosis from the GEO database. As a result,
GIMAP1, CD1B, ITGAL and CD53 were validated as positively
associated with prognosis and downregulation of these 4 genes was
associated with poor prognosis (Fig.
6).
Discussion
A number of different studies have applied the
ESTIMATE algorithm to breast, prostate and colon cancer, indicating
that it was a useful and effective tool for analyzing large
datasets (29–31). Previous studies also applied this
algorithm to mining TME-related genes and identified high number of
novel genes that were associated with prognosis in glioblastoma and
cutaneous melanoma (32,33). To the best of our knowledge, the
current study is the first study where the ESTIMATE scoring
algorithm was used to identify tumor-related genes that were
associated with the OS of patients with LUAD in the TCGA database.
A total of 281 prognosis-related genes were identified and were
found to be involved in the immune and inflammatory response and
were subsequently validated in two LUAD cohorts from the GEO
database. Finally, a total of 4 prognosis-related genes were
associated with OS in the TCGA and GEO database.
Firstly, patients with stage III–IV TNM LUAD had
lower immune and stromal scores compared with those with stage I–II
TNM LUAD. Patients with distant metastasis had lower immune and
stromal scores compared with those without distant metastasis. The
Kaplan-Meier survival curves revealed that patients with low
immune/stromal scores had a poorer prognosis compared with those
with high immune/stromal scores. All of the aforementioned results
suggested that high immune/stromal scores were associated with the
prognosis of patients with LUAD. This is consistent with previous
reports that the inflamed and immunogenic TME was associated with
favorable patient survival in lung adenocarcinoma (15,34).
Secondly, downregulated DEGs were identified from
the comparison of low vs. high immune score groups, and also from
the comparison of low vs. high stromal score groups. Subsequently,
519 DEGs that were downregulated in both the immune and the stromal
score groups were analyzed further. GO and KEGG pathway analysis of
these genes revealed that the majority were related to immune
response, inflammatory response, T cell co-stimulation and
chemokine-mediated signaling pathway. It is noteworthy that
Staphylococcus aureus infection was ranked first in the KEGG
pathway analysis. Pulmonary bacterial infections are frequently
found in advanced stages of lung cancer (35). Hattar et al (36) also reported that purified
lipoteichoic acids of S. aureus could induce growth of lung
adenocarcinoma cell lines and these effects were mediated by
inflammatory mediators such as ligation of Toll-like receptor 2 and
interleukin (IL)-8. Thus, infections with Gram-positive bacteria
might cause persistent inflammation and activated inflammatory
cascades, which contribute to tumor growth in lung cancer. The
results of GO and KEGG pathway analysis indicated that a number of
immune cells, chemokines, cytokines and extracellular matrix
components could be involved in regulating the relationship between
LUAD and TME (11).
Thirdly, the prognostic value of the 519 DEGs was
assessed and 281 DEGS were associated with OS in patients with
LUAD. GO and KEGG pathway analysis of the 281 genes of prognostic
value was performed, majority of which were related to immune and
inflammatory response (Fig. 5A). A
PPI network of the 281 genes was created and the top three modules
were selected using a MCODE plugin. There was high connectivity
between the nodes in these modules, including CCR7, HLA-DQB1 and
CTLA4, which have been reported to be associated with regulating
immune and inflammatory responses and OS of patients with LUAD
(3,37–39).
Finally, two independent LUAD cohorts from GEO
database were used to validate the prognostic value of genes
identified in TCGA database. Only 4 genes, GIMAP1, CD1B, ITGAL and
CD53, were found to have prognostic value in the TCGA database and
2 datasets from the GEO database. GIMAP1 belongs to the family of
guanosine triphosphatases of the immunity-associated proteins
(GIMAPs) (40). Previous studies
have shown that the deregulated expression of GIMAP genes is
associated with lymphomas (41).
GIMAP1 is required for the establishment and maintenance of B cell
survival and mature B cells do not survive without GIMAP1
expression (42). CD1B is a member
of the CD1 molecule family which specializes in presenting lipid
antigens to T cells (43). The
activation of CD1 restricts T cells in vivo leading to rapid
antitumor cytotoxicity and interferon-γ production, which could
prevent tumor metastasis (44).
Bagchi et al (45) reported
that CD1B-autoreactive T cells could recognize phospholipid
antigens and exert antitumor immunity against CD1B+ T
cell lymphoma. ITGAL, which is also known as LFA-1 and CD11a, is a
member of the integrins family and is expressed on all leukocytes.
LFA-1 is associated with myeloid cell function in the TME and plays
an important role in the function of regulatory T (T-reg) cells
that infiltrate tumors. A previous study indicated that neutrophils
were recruited into the tumor via increased expression of LFA-1,
activated by estradiol and transforming growth factor β1 in a mouse
model of estrogen receptor-positive breast cancer (46). In colon cancer, another study
reported that IL-18 was involved in eosinophil-mediated antitumor
activity by upregulating LFA-1 and ICAM-1 (47). LFA-1 is also necessary for the
development and function of T-reg cells and knockdown of LFA-1
leads to an increase in autoimmunity (48). CD53 is a member of the tetraspanin
family and participates in the formation of a complex with
integrins on the plasma membrane (49). It has been reported that CD53 is
associated with asthma risk in the general population and recurrent
bacteria, fungi and viruses infections (50,51);
however, the relationship between CD53 and cancer has rarely been
reported. Through reviewing the literature, these 4 genes were
found to be immune/inflammation-related genes; however, the
relationship between these 4 genes and lung cancer has also been
rarely reported. In the present study, downregulation of these 4
genes was associated with a worse OS in patients with LUAD,
therefore, they have potential to become novel cancer
biomarkers.
There are some limitations to the present study,
which are noteworthy. Firstly, the exploration occurred at a
bioinformatics level, thus further experiments are required to
validate the exact mechanism of these 4 genes in vitro and
in vivo. Secondly, patients with LUAD in TCGA database
received different treatments, such as chemotherapy, targeted
molecular therapy or immunotherapy. These treatments could affect
gene expression and OS rates of patients. Thus, clinical potential
of these 4 genes requires a larger sample size to be confirmed.
In conclusion, using the ESTIMATE algorithm to
calculate the immune and stromal scores of patients with LUAD in
TCGA database, it was found that low immune/stromal scores were
associated with a worse TNM stage, a greater likelihood of distant
metastasis and an unfavorable overall survival in patients with
LUAD. TME-related genes were identified and 4 of them were
validated in two independent LUAD cohorts from GEO database. These
4 genes were GIMAP1, CD1B, ITGAL and CD53, and were strongly
associated with immune response and inflammatory response. The
present study has obtained an improved comprehensive understanding
of TME by mining TCGA database and identified 4
immune/inflammatory-related genes, which have the potential to
predict prognosis of patients with LUAD.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Fundamental
Science Research Project of Xi'an Jiaotong University (grant no.
1191329849) and National Natural Science Foundation of China (grant
no. 81672300).
Availability of data and materials
The datasets analyzed during the present study are
available in The Cancer Genome Atlas repository (https://cancergenome.nih.gov/) and the GEO repository
(https://www.ncbi.nlm.nih.gov/geo/).
Authors' contributions
JY designed the study and wrote the manuscript. BY,
LZ and BXL contributed to the acquisition of data. YC, XM and RYS
performed the bioinformatics analysis. XL and WW analyzed data. SY
contributed to the experimental design and was responsible for
revising the manuscript and approving the version to be published.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Travis WD, Brambilla E, Nicholson AG,
Yatabe Y, Austin JHM, Beasley MB, Chirieac LR, Dacic S, Duhig E,
Flieder DB, et al: The 2015 World Health Organization
classification of lung tumors: Impact of genetic, clinical and
radiologic advances since the 2004 classification. J Thorac Oncol.
10:1243–1260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Osmani L, Askin F, Gabrielson E and Li QK:
Current WHO guidelines and the critical role of immunohistochemical
markers in the subclassification of non-small cell lung carcinoma
(NSCLC): Moving from targeted therapy to immunotherapy. Semin
Cancer Biol. 52:103–109. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reck M, Bondarenko I, Luft A, Serwatowski
P, Barlesi F, Chacko R, Sebastian M, Lu H, Cuillerot JM and Lynch
TJ: Ipilimumab in combination with paclitaxel and carboplatin as
first-line therapy in extensive-disease-small-cell lung cancer:
Results from a randomized, double-blind, multicenter phase 2 trial.
Ann Oncol. 24:75–83. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Horinouchi H, Nishio M, Hida T, Nakagawa
K, Sakai H, Nogami N, Atagi S, Takahashi T, Saka H, Takenoyama M,
et al: Three-year follow-up results from phase II studies of
nivolumab in Japanese patients with previously treated advanced
non-small cell lung cancer: Pooled analysis of ONO-4538-05 and
ONO-4538-06 studies. Cancer Med. 8:5183–5193. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ,
Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al:
Safety and activity of anti-PD-L1 antibody in patients with
advanced cancer. N Engl J Med. 366:2455–2465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hellmann MD, Rizvi NA, Goldman JW,
Gettinger SN, Borghaei H, Brahmer JR, Ready NE, Gerber DE, Chow LQ,
Juergens RA, et al: Nivolumab plus ipilimumab as first-line
treatment for advanced non-small-cell lung cancer (CheckMate 012):
Results of an open-label, phase 1, multicohort study. Lancet Oncol.
18:31–41. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fukumura D, Kloepper J, Amoozgar Z, Duda
DG and Jain RK: Enhancing cancer immunotherapy using
antiangiogenics: Opportunities and challenges. Nat Rev Clin Oncol.
15:325–340. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Datta M, Coussens LM, Nishikawa H, Hodi FS
and Jain RK: Reprogramming the tumor microenvironment to improve
immunotherapy: Emerging strategies and combination therapies. Am
Soc Clin Oncol Educ Book. 39:165–174. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li HY, McSharry M, Bullock B, Nguyen TT,
Kwak J, Poczobutt JM, Sippel TR, Heasley LE, Weiser-Evans MC,
Clambey ET and Nemenoff RA: The tumor microenvironment regulates
sensitivity of murine lung tumors to PD-1/PD-L1 antibody blockade.
Cancer Immunol Res. 5:767–777. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao D and Mittal V: The role of
bone-marrow-derived cells in tumor growth, metastasis initiation
and progression. Trends Mol Med. 15:333–343. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mittal V, El Rayes T, Narula N, McGraw TE,
Altorki NK and Barcellos-Hoff MH: The microenvironment of lung
cancer and therapeutic implications. Adv Exp Med Biol. 890:75–110.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Binnewies M, Roberts EW, Kersten K, Chan
V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI,
Ostrand-Rosenberg S, Hedrick CC, et al: Understanding the tumor
immune microenvironment (TIME) for effective therapy. Nat Med.
24:541–550. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rhee JK, Jung YC, Kim KR, Yoo J, Kim J,
Lee YJ, Ko YH, Lee HH, Cho BC and Kim TM: Impact of tumor purity on
immune gene expression and clustering analyses across multiple
cancer types. Cancer Immunol Res. 6:87–97. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pages F, Galon J, Dieu-Nosjean MC, Tartour
E, Sautes-Fridman C and Fridman WH: Immune infiltration in human
tumors: A prognostic factor that should not be ignored. Oncogene.
29:1093–1102. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Givechian KB, Garner C, Benz S, Song B,
Rabizadeh S and Soon-Shiong P: An immunogenic NSCLC
microenvironment is associated with favorable survival in lung
adenocarcinoma. Oncotarget. 10:1840–1849. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Patel SJ, Sanjana NE, Kishton RJ,
Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM,
Yamamoto TN, et al: Identification of essential genes for cancer
immunotherapy. Nature. 548:537–542. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brown SD, Warren RL, Gibb EA, Martin SD,
Spinelli JJ, Nelson BH and Holt RA: Neo-antigens predicted by tumor
genome meta-analysis correlate with increased patient survival.
Genome Res. 24:743–750. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lyssiotis CA and Kimmelman AC: Metabolic
interactions in the tumor microenvironment. Trends Cell Biol.
27:863–875. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Newman AM, Liu CL, Green MR, Gentles AJ,
Feng W, Xu Y, Hoang CD, Diehn M and Alizadeh AA: Robust enumeration
of cell subsets from tissue expression profiles. Nat Methods.
12:453–457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yoshihara K, Shahmoradgoli M, Martinez E,
Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW,
Levine DA, et al: Inferring tumour purity and stromal and immune
cell admixture from expression data. Nat Commun. 4:26122013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Botling J, Edlund K, Lohr M, Hellwig B,
Holmberg L, Lambe M, Berglund A, Ekman S, Bergqvist M, Pontén F, et
al: Biomarker discovery in non-small cell lung cancer: Integrating
gene expression profiling, meta-analysis, and tissue microarray
validation. Clin Cancer Res. 19:194–204. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie Y, Xiao G, Coombes KR, Behrens C,
Solis LM, Raso G, Girard L, Erickson HS, Roth J, Heymach JV, et al:
Robust gene expression signature from formalin-fixed
paraffin-embedded samples predicts prognosis of non-small-cell lung
cancer patients. Clin Cancer Res. 17:5705–5714. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ito K and Murphy D: Application of ggplot2
to pharmacometric graphics. CPT Pharmacometrics Syst Pharmacol.
2:e792013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tokumo M, Toyooka S, Kiura K, Shigematsu
H, Tomii K, Aoe M, Ichimura K, Tsuda T, Yano M, Tsukuda K, et al:
The relationship between epidermal growth factor receptor mutations
and clinicopathologic features in non-small cell lung cancers. Clin
Cancer Res. 11:1167–1173. 2005.PubMed/NCBI
|
|
29
|
Priedigkeit N, Watters RJ, Lucas PC,
Basudan A, Bhargava R, Horne W, Kolls JK, Fang Z, Rosenzweig MQ,
Brufsky AM, et al: Exome-capture RNA sequencing of decade-old
breast cancers and matched decalcified bone metastases. JCI
Insight. 2:957032017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shah N, Wang P, Wongvipat J, Karthaus WR,
Abida W, Armenia J, Rockowitz S, Drier Y, Bernstein BE, Long HW, et
al: Regulation of the glucocorticoid receptor via a BET-dependent
enhancer drives antiandrogen resistance in prostate cancer. ELife.
6:e278612017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alonso MH, Ausso S, Lopez-Doriga A,
Cordero D, Guinó E, Solé X, Barenys M, de Oca J, Capella G, Salazar
R, et al: Comprehensive analysis of copy number aberrations in
microsatellite stable colon cancer in view of stromal component. Br
J Cancer. 117:421–431. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jia D, Li S, Li D, Xue H, Yang D and Liu
Y: Mining TCGA database for genes of prognostic value in
glioblastoma microenvironment. Aging (Albany NY). 10:592–605. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang S, Liu T, Nan H, Wang Y, Chen H,
Zhang X, Zhang Y, Shen B, Qian P, Xu S, et al: Comprehensive
analysis of prognostic immune-related genes in the tumor
microenvironment of cutaneous melanoma. J Cell Physiol.
235:1025–1035. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Al-Shibli KI, Donnem T, Al-Saad S, Persson
M, Bremnes RM and Busund LT: Prognostic effect of epithelial and
stromal lymphocyte infiltration in non-small cell lung cancer. Clin
Cancer Res. 14:5220–5227. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Berghmans T, Sculier JP and Klastersky J:
A prospective study of infections in lung cancer patients admitted
to the hospital. Chest. 124:114–120. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hattar K, Reinert CP, Sibelius U,
Gökyildirim MY, Subtil FSB, Wilhelm J, Eul B, Dahlem G, Grimminger
F, Seeger W and Grandel U: Lipoteichoic acids from staphylococcus
aureus stimulate proliferation of human non-small-cell lung cancer
cells in vitro. Cancer Immunol Immunother. 66:799–809. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang L, Li M, Deng B, Dai N, Feng Y, Shan
J, Yang Y, Mao C, Huang P, Xu C and Wang D: HLA-DQB1 expression on
tumor cells is a novel favorable prognostic factor for relapse in
early-stage lung adenocarcinoma. Cancer Manag Res. 11:2605–2616.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Itakura M, Terashima Y, Shingyoji M, Yokoi
S, Ohira M, Kageyama H, Matui Y, Yoshida Y, Ashinuma H, Moriya Y,
et al: High CC chemokine receptor 7 expression improves
postoperative prognosis of lung adenocarcinoma patients. Br J
Cancer. 109:1100–1108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Leach DR, Krummel MF and Allison JP:
Enhancement of antitumor immunity by CTLA-4 blockade. Science.
271:1734–1736. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Krucken J, Stamm O, Schmitt-Wrede HP,
Mincheva A, Lichter P and Wunderlich F: Spleen-specific expression
of the malaria-inducible intronless mouse gene imap38. J Biol Chem.
274:24383–24391. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chadwick N, Zeef L, Portillo V, Boros J,
Hoyle S, van Doesburg JC and Buckle AM: Notch protection against
apoptosis in T-ALL cells mediated by GIMAP5. Blood Cells Mol Dis.
45:201–209. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Webb LM, Datta P, Bell SE, Kitamura D,
Turner M and Butcher GW: GIMAP1 Is essential for the survival of
naive and activated B cells in vivo. J Immunol. 196:207–216. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Brigl M and Brenner MB: CD1: Antigen
presentation and T cell function. Annu Rev Immunol. 22:817–890.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hayakawa Y, Godfrey DI and Smyth MJ:
Alpha-galactosylceramide: Potential immunomodulatory activity and
future application. Curr Med Chem. 11:241–252. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bagchi S, Li S and Wang CR:
CD1b-autoreactive T cells recognize phospholipid antigens and
contribute to antitumor immunity against a CD1b(+) T cell lymphoma.
Oncoimmunology. 5:e12139322016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Vazquez Rodriguez G, Abrahamsson A, Jensen
LD and Dabrosin C: Estradiol promotes breast cancer cell migration
via recruitment and activation of neutrophils. Cancer Immunol Res.
5:234–247. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gatault S, Delbeke M, Driss V, Sarazin A,
Dendooven A, Kahn JE, Lefèvre G and Capron M: IL-18 is involved in
eosinophil-mediated tumoricidal activity against a colon carcinoma
cell line by upregulating LFA-1 and ICAM-1. J Immunol.
195:2483–2492. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wohler J, Bullard D, Schoeb T and Barnum
S: LFA-1 is critical for regulatory T cell homeostasis and
function. Mol Immunol. 46:2424–2428. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mannion BA, Berditchevski F, Kraeft SK,
Chen LB and Hemler ME: Transmembrane-4 superfamily proteins CD81
(TAPA-1), CD82, CD63, and CD53 specifically associated with
integrin alpha 4 beta 1 (CD49d/CD29). J Immunol. 157:2039–2047.
1996.PubMed/NCBI
|
|
50
|
Lee H, Bae S, Jang J, Choi BW, Park CS,
Park JS, Lee SH and Yoon Y: CD53, a suppressor of inflammatory
cytokine production, is associated with population asthma risk via
the functional promoter polymorphism −1560 C>T. Biochim Biophys
Acta. 1830:3011–3018. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mollinedo F, Fontan G, Barasoain I and
Lazo PA: Recurrent infectious diseases in human CD53 deficiency.
Clin Diagn Lab Immunol. 4:229–231. 1997. View Article : Google Scholar : PubMed/NCBI
|