Introduction
Lung cancer is associated with one of the highest
rates of mortality worldwide (1).
Globally, lung cancer burden rose to 2.094 million new cases and
1.8 million cancer deaths in 2018 (2). Lung cancer is typically divided into
two major subtypes: Small cell lung cancer and non-small cell lung
cancer (NSCLC) (3). NSCLC accounts
for ~80–85% of all lung cancer cases, where >60% of patients
with NSCLC present with locally advanced or advanced stages of the
disease at the time of diagnosis (4). Platinum-based chemotherapy is currently
the treatment of choice for patients with NSCLC (5). Cisplatin is a small molecule that is
widely applied as a chemotherapeutic agent (6). Cisplatin binds to and crosslinks DNA,
thereby disrupting DNA function, inhibiting mitosis and
subsequently inducing apoptosis (7,8).
However, development of resistance to cisplatin has become a major
obstacle for lung cancer treatment, the mechanism of which remains
unknown, to the best of our knowledge. Accumulating evidence
suggests that cisplatin resistance occurs following the
dysregulation of gene transcription, which reduces the cellular
accumulation of cisplatin, in turn inhibiting apoptosis,
potentiating DNA repair and upregulating pro-survival signaling
pathways to increase cell proliferation (9,10).
Therefore, understanding cisplatin resistance is necessary for the
successful chemotherapeutic intervention of NSCLC.
Although a number of genes that are associated with
the induction of resistance have previously been identified from
differential gene expression profiles, identification of additional
genes is required to illustrate the mechanism underlying cisplatin
resistance. Bcl-2-associated transcription factor 1 (BCLAF1) was
initially identified as a protein that interacts with the
anti-apoptotic members of the Bcl-2 family (11). Subsequent studies have shown that
BCLAF1 serves a key role in a wide range of physiological
processes, including apoptosis, lung development, DNA repair and
transcriptional regulation (12–14).
Overexpression of BCLAF1 has previously been reported to induce
apoptosis in a manner that could be reversed by expression of the
anti-apoptotic protein Bcl-2 (15,16).
BCLAF1 has also been demonstrated to bind to BRCA1 in mediating
resistance to DNA damage and formation of BRCA1-mRNA splicing
complexes (17,18). The documented multifaceted function
of BCLAF1 in apoptosis, DNA repair and transcriptional regulation
raises the possibility that BCLAF1 may also serve a crucial role in
lung cancer cisplatin resistance. Therefore, in the present study,
the potential effect of BCLAF on the induction of cisplatin
resistance in NSCLC cells was explored.
Materials and methods
Cell lines
The human lung adenocarcinoma cell line
A549/wild-type (WT) (The Cell Bank of Type Culture Collection of
Chinese Academy of Sciences) and the cisplatin-resistant
counterpart (A549/DDP), which were obtained by high-dose cisplatin
shock ten times. When the fusion degree of A549 cells reached 70%,
100 µM cisplatin was added to the medium for 1 h and then these
cells were cultured in fresh medium. Repeat this ten times.
Cisplatin was purchased from Sigma-Aldrich; Merck KGaA. Both cell
lines were maintained in DMEM (Sigma-Aldrich; Merck KGaA)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 10 µl/ml penicillin-streptomycin solution
(Hyclone; GE Healthcare Life Sciences) and cultivated at 37°C in 5%
CO2. In addition, the A549/DDP cell medium contained 4
µM cisplatin to maintain the drug-resistant phenotype. All cells
used for the experiments were in logarithmic phases of growth.
Transfection of small interferring RNA
(siRNA) and plasmid
To inhibit the expression of BCLAF1 and USP22,
negative control siRNA, USP22-siRNA and BCLAF1-siRNA were purchased
from Shanghai GenePharma Co., Ltd. The same negative control siRNA
was used for both BCLAF1 and USP22 siRNA transfections. The
sequences were as follows: Negative control siRNA,
5′-UUACAGAGTAACTUCUUAUC-3′; BCLAF1-siRNA-1,
5′-UCACAUUCUUCAAGGUCAATT-3′; and BCLAF1-siRNA-2,
5′-CCGGUCAUAUAGAUCUUCUTT-3′. The sequences of USP22-siRNA-1 and
USP22-siRNA-2 were as follows: 5′-CACGGACAGUCUCAACAAUTT-3′ and
5′-GGAGAAAGAUCACCUCGAATT-3′. In total, 200 pmol BCLAF1-siRNA or
ubiquitin-specific peptidase 22 (USP22)-siRNA was transfected into
A549/WT and A549/DDP cells using Lipofectamine® 2000
(Thermo Fisher Scientific, Inc.) according to manufacturer's
protocol. 200 pmol negative control siRNA was also transfected into
A549/WT cells and A549/DDP using Lipofectamine® 2000
(Thermo Fisher Scientific, Inc.). For overexpression of BCLAF1,
full length cDNA of BCLAF1 was cloned into the pS-Flag-SBP vector
(Addgene, Inc.). An empty vector was used as the negative control.
pS-Flag-SBP vector was obtained as previously described (19). A solution containing 5 µg plasmid and
10 µl ViaFect™ Transfection Reagent (Promega Corporation) was
transfected into A549 or A549/DDP cells. Follow up experiments were
performed 24 h after transfection.
Cell viability assay
The in vitro chemosensitivity of cells was
analyzed using MTT assay. Briefly, cells were seeded into sterile
96-well plates at a density of 5×103 cells/well followed
by attachment overnight. The cells were then treated with different
concentrations (6.25, 12.5, 25, 50 and 100 µM) of cisplatin
for 36 h at room temperature, following which 10 µl MTT was added
to each well. Following 4 h MTT incubation, 10% SDS was added into
each well and the cells were cultivated at 37°C in 5%
CO2 overnight. A spectrophotometer was then used to
measure absorbance in each well at 570 nm. MTT experiments were
performed at least three times.
Colony formation assays
A549/WT and A549/DDP cells transfected with NC or
BCLAF1 siRNA were plated in 6-well culture dishes at a density of
5×104 cells/ml and exposed to 40 µM cisplatin
continuously for 36 h at room temperature. The cells were fixed
with methanol for 15 min at roon temperature and stained with 1%
crystal violet for 15 min at room temperature. Cells were observed
with a fluorescence microscope (Olympus-BX53; Olympus Corporation;
magnification, ×40).
Reverse transcription-quantitative PCR
(RT-qPCR)
For RNA extraction and RT-qPCR, total RNA was
isolated from A549/WT and A549/DDP cultured cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). cDNA was synthsized from total RNA using M-MLV Reverse
Transcriptase (Promega Corporation) according to the manufacturer's
protocols. Subsequent qPCR reactions were then performed in a
volume of 10 µl that included 1 µl cDNA template, 0.8
µl 10 µM primers (contains forward and reverse primers), 0.8
µl 50X ROX Reference Dye (used to calibrate the instrument),
5 µl 2X TB Green® Premix Ex Taq™ II (Tli RNaseH
Plus; Takara Bio, Inc.) and 3 µl ddH2O. The
thermocycling conditions were 95°C for 5 sec and 60°C for 30 sec
for 45 cycles. The sequences of primers used were as follows:
Homo-GAPDH forward, 5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse,
5′-TGGTGAAGACGCCAGTGGA-3′; USP22 forward,
5′-GGAAAATGCAAGGCGTTGGAGA-3′ and reverse,
5′-GTGCAGTTCGAGGTGATCTTT-3′; p21 forward,
5′-CATGCCAGCTACTTCCTCCT-3′ and reverse, 5′-CAGGTCTGAGTGTCCAGGAA-3′;
and BCLAF1 forward, 5′-TCTGGAATAGAAGGCACTCTAGG-3′ and reverse,
5′-ACCCTCGTCTTTTAGAAACAGGA-3′. The instrument used was CFX96™
Real-Time PCR Detection system (Bio-Rad Laboratories, Inc.). The
relative mRNA expression was quantified using the 2−ΔΔCq
method (20), where the expression
level of GAPDH mRNA was used as an internal control for
normalization. All experiments were performed in duplicate, each
being repeated a minimum of three times.
Immunofluorescence
A549/WT and A549/DDP cells treated with 4 µM
cisplatin were incubated at 37°C in 5% CO2 for 24 h,
before being fixed with 3% paraformaldehyde for 20 min at room
temperature. The cells were then incubated with 0.5% Triton X-100
for 10 min at room temperature, following which they were blocked
with 10% goat serum (Sangon Biotech) for 1 h at room temperature
before incubation with the anti-γH2A histone family member X
(γH2AX) antibody (1:1,000; cat. no. D7T2V; Cell Signaling
Technology, Inc.) or the anti-BCLAF1 (1:1,000; cat. no. A300-608A;
Bethyl Laboratories) antibody dissolved in PBS containing 5% goat
serum at 4°C overnight. The next day, the cells were stained
with the secondary antibodies FITC AffiniPure goat anti-rabbit IgG
(H+L) (1:200; cat. no. 111-095-144; Jackson Immunoresearch) and
AffiniPure goat anti-mouse IgG (H+L) (1:200; cat. no. 115-025-146;
Jackson Immunoresearch) dissolved in PBS containing 5% goat serum
at room temperature for 1 h. Nuclei were then stained using DAPI
(Invitrogen; Thermo Fisher Scientific, Inc.) for 5 min at room
temperature. A fluorescence microscope was used (magnification,
×600). Quantitative image analysis was performed using Image J
version 1.8.0 (National Institutes of Health).
Western blot analysis
Total proteins of A549 or A549/DDP cells were
extracted using an effective lysing fraction of 1% Triton X-100
dissolved in RIPA lysis buffer (EpiZyme Biotech). An appropriate
amount of lysate was incubated with a protease inhibitor (1:100)
for 1 min before use. The protein concentration was determined
using a bicinchonininc acid (BCA) assay (Thermo Fisher Scientific
Inc.). The protein samples (20 µg of protein loaded per lane) were
separated by 7.5 or 15%; SDS-PAGE (90V, 90 min) and transferred
(70V, 90 min) onto PVDF membranes (Immobilon-P; EMD Millipore).
Membranes were blocked with a TBS buffer (50 mM Tris-HCl, 150 mM
NaCl, pH 7.4) supplemented with 5% skimmed milk and 0.1% Tween-20
for 30 min at room temperature, followed by incubation with primary
antibodies against BCLAF1 (1:2,500; cat. no. A300-608A; Bethyl
Laboratories), p21 (1:2,500; cat. no. ab109520; Abcam), BTB domain
and CNC homolog 1 (BACH1; 1:2,500; cat. no. 4578s; Cell Signaling
Technology, Inc.), cyclin D1 (1:2,500; cat. no. 55506; Cell
Signaling Technology, Inc.), BRCA1 (1:2,500; cat. no. 14823; Cell
Signaling Technology, Inc.), γH2AX (1:2,500; cat. no. D7T2V; Cell
Signaling Technology, Inc.), USP22 (1:2,500; cat. no. EPR18945;
Abcam) or β-actin (1:2,500; cat. no. IPSC1030, Sigma-Aldrich, Merck
KgaA) at 4°C overnight. Membranes were incubated with the secondary
antibodies goat anti-mouse IgG (H+L)-HRP (1:5,000; cat. no. PB001;
CASICO) and mouse anti-rabbit IgG, light chain specific (1:5,000;
cat. no. 211-032-171; Jackson Immunoresearch) at room temperature
for 1 h. For visualization, membranes were incubated with the Light
Chemiluminescence (Omni-ECL™ Femto Light Chemiluminescence kit;
cat. no. SQ201; EpiZyme Biotech). Western blot results were
analyzed by Image J version 1.8.0 (National Institutes of
Health).
Cell cycle analysis
A549/DDP cells transfected with either BCLAF1 siRNA
or negative control siRNA were used for cell cycle analysis. Cell
cycle progression was determined by propidium iodide (PI) staining
using a flow cytometer. Briefly, cells were fixed with 70% cold
ethanol at 4°C overnight, washed twice with ice-cold PBS, and
incubated with 10 mg/ml RNase at 37°C. Cell cycle was monitored by
PI staining of nuclei for 30 min at room temperature, and PI uptake
was analyzed by fluorescence-activated cell sorting using a flow
cytometer (CyAn™ ADP Analyzer; Beckman Coulter, Inc.). Results were
analyzed by Modfit LT version 5.0 (Verity Software House,
Inc.).
Statistical analysis
All statistical analyses were performed using
GraphPad Prism software (version 7; GraphPad Software, Inc.). Data
are presented as the mean ± standard devaition from at least three
independent experiments. Statistical significance was determined by
one-way analysis of variance followed by Bonferroni's test.
Comparisons between two group were performed by unpaired Student's
t-test. P<0.05 was considered to indicate a significantly
significant difference.
Results
BCLAF1 expression is upregulated in
the cisplatin-resistant A549/DDP cell line
To study the chemoresistance of NSCLC,
cisplatin-resistant NSCLC cell line models (A549/DDP) were first
established by treating the A549 cell line with cisplatin for 2
months. Surviving cells after this period were then tested for
cisplatin sensitivity using a MTT assay. Results from the MTT assay
demonstrated that A549/DDP cells exhibited a significantly
increased cisplatin resistance compared with that of the parental
A549 cell line (Fig. 1A), and the
IC50 value of cisplatin for A549/DDP was 1.2-fold higher
compared with that for A549/WT (119.6 µM vs. 54 µM).
 | Figure 1.BCLAF1 expression is upregulated in
A549/DDP cells. (A) MTT assays were used to measure the sensitivity
of A549/WT and A549/DDP cells to cisplatin. Cells were treated with
different concentrations of cisplatin for 36 h. *P<0.05,
**P<0.01, ***P<0.001 vs. A549/WT. (B) BRCA1, BACH1 and BCLAF1
protein expression levels were measured in A549/WT and A549/DDP
cells by western blotting and (C) were quantified. *P<0.05,
***P<0.001. (D) A549/WT and A549/DDP cells were treated
continuously with either 0 or 4 µM cisplatin for 10 h, following
which BRCA1, BACH1 and BCLAF1 protein expression were examined by
western blotting. (E) BCLAF1 mRNA expression was measured using
reverse transcription- quantitative PCR in A549/WT and A549/DDP
cells. Data are presented as the means ± SD from three independent
experimental repeats. *P<0.05, ***P<0.001. DDP, cisplatin
resistance; WT, wild-type; BCLAF1, bcl-2- associated transcription
factor 1; BACH1, BTB domain and CNC homolog 1. |
It has previously been reported that increased
tolerance to DNA damage is one of the major mechanisms of cisplatin
resistance (10,21,22).
Therefore, the expression levels of several key components of the
DNA damage response, including BRCA1, BACH1 and BCLAF1, were
examined in A549/DDP cells. No change in the expression level of
BACH1 was observed in A549/DDP cells compared with in A549 cells,
whilst both BRCA1 and BACH1 protein expression was increased in
A549/DDP cells compared with A549/WT cells (Fig. 1B-D). When compared with A549/WT
cells, in A549/DDP cells the protein expression level of BCLAF1 is
significantly higher than that of BRCA1, hence the functional link
between BCLAF1 and cisplatin resistance in NSCLC was further
explored. When BCLAF1 levels were investigated further it was found
that BCLAF1 mRNA level also significantly higher in the
cisplatin-resistant A549/DDP cells compared with A549/WT cells
(Fig. 1E).
Elevated BCLAF1 expression levels are
associated with acquired cisplatin resistance in A549 cells
BCLAF1 is a multifunctional protein that has been
implicated in a number of cellular functions, including apoptosis
(16), lung development and
transcriptional regulation (14,23). As
aforementioned, the present study demonstrated that BCLAF1
expression levels were significantly higher in A549/DDP cells
compared with in A549 cells, prompting further examination into
whether BCLAF1 exerts an effect on cisplatin resistance in A549/DDP
cells. Therefore, the association between BCLAF1 expression and
cisplatin sensitivity in NSCLC cell lines was next evaluated by
first testing the effects of BCALF1 overexpression on cisplatin
resistance in A549 cells. A549 cells transfected with the BCLAF1
overexpression vector or the empty vector were treated with
cisplatin, following which cisplatin sensitivity was determined
using MTT assay. The IC50 for cisplatin in A549 cells
transfected with the empty vector was calculated to be 52.3
µM. By contrast, following overexpression of BCLAF1 the
IC50 was 84.61 µM, and cell viability was found
to be significantly increased (Fig.
2A). These observations suggest that the upregulation of BCLAF1
can promote cisplatin resistance in A549 cells.
 | Figure 2.Elevated BCLAF1 expression levels are
associated with acquired cisplatin resistance in A549 cells. (A)
MTT assay was used to measure the sensitivity to cisplatin in
A549/WT cells transfected with either the SBP-vector or SBP-BCLAF1.
Cells stably expressing the SBP-vector or SBP-BCLAF1 were treated
with different concentrations of cisplatin for 36 h. *P<0.05 vs.
A549/WT + SBP-vector. (B) MTT assays were used to measure the
sensitivity to cisplatin in A549/WT cells transfected with siCTRL
or siBCLAF1 and in A549/DDP cells transfected with siCTRL or
siBCLAF1, following which they were treated with different
concentrations of cisplatin for 36 h. *P<0.05, **P<0.01. (C)
The colony-forming ability of A549/WT cells transfected with siCTRL
or siBCLAF1 and in A549/DDP cells transfected with siCTRL or
siBCLAF1 to cisplatin was examined after exposure to 40 µM
cisplatin continuously for 36 h. (D) BCLAF1 protein expression were
measured by western blotting in A549/WT cells transfected with
siCTRL, siBCLAF1, SBP-vector or SBP-BRCAF1 and in A549/DDP cells
transfected with siCTRL or siBCLAF1. Data are presented as the
means ± SD from three independent experimental repeats. SBP,
streptavidin-binding peptide; DDP, cisplatin resistance; WT,
wild-type; BCLAF1, bcl-2-associated transcription factor 1; si,
small interfering; CTRL, control. |
MTT assays subsequently demonstrated that knockdown
of BCLAF1 expression using siRNA reduced the IC50 of
A549/WT cells from 66.52 to 39.9 µM, and cell viability was
significantly reduced (Fig. 2B),
supporting the notion further that the depletion of BCLAF1 can
reverse the cisplatin resistance of A549/DDP cells. Data from the
colony formation assays following cisplatin treatment showed that
BCLAF1-knockdown significantly reduced A549 cell viability
(Fig. 2C and D), proving that BCLAF1
can promote A549 cell viability. Furthermore, in A549/DDP cells,
knockdown of BCLAF1 expression reduced the IC50 for
cisplatin from 122.59 to 38.58 µM, and significantly
inhibited cell viability (Fig. 2B).
These results suggested that BCLAF1 is a key component in mediating
cisplatin sensitivity in A549/DDP cells.
DNA damage repair in A549/DDP cells is
under the regulation of BCLAF1
It has previously been reported that BCLAF1 is
involved in the γH2AX-mediated regulation of apoptosis and
DNA repair (16). Therefore, the
effects of BCLAF1 on DNA damage repair and γH2AX foci
formation were examined in A549/DDP cells. Since phosphorylation of
γH2AX at the ser-139 residue is a sensitive early genotoxic
biomarker in cisplatin-induced DNA double strand breaks (DSBs)
(24,25), the relationship between γH2AX foci
formation and the levels of BCLAF1 expression were examined further
to explore the effects of BCLAF1 on cisplatin-induced DNA damage
repair.
Following cisplatin treatment, the number of γH2AX
foci in A549/DDP cells was found to be significantly lower compared
with that in A549/WT cells, though the intensity of nuclear BCLAF1
staining was observed to be significantly higher in A549/DDP cells
(Fig. 3A-B). This suggests that
elevated BCLAF1 expression in A549/DDP cells enhances DSB repair
and thus induces cisplatin resistance. Next, to verify the
association between BCLAF1 expression and DNA damage repair
following treatment with cisplatin, γH2AX foci formation were
examined in A549/DDP cells following the depletion of BCLAF1
expression (Fig. 3A and B). The
results of western blotting further verified the aforementioned
result (Fig. 3C and D). The results
demonstrated that BCLAF1-knockdown in A549/DDP cells significantly
increased the number of cisplatin-induced γH2AX foci, suggesting
that BCLAF1 induces cisplatin resistance by facilitating DSB repair
in A549/DDP cells.
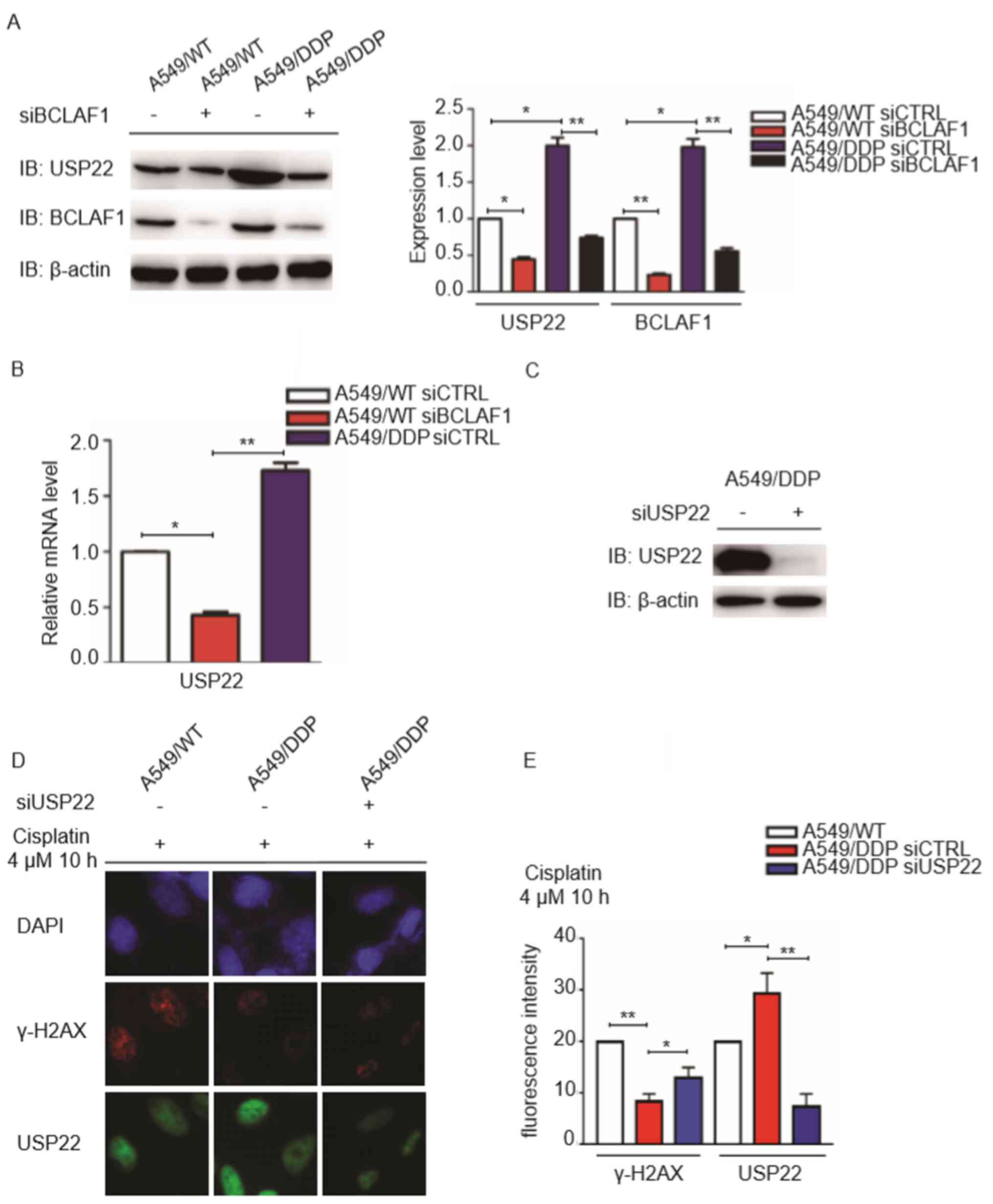
Expression of BCLAF1 associates with
that of USP22 in A549/DDP cells
It has been reported previously that USP22
overexpression can mediate cisplatin resistance in A549 cells,
where it modulates Ku70/BAX-dependent apoptosis and γH2AX-mediated
DNA damage repair (26). Since
BCLAF1 overexpression was also observed to enhance cisplatin
resistance in A549 cells, the association between the expression
levels of BCLAF1 and USP22 was next analyzed in A549/DDP cells. The
results demonstrated that the protein expression levels of USP22
and BCLAF1 were significantly higher in A549/DDP cells compared
with those in A549/WT cells (Fig. 4A and
B). Notably, following knockdown of BCLAF1 expression, USP22
protein expression was also found to be significantly reduced in
A549/DDP cells (Fig. 4A). These
results indicated a positive association between BCLAF1 and USP22
expression. As BCLAF1 can also function as a transcriptional factor
to regulate target gene expression (27), the hypothesis that BCLAF1 can
directly regulate USP22 expression was considered. Supporting this,
USP22 mRNA expression was found to be significantly reduced
following BCLAF1 downregulation in A549/WT cells. By contrast,
USP22 mRNA expression was observed to be markedly elevated in
A549/DDP cells with higher expression of BCLAF1 (Fig. 4A and B), suggesting that BCLAF1
regulates USP22 expression at the mRNA level. Following USP22
knockdown in A549/DDP (Fig. 4C), it
was subsequently found that USP22-knockdown in A549/DDP cells
significantly increased the number of cisplatin-induced γH2AX foci
(Fig. 4D and E), suggesting that
BCLAF1 can modulate USP22 expression to facilitate DNA damage
repair.
BCLAF1 regulates G1 phase
cell cycle arrest by targeting p21 expression
It has been shown that BCLAF1 co-ordinates with
BRCA1 as a component of the RNA splicing complex, which regulates
the stability of cyclin D1 and BRCA1 mRNA (17). Since BCLAF1 expression was found to
be increased in A549/DDP cells, the expression levels of cyclin D1
and p21 in A549/WT and A549/DDP cells with or without cisplatin
treatment were compared. Following cisplatin treatment, the
expression levels of BCLAF1 and p21 were revealed to be increased
in A549/DDP cells, whilst cyclin D1 expression were demonstrated to
be reduced (Fig. 5A). Depletion of
BCLAF1 expression in A549/DDP cells lead to a reduction in p21
expression but increase in cyclin D1 expression (Fig. 5B. These findings suggest that BCLAF1
serves an important role in the regulation of p21 and cyclin D1
gene expression.
Elevated p21 protein levels have previously been
documented to inhibit cyclin D-Cyclin-dependent kinase (CDK) 4/6
activity, contributing to G1 phase cell cycle arrest
(28–30). The p21 mRNA level was decreased in
A549/DDP cells depletion of BCLAF1 (Fig.
5C). Therefore, the cell cycle status of A549/WT and A549/DDP
cells was subsequently characterized. A549/DDP cells compared with
A549/WT cells were more arrested at the G1 phase
(Fig. 5D). To assess if BCLAF1 may
also serve a role in regulating cell cycle progression in A549/DDP
cells, the effect of BCLAF1-knockdown on cell cycle progression was
also examined. Notably, depletion of BCLAF expression was
demonstrated to reduce G1 arrest by 0.86-fold in
A549/DDP cells (Fig. 5D). These
results suggested that BCLAF1 can induce G1 phase cell
cycle arrest by regulating p21 and cyclin D1 expression.
Discussion
Lung cancer has one of the highest rates of
incidence and is the leading cause of cancer-associated mortality
worldwide (1). Although chemotherapy
can be an effective therapeutic intervention strategy for the
treatment of NSCLC, NSCLC gradually develops resistance to
chemotherapeutic agents, such as cisplatin (3,5); the
underlying molecular mechanism of which remains poorly understood.
In the present study, elevated BCLAF1 expression was observed in
cisplatin-resistant A549/DDP cells, whilst downregulation of BCLAF1
expression was found to reverse cisplatin resistance in A549/DDP
cells, suggesting that BCLAF1 serves an important role in the
regulation of cisplatin resistance in NSCLC.
BCLAF1 has previously been reported to be involved
in a number of biological processes (12,13,31). It
regulates gene transcription by mediating the formation of
BRCA1-mRNA splicing complexes and modulating the DNA damage
response by stabilizing the Ku70/DNA-dependent protein kinase
complex during non-homologous end joining (NHEJ) (17). Since increased repair of drug-induced
DNA damage and cell cycle alterations are two major mechanisms
underlying chemotherapeutic resistance (32–34), the
potential function of BCLAF1 on these two processes was
investigated in A549/DDP cells in the present study. The key
finding of the present study is that increased BCLAF1 expression in
A549/DDP cells accelerated DNA damage repair and cell cycle
progression. These observations suggest that BCLAF1 can induce
cisplatin resistance in lung cancer cells by regulating the cell
cycle and DNA damage repair.
It has been demonstrated that BCLAF1 interacts with
γH2AX to stabilize the complex, promoting NHEJ-based DSB repair in
cancer cells following irradiation (14,16). A
previous study revealed that BCLAF1 interacts with BRCA1 and
thyroid hormone receptor associated protein 3, forming a
BRCA1-interacting RNA splicing complex in response to DNA damage
(17). This complex functions as a
transcriptional regulator to selectively control the expression of
a subset of genes associated with the DNA damage response,
promoting efficient homologous recombination (HR)-mediated repair
and repair of DNA interstrand crosslinks (14). These previous findings suggest that
BCLAF1 serves a crucial role in the repair of double stranded
breaks not only by NHEJ repair but also by HR repair. The current
study further suggested elevated expression of BCLAF1 conferred
cisplatin resistance in A549/DDP cells by increasing DNA repair
capacity.
In addition to its role in the DNA damage response,
BCLAF1 can also indirectly promote changes in cell cycle
progression or the DNA damage response through transcriptional
regulation (27). BCLAF1 is an
essential component of a BRCA1-mRNA splicing complex (14,17). In
the present study, it was found that the loss of BCLAF1 expression
in A549/DDP cells lead to reductions in USP22 and p21 expression,
which partially supports the role of BCLAF1 in selective mRNA
splicing and the export of mRNA encoding key DNA damage response
proteins. Increasing BCLAF1 expression was revealed to contribute
to cisplatin resistance by targeting p21 and cyclin D1 expression.
p21 is a negative regulator of cyclin D-CDK4/6 activity and is
sufficient to inhibit cell cycle progression during the
G1 and S phases (30,35,36).
Elevated p21 expression protected A549/DDP cells from
cisplatin-induced apoptosis by inhibiting DNA synthesis during S
phase, suggesting that one potentially important mechanism for
acquired cisplatin resistance is that high expression levels of
BCLAF1 can inhibit DNA synthesis through p21-induced G1
arrest. The present study had limitations. No clinical samples and
clinically relevant conditions were investigated hence clinical
relevance was not assessed. The findings of the present study
indicate that BCLAF1 is likely a novel target mediating cisplatin
resistance. Future studies targeting BCLAF1 in therapeutic practice
are required to determine the true role of BCLAF1 in tumor
suppression.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from National
Natural Science Foundation of China (grant nos. 81572775 and
81773004 to FZ) and the Program for Professor of Special
Appointment (Eastern Scholar) at Shanghai Institutions of Higher
Learning (grant no. TP2014055 to FZ).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TJ and BL performed the experiments, analyzed the
data, created the figures and wrote the manuscript. FZ and DW
conceived the project, designed the experiments and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang J, Zhang ZQ, Li FQ, Chen JN, Gong X,
Cao BB and Wang W: Triptolide interrupts rRNA synthesis and induces
the RPL23MDM2p53 pathway to repress lung cancer cells. Oncol Rep.
43:1863–1874. 2020.PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morgensztern D, Campo MJ, Dahlberg SE,
Doebele RC, Garon E, Gerber DE, Goldberg SB, Hammerman PS, Heist
RS, Hensing T, et al: Molecularly targeted therapies in
non-small-cell lung cancer annual update 2014. J Thorac Oncol. 10
(Suppl 1):S1–S63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Janssen-Heijnen ML, van Erning FN, De
Ruysscher DK, Coebergh JW and Groen HJ: Variation in causes of
death in patients with non-small cell lung cancer according to
stage and time since diagnosis. Ann Oncol. 26:902–907. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zappa C and Mousa SA: Non-small cell lung
cancer: Current treatment and future advances. Transl Lung Cancer
Res. 5:288–300. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fennell DA, Summers Y, Cadranel J, Benepal
T, Christoph DC, Lal R, Das M, Maxwell F, Visseren-Grul C and Ferry
D: Cisplatin in the modern era: The backbone of first-line
chemotherapy for non-small cell lung cancer. Cancer Treat Rev.
44:42–50. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Perse M and Večerić-Haler Z:
Cisplatin-induced rodent model of kidney injury: Characteristics
and challenges. Biomed Res Int. 2018:14628022018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kartalou M and Essigmann JM: Mechanisms of
resistance to cisplatin. Mutat Res. 478:23–43. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rocha CRR, Silva MM, Quinet A, Cabral-Neto
JB and Menck CFM: DNA repair pathways and cisplatin resistance: An
intimate relationship. Clinics (Sao Paulo). 73 (Suppl 1):e478s2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kasof GM, Goyal L and White E: Btf, a
novel death-promoting transcriptional repressor that interacts with
Bcl-2-related proteins. Mol Cell Biol. 19:4390–4404. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sarras H, Azami SA and McPherson JP: In
search of a function for BCLAF1. ScientificWorldJournal.
10:1450–1461. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wen Y, Zhou X, Lu M, He M, Tian Y, Liu L,
Wang M, Tan W, Deng Y, Yang X, et al: Bclaf1 promotes angiogenesis
by regulating HIF-1α transcription in hepatocellular carcinoma.
Oncogene. 38:1845–1859. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vohhodina J, Barros EM, Savage AL,
Liberante FG, Manti L, Bankhead P, Cosgrove N, Madden AF, Harkin DP
and Savage KI: The RNA processing factors THRAP3 and BCLAF1 promote
the DNA damage response through selective mRNA splicing and nuclear
export. Nucleic Acids Res. 45:12816–12833. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li X, He Z, Cheng B, Fang Q, Ma D, Lu T,
Wei D, Kuang X, Tang S, Xiong J and Wang J: Effect of BCLAF1 on
HDAC inhibitor LMK-235-mediated apoptosis of diffuse large B cell
lymphoma cells and its mechanism. Cancer Biol Ther. 19:825–834.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee YY, Yu YB, Gunawardena HP, Xie L and
Chen X: BCLAF1 is a radiation-induced H2AX-interacting partner
involved in γH2AX-mediated regulation of apoptosis and DNA repair.
Cell Death Dis. 3:e3592012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Savage KI, Gorski JJ, Barros EM, Irwin GW,
Manti L, Powell AJ, Pellagatti A, Lukashchuk N, McCance DJ,
McCluggage WG, et al: Identification of a BRCA1-mRNA splicing
complex required for efficient DNA repair and maintenance of
genomic stability. Mol Cell. 54:445–459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Carmody SR and Wente SR: mRNA nuclear
export at a glance. J Cell Sci. 122:1933–1937. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang F, Wu J and Yu X: Integrator3, a
partner of single-stranded DNA-binding protein 1, participates in
the DNA damage response. J Biol Chem. 284:30408–30415. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen P, Li J, Chen YC, Qian H, Chen YJ, Su
JY, Wu M and Lan T: The functional status of DNA repair pathways
determines the sensitization effect to cisplatin in non-small cell
lung cancer cells. Cell Oncol (Dordr). 39:511–522. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qin C, Zhang R, Lang Y, Shao A, Xu A, Feng
W, Han J, Wang M, He W, Yu C and Tang J: Bclaf1 critically
regulates the type I interferon response and is degraded by
alphaherpesvirus US3. PLoS Pathog. 15:e10075592019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kinner A, Wu W, Staudt C and Iliakis G:
Gamma-H2AX in recognition and signaling of DNA double-strand breaks
in the context of chromatin. Nucleic Acids Res. 36:5678–5694. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fragkos M, Jurvansuu J and Beard P: H2AX
is required for cell cycle arrest via the p53/p21 pathway. Mol Cell
Biol. 29:2828–2840. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang A, Ning Z, Lu C, Gao W, Liang J, Yan
Q, Tan G and Liu J: USP22 induces cisplatin resistance in lung
adenocarcinoma by regulating γH2AX-mediated DNA damage repair and
Ku70/Bax-mediated apoptosis. Front Pharmacol. 8:2742017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Meng X, Yang S and Camp VJA: The interplay
between the DNA damage response, RNA processing and extracellular
vesicles. Front Oncol. 9:15382020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang Y, Liu C, Zeng WC, Xu GY, Wu JM, Li
ZW, Huang XY, Lin RJ and Shi X: Isoliquiritigenin inhibits the
proliferation, migration and metastasis of Hep3B cells via
suppressing cyclin D1 and PI3K/AKT pathway. Biosci Rep.
40:BSR201927272020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen S, Zhou Q, Guo Z, Wang Y, Wang L, Liu
X, Lu M, Ju L, Xiao Y and Wang X: Inhibition of MELK produces
potential anti-tumour effects in bladder cancer by inducing G1/S
cell cycle arrest via the ATM/CHK2/p53 pathway. J Cell Mol Med.
24:1804–1821. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kreis NN, Louwen F and Yuan J: The
multifaceted p21 (Cip1/Waf1/CDKN1A) in cell differentiation,
migration and cancer therapy. Cancers (Basel). 11:12202019.
View Article : Google Scholar
|
|
31
|
Dell'Aversana C, Giorgio C, D'Amato L,
Lania G, Matarese F, Saeed S, Costanzo AD, Petrizzi VB, Ingenito C,
Martens JHA, et al: miR-194-5p/BCLAF1 deregulation in AML
tumorigenesis. Leukemia. 31:2315–2325. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li HL, Wang CY, Fu J, Yang XJ, Sun Y, Shao
YH, Zhang LH, Yang XM, Zhang XL and Lin J: PTEN expression in U251
glioma cells enhances their sensitivity to ionizing radiation by
suppressing DNA repair capacity. Eur Rev Med Pharmacol Sci.
23:10453–10458. 2019.PubMed/NCBI
|
|
33
|
Kamarudin MNA, Sarker MMR, Zhou JR and
Parhar I: Metformin in colorectal cancer: Molecular mechanism,
preclinical and clinical aspects. J Exp Clin Cancer Res.
38:4912019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yilmaz TE, Taşdemir M, Kaya M, Arican N
and Ahishali B: The effects of magnesium sulfate on
cyclophosphamide-induced ovarian damage: Folliculogenesis. Acta
Histochem. 122:1514702020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Warfel NA and El-Deiry WS: p21WAF1 and
tumourigenesis: 20 years after. Curr Opin Oncol. 25:52–58. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|