Introduction
Surfactant protein D (SP-D) is a member of the
collectin family of proteins secreted by pulmonary epithelial
cells. SP-D reduces the surface tension at the alveolar air-liquid
interface and prevents the lungs from collapsing at the end of
expiration (1). There are four types
of surfactant proteins: SP-A, SP-B, SP-C, and SP-D. SP-A and SP-D
are hydrophilic proteins that are involved in the immune and
inflammatory regulation of the lungs (2,3).
The expression of SP-D is highest in the lungs
(4). SP-D is also found in the
systemic circulatory system, but its role in this system is unknown
(5). Smoking damages the alveolar-
capillary barrier, allowing SP-D to leak into the systemic
circulatory system. Consequently, SP-D levels decrease in the lung
and increase in the serum with smoking (6). A similar phenomenon occurs in other
lung inflammatory diseases, such as chronic obstructive pulmonary
disease (COPD) (7), idiopathic
pulmonary fibrosis (8), and
community-acquired pneumonia (9).
Lung cancer related to smoking is promoted by pulmonary
inflammation (10). This raises the
possibility that SP-D may have a regulatory role in the
pathogenesis of lung cancer.
Consistent with this notion, SP-D was recently shown
to antagonize epidermal growth factor receptor (EGFR) by blocking
ligand binding and inhibiting EGFR signaling, which suppressed the
proliferation, migration, and invasion of A549 human lung
adenocarcinoma cells (11). In
vivo lung expression of SP-D is inversely related to the
progression of bronchial dysplasia in smokers (12), while a retrospective clinical study
revealed that higher serum SP-D levels correlated with better
prognosis in selective patients with non-small cell lung cancer
(13). Based on these studies, we
hypothesized that SP-D could also affect pulmonary metastases from
colon cancer.
In this study, we investigated whether SP-D could
inhibit the malignant potential of colon cancer cells in
vitro and in vivo. We developed an endogenous pulmonary
metastasis model of colon cancer in mice and investigated the
ability of SP-D to suppress pulmonary metastases from colon cancer
in vivo. Furthermore, we established novel cell lines from
the mouse rectal cancer cell, CMT-93, which are more susceptible to
pulmonary metastases, and investigated the impact of SP-D on
pulmonary metastases from colon cancer.
Materials and methods
Cell culture
The mouse CMT-93 rectal carcinoma cell line
(CCL-223; ATCC) and the human HCT116 (CCL-247, ATCC) were
maintained in Dulbecco's modified Eagle's medium (DMEM;
Sigma-Aldrich; Merck KGaA) with 10% (v/v) fetal bovine serum (FBS)
and 1% penicillin/streptomycin (Thermo Fisher Scientific, Inc.).
The cells were cultured at 37°C with 5% CO2.
Wound healing assay
The wound healing assay was conducted using a
six-well culture cluster (Corning Incorporated). CMT-93 cells were
seeded into the insert in DMEM with 0.1% (v/v) FCS and grown to
confluence (100%). A wound was introduced into the cells using a
200 µl pipette tip and the recombinant Mouse SP-D Protein (R&D
Systems) was added at concentrations of 0, 5, 10, 15, and 20 µg/ml.
Twenty-four hours later, cells that had migrated into the scraped
areas were counted under a microscope. Five images (2.8×2.0 mm) per
well were captured randomly and the wound areas were calculated
using the ImageJ public domain software (14). The same assay using concentrations of
0, 5, and 10 µg/ml of the recombinant Human SP-D Protein (R&D
Systems) was performed using HCT116 cells.
Cell invasion assays
Cell invasion assays were conducted using multiwall
24-well plates (Corning Incorporated) (15). The upper insert was coated with
Matrigel (200 µg/ml; cat. no. 354234; Corning Life Sciences) and
incubated for 12 h. CMT-93 cells were seeded into the upper insert
in DMEM with 10% (v/v) FBS and 1% penicillin/streptomycin. In the
SP-D group, recombinant SP-D protein was added to the upper insert
at a concentration of 0, 5, 10, 15, and 20 µg/ml. The upper insert
was placed into the lower outer well in DMEM. The set was incubated
at 37°C with 5% CO2 for 36 h. The cells that passed the
Matrigel were stained using the diff-quick method and counted with
a microscope. The invasion ability was defined as the ratio of the
number of the cells that passed through the Matrigel to the number
of the cells that did not pass through the Matrigel. The same assay
using the concentrations of 0, 5, 10 µg/ml SP-D was performed using
HCT116 cells.
Animals
We used C57BL/6 female mice that were purchased from
CLEA Japan. SP-D knockout mice were kindly provided by Dr Don D.
Sin in Vancouver, Canada (16). DNA
was extracted from tail biopsies of mice using DNeasy blood and
tissue kit (Qiagen, Inc.), according to the manufacturer's
protocol. GAPDH was identified by PCR using the following primers:
Forward, 5′-CGACTTCAACAGCAACTCCCACTCTTCC-3′ and reverse,
5′-TGGGTGGTCCAGGGTTTCTTACTCCTT-3′ (Thermo Fisher Scientific, Inc.).
SP-D genotypes were identified by multiplex PCR using the following
primers: Forward, 5′-TGGTTTCTGAGATGGAGTCGT-3′ and reverse,
5′-TGGGGCAGTGGATGGAGTGTGC-3′ and 5′-GTGGATGTGGAATGTGTGCGAG-3′
(Thermo Fisher Scientific, Inc.). PCR was performed with Blend Taq
(Toyobo Life Science) under the following conditions: 95°C for 15
min, followed by 38 cycles at 94°C for 30 sec, 57°C for 30 sec and
72°C for 30 sec, and a final extension step at 72°C for 10 min. The
PCR products were separated by 2% agarose gel electrophoresis and
visualized using ethidium bromide. Wild-type (C57BL/6) mice
contained 0.4 kb PCR products but SP-D knockout mice did not
(Fig. 1). These animals were
acclimatized for at least 7 days before use and were 6 weeks old at
the start of the experimental protocol. All animals were housed in
a controlled environment at the Keio University School of Medicine
under standard temperature and light and dark cycles. All
procedures were performed under the approval of the Laboratory
Animal Care and Use Committee at the Keio University School of
Medicine (approval no. 15006).
Pulmonary metastasis mouse model
The experimental protocol is summarized in Fig. 2. Six-week-old female mice (C57BL/6
mice or SP-D knockout mice) were anesthetized with 2.0% isoflurane,
and 3–5×106 cells of CMT-93 in 100 µl PBS were injected
into the tail vein with a 26-gauge needle (17). Twenty-five of both the control and
SP-D KO mice were used for this experiment. At 8 and 10 weeks
following the tail vein injection, the presence of pulmonary
metastases was evaluated using an in vivo micro CT
(CosmoScan FX, Rigaku) under mild inhalation anesthesia using
isoflurane (Fig. 3).
Three-dimensional microstructural image data were reconstructed
using Tri/3D-BON software (Ratoc System Engineering) (Fig. 3). We compared the number of mice
affected by pulmonary metastases, and the number of pulmonary
metastases in each mouse. All mice were sacrificed at 12 weeks by
exsanguination under systemic anesthesia using 2.0% isoflurane
(Figs. 2 and 3). The lungs were extracted, fixed by
injecting methanol (1 ml) through the trachea, and then embedded
using paraffin.
Hematoxylin-eosin staining
Lung tissues from the mice were embedded in paraffin
and the paraffin blocks were cut into 4-µm thick sections and
stained with hematoxylin-eosin. Pathological evaluation for
pulmonary metastases was performed using the maximum longitudinal
section in the left lobe.
Establishment of the
pulmonary-metastasis-prone colon cancer cells
The pulmonary metastasis mouse model was generated
as described above. All 15 mice were sacrificed and dissected at 10
weeks. Pulmonary metastases were cut into small pieces on the
membrane. Filtered cells from the lung pieces were stirred with
DMEM and incubated at 37°C with 5% CO2. Cell culture was
performed as described for CMT-93. The cells were grown to
confluency and then passaged. After five passages, the cells were
diluted to form monoclonal colonies in 96-well culture clusters
(Corning Incorporated). Cells were further cultured and passaged
more than 10 times. Finally, a novel cell line was established,
which we called CMT-93 pulmonary metastasis (CMT-93 PM). The
molecular characteristics of CMT-93 PM were assessed by a wound
healing assay and invasion assay with or without SP-D. Moreover,
CMT-93 PM and CMT-93 were injected into normal mice and the
incidence of pulmonary metastases was compared.
ELISA
ELISA was used to determine the expression of Akt,
which is downstream of the EGFR signaling pathway, in CMT-93 and
CMT-93 PM. The cells (1×106/sample) were serum starved
overnight and incubated with SP-D (10 µg/ml) for 2 hours at 37°C.
The cell lysate was prepared and was subjected to ELISA using the
Akt (pS473) + total Akt ELISA kits (cat. no. ab126433; Abcam).
Statistical analyses
All results were expressed as the mean value (mean ±
SE). All statistical analyses were performed using Stata software
(Stata Corp.). P<0.05 was considered to indicate a statistically
significant difference. All procedures were performed in
triplicate. Differences in the wound healing assay, cell invasion
assay and the number of metastases per mouse were statistically
analyzed by Mann-Whitney U test or one-way ANOVA. The development
of pulmonary metastases in mice was analyzed using χ2
tests.
Results
SP-D suppresses the proliferation,
migration, and invasion of CMT-93
First, we determined whether the efficacy of SP-D on
the malignant potential of CMT-93 is equivalent to primary lung
adenocarcinoma cells. SP-D significantly suppressed CMT-93 compared
to the control in a wound healing assay, but not in a
dose-dependent manner (Fig. 4). In
an invasion assay, treatment with SP-D significantly decreased the
number of CMT-93 cells that passed through the membrane compared to
untreated cells, but not in a dose-dependent manner (Fig. 5). SP-D similarly suppressed migration
and invasion ability in human HCT116 cells (Fig. 6).
 | Figure 4.Wound healing assay of CMT-93 cells
using SP-D. CMT-93 cells were plated in six-well culture cluster
plates. Wounds were introduced using the 200 µl pipette tips, and
SP-D was added at concentrations of 0, 5, 10, 15 and 20 µg/ml.
After 24 h, cells that had migrated into the wounded areas were
counted under a microscope. Five images (2.8×2.0 mm) per well were
randomly captured, and the wound area was measured using ImageJ.
(A) SP-D significantly suppressed the increase in CMT-93 cells
compared to the control, but the effect was not dose-dependent. The
data shown are mean ± SD. *P<0.05, **P<0.01 compared with the
control. (B) Representative image of cells at 0 h, the scraped
areas without SP-D and the scraped area with 10 µg/ml SP-D. SP-D,
surfactant protein D. |
SP-D suppresses pulmonary metastases
from colon cancer
We used a pulmonary metastasis mouse model with an
SP-D KO to determine if SP-D suppresses pulmonary metastases from
colon cancer. The incidence of pulmonary metastases between SP-D KO
mice and control C57Bl/6 mice was compared by micro-CT and
pathological examination (Fig. 3).
Significantly more SP-D KO mice were affected by pulmonary
metastases compared to the C57BL/6 mice (Table I) and the average number of
metastases per mouse was greater in SP-D KO mice than in C57BL/6
mice in both the CT scans and pathological examinations (Fig. 7).
 | Table I.Number of mice with pulmonary
metastasis, evaluated using CT scan and pathological
examination. |
Table I.
Number of mice with pulmonary
metastasis, evaluated using CT scan and pathological
examination.
| Method | SP-D KO mice, n=24
(%) | C57BL/6 mice, n=23
(%) | P-value |
|---|
| CT scan | 11 (45.8) | 4 (17.4) | 0.037 |
| Pathological
examination | 15 (62.5) | 5 (21.7) | 0.005 |
Establishment of a novel colon cell
line, CMT-93 PM
To clarify the mechanism of pulmonary metastasis
development and the influence of SP-D, we established a novel colon
cancer cell line, CMT-93 PM, which was predisposed to developing
pulmonary metastases. Two clones of CMT-93 PM, called CMT-93 PM
(1) and CMT-93 PM (2), were identified. A wound healing assay
showed that the proliferative capacity of CMT-93 PM was higher than
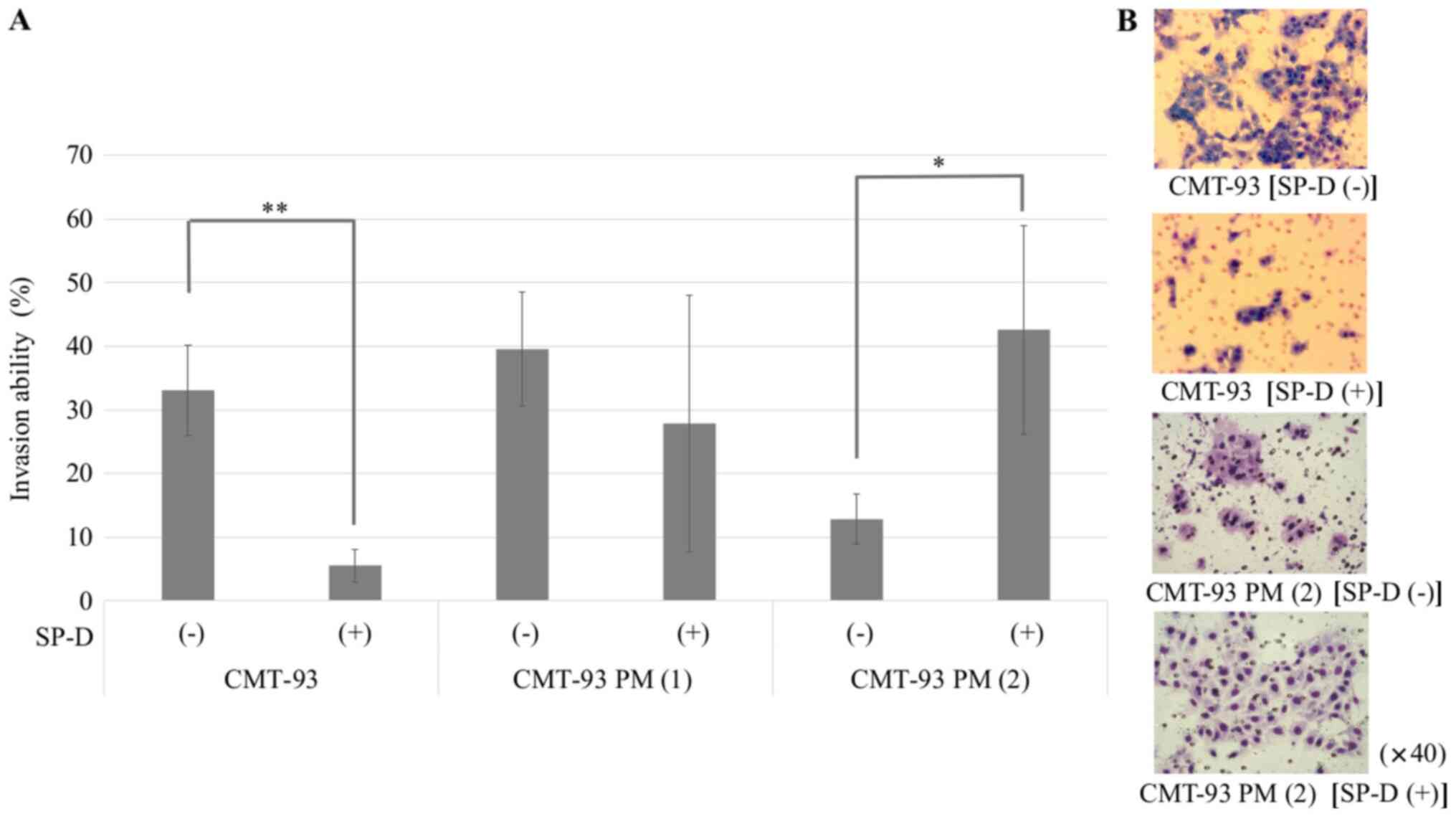
CMT-93 (Fig. 8). An invasion assay
showed that the SP-D-induced suppression of invasion ability was
weaker in CMT-93 PM (1) than that of
CMT-93PM. Moreover, SP-D reversely enhanced the invasion ability of
CMT-93 PM (2), while it was lower
than that of CMT-93 without SP-D (Fig.
9). In vivo, we confirmed that injection of CMT-93 PM
(2) cells into the tail vein
resulted in the development of significantly more pulmonary
metastases compared to CMT-93 (80.0 vs. 20.0%; P=0.025).
Change of Akt due to SP-D
SP-D suppressed the malignant potential through the
EGF-EGFR signaling pathway, in which SP-D played antagonist to the
EGFR and downregulated its signal according to a previous study.
Thus, we evaluated the change in Akt level by SP-D in CMT-93 and
CMT-93PM (2), which were the most
resistant cells against SP-D. Both cell types demonstrated a
decrease in Akt level due to SP-D (Fig.
10). CMT-93PM showed a relatively small change in Akt level due
to the SP-D treatment compared to CMT-93 (0.20 vs. 0.11; P=0.10)
(0.24 vs. 0.18; P=0.88).
Discussion
SP-D is an important protein for the maintenance of
the alveolar structure and management of the immune system in the
lung (1–3). In addition, leakage of SP-D from the
lung into the systemic circulatory system causes increased levels
of serum SP-D. Therefore, SP-D is a surrogate marker of COPD
(7). Moreover, SP-D is an antagonist
of EGFR and suppresses the signals downstream of EGFR, which
inhibits the progression of primary lung adenocarcinoma cells
(11). We hypothesized that SP-D
suppresses pulmonary metastases from colon cancer in the same
manner. In this study, we found that SP-D impacts pulmonary
metastases from colon cancer.
We found that SP-D suppressed the progression of
CMT-93 colon cancer cells, which was comparable to a previous study
of primary lung adenocarcinoma cells (11). Therefore, we hypothesized that SP-D
would also play a role in the pathogenesis and development of
pulmonary metastases from colon cancer. After the injection of
CMT-93 into the tail vein of mice, significantly larger and more
numerous pulmonary metastases were found in SP-D KO mice compared
to the control. Consequently, loss of SP-D in the lung enhanced and
exacerbated the pulmonary metastases from colon cancer through
increased cell proliferation.
As the influence of SP-D in tissues other than the
lung was removed, we determined that the serum SP-D protein level
was 6.89±1.4 ng/ml. Our result was almost the same as one
previously reported (18,19). Thus, the influence of SP-D in serum
could be negligible during the development of pulmonary metastasis
in our model.
Smoking causes inflammation in the lung, which
damages the membrane between the alveolar and vasculature and
allows SP-D to leak into the systemic circulatory system.
Consequently, the SP-D levels decrease in the lung and increase in
the circulatory system. Our previous study demonstrated that
smoking is a risk factor for pulmonary metastasis from colon cancer
(20) and SP-D is a potential
player. The reduction of SP-D in the lung caused by smoking may
also be a risk factor for pulmonary metastasis from colon cancer.
The results of this study support our hypothesis that SP-D in the
lung suppresses the development and progression of pulmonary
metastasis from colon cancer by blocking EGFR signals (11).
In addition, we established a novel pulmonary
metastasis prone cell line, CMT-93 PM, to increase the impact of
SP-D on pulmonary metastasis. Interestingly, CMT-93 PM was more
resistant to SP-D than CMT-93. Therefore, resistance to SP-D is
required for development of pulmonary metastases. CMT-93 PM
increased the incidence of pulmonary metastases following the
injection of the cells into the tail vein. Similarly, more
pulmonary metastases were found in SP-D KO mice than C57Bl/6
mice.
We used a micro-CT scan and 3D rearrangement to
evaluate the pulmonary metastasis. In addition, we performed two
examinations within 2 weeks, which allowed for more accurate
identification of pulmonary metastases from vasculature or minor
bronchus. We also performed a pathological evaluation, which
confirmed the radiological results.
Since SP-D inhibits EGFR signaling in A549 human
lung adenocarcinoma cells, the same pathway is likely to be
involved in the growth of pulmonary metastases from colon cancer.
In this study, we did not directly check SP-D binding with the
EGFR, but we initially confirmed expression of EGFR in both CMT-93
and CMT-93PM. We also evaluated the change in the Akt level to
determine whether the EGFR signaling pathway was involved in the
different behavior of SP-D between CMT93 and CMT-93PM. As a result,
both cells demonstrated a decrease in Akt level due to SP-D, but
the change in CMT-93PM was smaller. Therefore, downregulation of
the EGF-EGFR signaling pathway might contribute to the resistance
to SP-D. SP-D is a collectin protein that binds cytokine ligands.
Further experiments including identification of its ligands would
contribute to our results.
Although further experiments are needed, our results
are promising for managing pulmonary metastasis from colorectal
cancer. In the future, SP-D might be a feasible biomarker for
monitoring, and restitution of SP-D into the lung could be a novel
strategy for preventing or treating pulmonary metastases, though
the effect of restoration of SP-D on this mouse model needs to be
evaluated.
In conclusion, we demonstrated that SP-D suppressed
the growth of pulmonary metastases from colon cancer in vivo
and in vitro using a pulmonary metastasis mouse model.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YT and MT participated in the study design and
coordination, and drafting of the manuscript. MY, AM, KK and SA
performed the experiments and participated in the acquisition of
data. KO and TI participated in the design of the study and
performed the statistical analysis. HH, DDS and YK conceived the
study, participated in its design and coordination, and helped to
draft the manuscript. All authors read and approved the final
manuscript and agree to be accountable for all aspects of the
research.
Ethics approval and consent to
participate
All the procedures were performed under the approval
of the Laboratory Animal Care and Use Committee at Keio University
School of Medicine (approval no. 15006).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Johansson J and Curstedt T: Molecular
structures and interactions of pulmonary surfactant components. Eur
J Biochem. 244:675–693. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crouch E and Wright JR: Surfactant
proteins A and D and pulmonary host defense. Ann Rev Physiol.
63:521–524. 2001. View Article : Google Scholar
|
|
3
|
Kuroki Y, Takahashi M and Nishitani C:
Pulmonary collectins in innate immunity of the lung. Cell
Microbiol. 9:1871–1879. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mori K, Kurihara N, Hayashida S, Tanaka M
and Ikeda K: The intrauterine expression of surfactant protein D in
the terminal airways of human fetuses compared with surfactant
protein A. Eur J Pediatr. 161:431–434. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sørensen GL, Hjelmborg Jv, Kyvik KO,
Fenger M, Høj A, Bendixen C, Sørensen TI and Holmskov U: Genetic
and environmental influences of surfactant protein D serum levels.
Am J Physiol Lung Cell Mol Physiol. 290:L1010–L1017. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sin DD, Pahlavan PS and Man SF: Surfactant
protein D: A lung specific biomarker in COPD? Ther Adva Respir Dis.
2:65–74. 2008. View Article : Google Scholar
|
|
7
|
Ozyurek BA, Ulasli SS, Bozbas SS,
Bayraktar N and Akcay S: Value of serum and induced sputum
surfactant protein-D in chronic obstructive pulmonary disease.
Multidiscip Respir Med. 8:362013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Greene KE, King TE Jr, Kuroki Y,
Bucher-Bartelson B, Hunninghake GW, Newman LS, Nagae H and Mason
RJ: Serum surfactant proteins-A and -D as biomarkers in idiopathic
pulmonary fibrosis. Eur Respir J. 19:439–446. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leth-Larsen R, Nordenbaek C, Tornoe I,
Moeller V, Schlosser A, Koch C, Teisner B, Junker P and Holmskov U:
Surfactant protein D (SP-D) serum levels in patients with
community-acquired pneumonia. Clin Immunol. 108:29–37. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smith CJ, Perfetti TA and King JA:
Perspectives on pulmonary inflammation and lung cancer risk in
cigarette smokers. Inhal Toxicol. 18:667–77. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hasegawa Y, Takahashi M, Ariki S, Asakawa
D, Tajiri M, Wada Y, Yamaguchi Y, Nishitani C, Takamiya R, Saito A,
et al: Surfactant protein D suppresses lung cancer progression by
downregulation of epidermal growth factor signaling. Oncogene.
6:828–845. 2014.
|
|
12
|
Sin DD, Man SF, McWilliams A and Lam S:
Surfactant protein D and bronchial dysplasia in smokers at high
risk of lung cancer. Chest. 134:582–588. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Umeda Y, Hasegawa Y, Otsuka M, Ariki S,
Takamiya R, Saito A, Uehara Y, Saijo H, Kuronuma K, Chiba H, et al:
Surfactant protein D inhibits activation of non-small cell lung
cancer-associated mutant EGFR and affects clinical outcomes of
patients. Oncogene. 36:6432–6445. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Grada A, Otero-Vinas M, Prieto-Castrillo
F, Obagi Z and Falanga V: Research techniques made simple: Analysis
of collective cell migration using the wound healing assay. J
Invest Dermatol. 137:e11–e16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao X, Yang W, Pei F, Ma W and Wang Y:
Downregulation of matrix metalloproteinases contributes to the
inhibition of cell migration and invasion in HepG2 cells by sodium
valproate. Oncol Lett. 10:531–535. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hirano Y, Choi A, Tsuruta M, Jaw JE, Oh Y,
Ngan D, Moritani K, Chen YR, Tam S, Li Y, et al: Surfactant
protein-D deficiency suppresses systemic inflammation and reduces
atherosclerosis in ApoE knockout mice. Cardiovasc Res.
113:1208–1218. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eberting CL, Shrayer DP, Butmarc J and
Falanga V: Histologic progression of B16 F10 metastatic melanoma in
C57BL/6 mice over a six week time period: Distant metastases before
local growth. J Dermatol. 31:299–304. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gaunsbaek MQ, Rasmussen KJ, Beers MF,
Atochina- Vasserman EN and Hansen S: Lung surfactant protein D
(SP-D) response and regulation during acute and chronic lung
injury. Lung. 191:295–303. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hansen S, Schmidt V, Steffensen MA, Jensen
PH, Gjerstorff M, Thiel S and Holmskov U: An enzyme-linked
immunosorbent assay (ELISA) for quantification of mouse surfactant
protein D (SP-D). J Immunol Methods. 31:75–85. 2008. View Article : Google Scholar
|
|
20
|
Yahagi M, Tsuruta M, Hasegawa H,
Okabayashi K, Toyoda N, Iwama N, Morita S and Kitagawa Y: Smoking
is a risk factor for pulmonary metastasis in colorectal cancer.
Colorectal Dis. 19:O322–O328. 2017. View Article : Google Scholar : PubMed/NCBI
|