Introduction
Lung cancer is currently the leading cause of
cancer-associated mortalities worldwide. Up to 80% of patients with
lung cancer are diagnosed with non-small cell lung cancer (NSCLC)
(1). Over the past decade, epidermal
growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs),
including gefitinib, have been widely used for the treatment of
advanced NSCLC (2). Gefitinib exerts
its antitumor effects by abrogating the autophosphorylation of EGFR
and subsequently inhibits tumor growth, angiogenesis,
vasculogenesis and metastasis (3).
Although the efficacy of gefitinib therapy is impressive initially,
the majority of patients treated with gefitinib eventually develop
gefitinib-resistance, rendering the overall survival of patients
with NSCLC unsatisfactory. Therefore, there is an urgent
requirement to investigate the underlying molecular mechanisms for
advanced NSCLC with acquired gefitinib resistance.
COOH-terminus tensin-like molecule (CTEN, also known
as TNS4), a member of the tensin gene family, is involved in cell
adhesion, migration and signal transduction (4). Although CTEN is downregulated or absent
in advanced prostate cancer, CTEN has been reported to be
significantly increased in a variety of malignant tumors, including
colorectal (5), breast (6), prostate (7), pancreatic (8), lung cancer (9) and melanoma (10). It has been demonstrated that
increased expression of CTEN facilitates cell motility, apoptosis
and renal tubulogenesis, suggesting that CTEN may serve important
roles in tumorigenesis. In recent years, studies have indicated
that CTEN is a promising biomarker and therapeutic target for
numerous malignant cancer types (11).
Although CTEN has been demonstrated to be
upregulated in NSCLC, the mechanism by which CTEN confers acquired
gefitinib resistance in NSCLC remains poorly understood. The
present study aimed to investigate the roles of CTEN in acquired
gefitinib resistance in NSCLC, which may facilitate the development
of a novel therapeutic strategy for the treatment of patients with
NSCLC with acquired gefitinib resistance.
Materials and methods
Patient samples
Available plasma samples were collected from
patients with EGFR-mutant NSCLC (n=10), including 5 patients with
an L858R mutation and 5 patients with an exon 19 deletion. The
plasma of patients with NSCLC (n=10) was collected prior to and
following gefitinib treatment. All patients with NSCLC provided
written informed consent and the study was approved by the Ethics
Committee of the Jiangyin Hospital Affiliated to Medical College of
Southeast University. Detailed clinical information of the enrolled
patients are provided in Table
SI.
Cell culture and transfection
Human NSCLC PC9 cells were obtained from the Chinese
Academy of Sciences Cell Bank of Type Culture Collection and
cultured with Dulbecco's modified Eagle's medium (DMEM; Gibco;
Thermo Fisher Scientific, Inc.), containing 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.) plus antibiotics (100
U/ml penicillin + 100 µg/ml streptomycin) at 37°C with 5%
CO2. The gefitinib-resistant NSCLC cell model (PC9/GR)
was successfully established by continually exposing PC9 cells to
gefitinib for 6 months. For transient transfection, synthesized
negative control (NC; GenePharm, Inc.; 5′-UUCUCCGAACGUGUCACGUTT-3′;
100 nM), CTEN small interfering RNA (siRNA) mimics (GenePharm;
5′-GUGUCUGAUGUCAGCUAUAUG-3′; 100 nM) and CTEN-overexpressed pCMV
plasmid (Clontech Laboratories, Inc.) were transfected using
Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocols. A total of 4 h
after transfection, serum-free medium was changed to fresh DMEM
medium containing 10% FBS plus antibiotics at 37°C with 5%
CO2.
In vitro analysis of cell
proliferation
To assess the cell proliferation ability, a Cell
Counting kit-8 (CCK-8) assay (C0038; Beyotime Institute of
Biotechnology) was performed in vitro. PC9 and PC9/GR cells
were implanted into 96-well plates (3×103 cells/well).
At the times indicated (days 0, 1, 2 and 3), absorption at 450 nm
(OD450) was determined following incubation with 10 µl CCK-8
reagent by spectrophotometry (Bio Rad Laboratories, Inc.).
In vitro colony formation assay
In vitro colony-formation assay was performed
to investigate the in vitro colony-formation ability. PC9
and PC9/GR cells were seeded onto a 6-well plate (1,000
cells/well), and cultured with DMEM medium (Gibco; Thermo Fisher
Scientific, Inc.) containing 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) for two weeks at 37°C with 5% CO2.
Cells were fixed with 100% methanol for 20 min at room temperature
and stained with 0.25% crystal violet (dissolved in 20% methanol)
for 30 min at room temperature. The images of colonies were
captured with a GT-X700 scanner (Epson Corp.).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using the standard TRIzol
method (Thermo Fisher Scientific, Inc.) and the cDNA synthesis was
performed using the HiScript Q RT SuperMix for qPCR (+gDNA wiper;
R123-01, Vazyme Biotech Co., Ltd.) under the following conditions:
42°C for 2 min, 50°C for 15 min and 85°C for 2 min. qPCR was
performed using AceQ Universal SYBR qPCR Master Mix (Q511-02;
Vazyme Biotech Co., Ltd.) using the StepOnePlus Real-Time PCR
System (Bio-Rad Laboratories, Inc.). All reactions were run in
duplicate and 10 µl aliquots of the reaction mixture were pipetted
into each well of a 96-well PCR plate (Bio-Rad Laboratories, Inc.).
The thermocycling conditions were as follows: 95°C for 10 min, then
40 cycles of 95°C for 30 sec, 60°C for 30 sec and 72°C for 60 sec.
Relative mRNA amounts of target genes were calculated following
normalization to an endogenous reference gene (GAPDH) according to
the arithmetic formula 2−ΔΔCq (12). The real-time PCR primer sequences
were: CTEN forward, 5′-ACTGATGTCCAGAGGAAGGTG-3′ and reverse,
5′-ATGTCATACTCCGCAAAGAGG-3′; GAPDH forward,
5′-GGTCTCCTCTGACTTCAACA-3′ and reverse,
5′-AGCCAAATTCGTTGTCATAC-3′.
Protein extraction and Western blot
analysis
Cells were washed twice with 1X PBS, and lysed with
RIPA buffer (25 mM Tris, 150 mM NaCl, 0.1% SDS, 1% NP40, 2 mM EDTA
and 1% Triton X-100; pH 7.4). Protein concentration was determined
by the bicinchoninic acid (BCA) protein assay kit (Pierce; Thermo
Fisher Scientific Inc.). Cell lysates were mixed with 5X loading
buffer and denatured at 100°C for 10 min. Next, protein samples (40
µg per lane) were subjected to 10% SDS-PAGE and transferred onto a
45-µm polyvinylidene fluoride membrane (EMD Millipore) using a
semi-dry electrophoresis transfer apparatus (Bio-Rad Laboratories,
Inc.). Next, the membrane was blocked with PBST (1X PBS + 0.1%
Tween-20) containing 5% (w/v) skimmed milk for 1 h at room
temperature. The blots were then incubated with anti-CTEN (cat. no.
ab192247; dilution, 1:1,000; Abcam) and anti-b-actin (cat. no.
ab8227; dilution, 1:3,000; Abcam) antibodies overnight at 4°C.
Following washing with PBST and incubation with a horseradish
peroxidase-conjugated secondary rabbit antibody for 1 h (cat. no.
sc-2357; dilution, 1:3,000; Santa Cruz Biotechnology) at room
temperature, the protein bands were visualized using an ECL
detection kit (Thermo Fisher Scientific, Inc.).
In vivo xenograft assays
All animal experimental protocols were approved by
the Animal Research Ethics Committee of Jiangyin Hospital
affiliated to Medical College of Southeast University. BALB/c nude
mice (6-week-old, female, n=10) were purchased from the Animal
Laboratory Center of Nanfang Medical University (Guangdong, China).
All mice were housed under a fixed light-dark cycle (12 h of light
and 12 h of darkness) in a laminar flow room at constant
temperature and humidity, with ad libitum access to
sterilized food and water. CTEN-short hairpin RNA (shRNA) and
negative control shRNA (shRNA-NC) were designed and synthesized,
prior to being inserted into the lentiviral vector pLKO.1-TRC (1st
generation; cat. no. 10879; Addgene Inc.). Control and
pLKO.1-CTEN-shRNA were co-transfected into HEK-293T cells together
with lentivirus packaging plasmids (PMD2G, psPAX2)
(pLKO.1-CTEN-shRNA:PMD2G:psPAX2=1:1:1) to prepare the lentiviruses
using Lipofectamine 2000 (Thermo Fisher Scientific, Inc.).
Subsequently, PC9/GR cells (2×106 cells in PBS/100 µl)
were infected with shRNA-NC or CTEN shRNA-viruses and selected
using puromycin, prior to being injected subcutaneously into the
flanks of BALB/c nude mice (5 mice per group). Mice were euthanized
when tumors (shRNA-NC) reached >1 cm in diameter. All mice were
weighed and the tumor size was measured every 3 days using an
electronic-caliper. Tumor volume was calculated using the equation:
V=(LxW2)/2, where V represents the tumor volume, L
represents the length and W represents the width. Mice were
sacrificed at day 21 and tumors were surgically excised from the
mice.
Statistical analysis
The results were performed in triplicate, and the
results are expressed as the mean ± standard deviation. Student's
t-test was performed to analyze the data. Furthermore,
paired t-tests were performed to compare the expression of CTEN
mRNA in plasma samples collected prior to and following gefitinib
treatment. P<0.05 was considered to indicate a statistically
significant difference.
Results
CTEN is upregulated in
gefitinib-resistant NSCLC cells
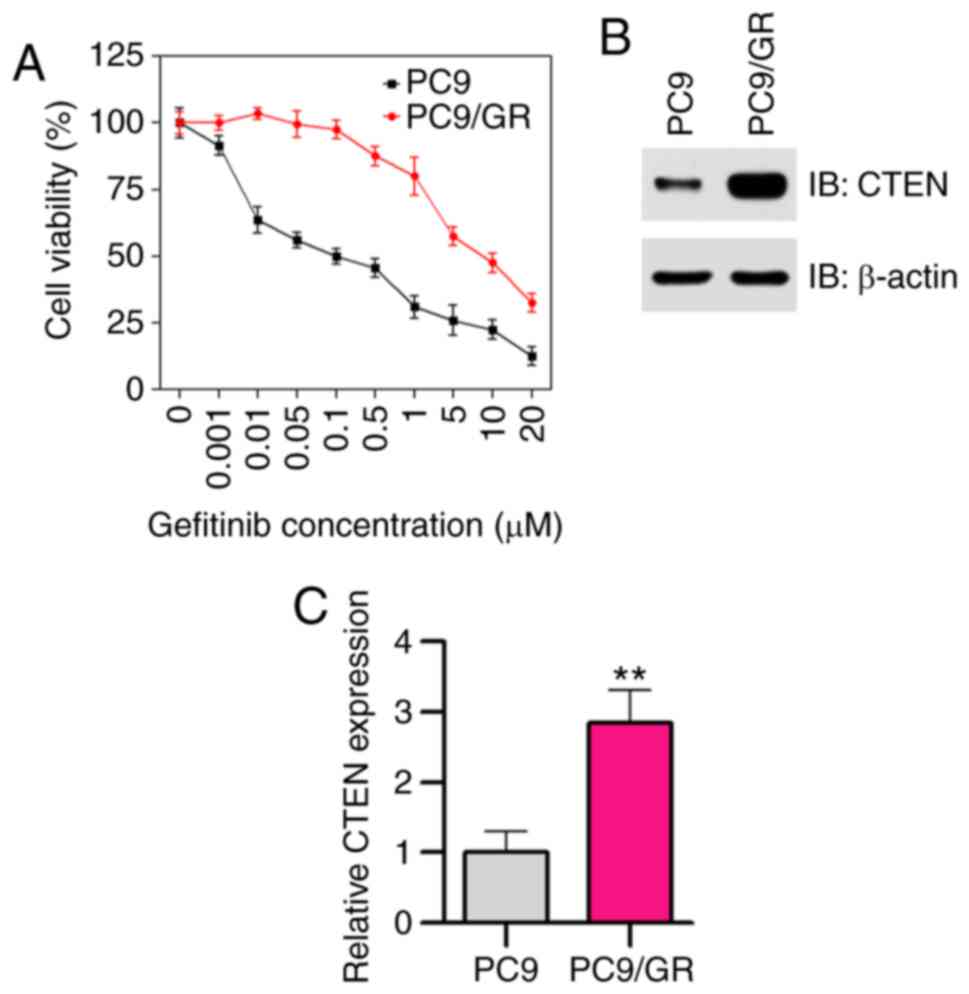
In the present study, a gefitinib-resistant cell
line, PC9/GR, was established by increasing the concentrations of
gefitinib in a stepwise manner in vitro. Half inhibition
concentration (IC50) values of gefitinib in PC9 and PC9/GR cells
treated with different concentrations of gefitinib ranging between
0.001 and 20 µM for 48 h were evaluated using CCK-8 assays. As
expected, PC9/GR cells were found to be resistant to gefitinib
(IC50=8.6 µM), and PC9 cells were found to be sensitive
to gefitinib (IC50=0.10 µM; Fig.
1A). To examine the possible role of CTEN in acquired
resistance to gefitinib, the expression of CTEN in
gefitinib-sensitive PC9 and gefitinib-resistant PC9/GR cells was
investigated. As shown in Fig. 1B and
C, the protein and mRNA expression levels of CTEN were higher
in the PC9/GR cells compared with those in the PC9 cells
(P<0.01), suggesting that CTEN may serve potential roles of in
acquired gefitinib resistance.
CTEN attenuates the sensitivity of
PC9/GR cells to gefitinib
To elucidate the mechanistic and functional aspects
of CTEN in modulating the sensitivity of NSCLC cells to gefinib
in vitro, the expression of CTEN in PC9/GR cells was
upregulated by transfection using the pCMV-CTEN plasmid, following
which the gefitinib-sensitivity of transfected PC9/GR cells was
determined using CCK-8 assays. Notably, as shown in Fig. 2A, the sensitivity of PC9/GR cells to
gefitinib was decreased when CTEN was overexpressed (P<0.05,
P<0.01). Next, the expression of CTEN in PC9/GR cells was
downregulated by siRNA transfection, and the sensitivity of
transfected PC9/GR cells to gefitinib was investigated using CCK-8
assays. As expected, PC9/GR cells with CTEN downregulation showed
markedly enhanced sensitivity to gefitinib when the cells were
treated with different concentrations of gefitinib (Fig. 2B; P<0.05, P<0.01). Taken
together, these results demonstrated that CTEN-overexpression may
attenuate the sensitivity of PC9/GR cells to gefitinib.
Overexpression of CTEN promotes the
proliferation of NSCLC cells
The effect of CTEN-overexpression on the
proliferative ability of gefitinib-sensitive (PC9) and
gefitinib-resistant (PC9/GR) cells was investigated. Western blot
analysis revealed that CTEN was markedly upregulated in PC9 and
PC9/GR cells transfected with the pCMV-CTEN plasmid (Fig. 3A). The proliferative capabilities of
PC9 and PC9/GR cells were significantly augmented in vitro
(Fig. 3B; P<0.05, P<0.01) when
the expression of CTEN was upregulated. Furthermore, the
proliferative capacity of NSCLC cells in vitro was
investigated using a colony-forming assay. The results of the
present study suggested that overexpression of CTEN significantly
promoted the colony-forming abilities of PC9 and PC9/GR cells
(Fig. 3C; P<0.01). Taken
together, these results suggested that CTEN serves a role in
facilitating the proliferation of NSCLC cells in vitro.
Knockdown of CTEN inhibits the
proliferation of NSCLC cells
In order to silence CTEN, PC9 and PC9/GR cells were
transfected with CTEN-siRNA. As shown in Fig. 4A, cells transfected with CTEN-siRNA
resulted in a significant loss of CTEN expression compared with
that in cells transfected with NC-siRNA. Next, the effect of
CTEN-knockdown on cell proliferation was investigated using CCK-8
assays. As shown in Fig. 4B, the
proliferative capabilities of gefitinib-sensitive and
gefitinib-resistant NSCLC cells were observed to be significantly
decreased (P<0.01) when the expression of CTEN was
downregulated. Consistently, siRNA-mediated knockdown of CTEN
significantly suppressed the colony-forming abilities of PC9 and
PC9/GR cells (Fig. 4C;
P<0.01).
Depletion of CTEN suppresses the
growth of PC9/GR xenografts in vivo
The aforementioned phenotypic analysis demonstrated
that CTEN-knockdown significantly decreased the proliferative and
colony-formative capabilities of PC9/GR cells in vitro. To
further confirm the oncogenetic roles of CTEN in NSCLC, the effects
of lentivirus-mediated CTEN depletion on the growth of PC9/GR cells
in vivo were investigated. Notably, as shown in Fig. 5A and B (P<0.01), CTEN depletion
suppressed the growth of PC9/GR xenografts in vivo.
Furthermore, there was no significant difference observed between
the body weight of shRNA-NC- and CTEN shRNA-treated mice (Fig. 5C). Based on the results of RT-qPCR,
the mRNA expression CTEN in CTEN-depleted PC9/GR xenografts was
significantly lower than that in the control PC9/GR xenografts
(Fig. 5D; P<0.01). Taken
together, these results further suggested that CTEN is required for
the growth of PC9/GR xenografts, emphasizing the important role of
CTEN in promoting NSCLC progression.
CTEN is upregulated in patients with
NSCLC with acquired resistance to gefitinib
To determine the expression levels of CTEN in
patients with NSCLC treated with EGFR-TKI, plasma samples from 10
patients with NSCLC were collected prior to and following gefitinib
treatment, following which total RNA was isolated for CTEN
expression evaluation. As shown in Fig.
6, the mRNA expression of CTEN was significantly increased in
patients with NSCLC with acquired gefitinib resistance
(P<0.05).
Discussion
Somatic mutations of EGFR may facilitate the
development of effective EGFR-TKIs for successfully treating
patients with NSCLC. However, chemoresistance development is
inevitable, and the majority of patients with NSCLC experience
highly malignant and aggressive relapses. Therefore, there is an
urgent requirement to identify the prognostic indicators and to
elucidate the underlying molecular mechanisms for advanced NSCLC
with acquired gefitinib resistance. The results of the present
study demonstrated that upregulation of CTEN may promote cell
proliferation and colony formation of gefitinib-resistant NSCLC
cells, suggesting that CTEN may contribute toward gefitinib
resistance in NSCLC.
Lo et al (11)
have suggested that CTEN expression is frequently increased in a
variety of malignant tumors, including colorectal, gastric,
pancreatic and lung cancer (11).
Thorpe et al (13)
demonstrated that CTEN may facilitate epithelial-mesenchymal
transition (EMT) processes by preventing the degradation of Snail
protein in colorectal cancer. Asiri et al (14) demonstrated that CTEN is a positive
modulator of Src, and that CTEN may promote EMT and metastasis via
post-transcriptional stabilization of Src protein in colorectal
cancer. Notably, Hong et al (15) revealed that a proportion of
nuclear-localized CTEN contributes toward cell proliferation in
cancer cells. Wu et al (16)
demonstrated that knockdown of CTEN inhibits prostate cancer cell
proliferation and results in G1/S cell cycle arrest,
suggesting that inhibition of CTEN expression is required for
luminal differentiation and acinar formation. Consistently, our
previous study demonstrated that CTEN may activate the expression
of TGF-β1, which promotes EMT and metastasis in NSCLC cells,
thereby strongly supporting the role of CTEN as an oncoprotein in
NSCLC (17).
Over the last decade, a number of potential
mechanisms that may lead to acquired resistance to gefitinib in
NSCLC have been proposed. Using a genome-wide RNAi screening
approach, Cho et al (18)
examined a synthetic lethality with gefitinib in EGFR-mutated
TKI-resistant NSCLC cells, and identified RNF25 as an important
modulator that may enhance the acquired gefitinib resistance.
Furthermore, they revealed that RNF25 may serve crucial roles in
the contribution of acquired gefitinib resistance by facilitating
the cross-talk between NF-κB and ERK signaling pathways in NSCLC
(18). Li et al (19) suggested that downregulation of
RHPN1-AS1 lncRNA contributes toward acquired gefitinib resistance
by targeting the miR-299-3p/TNFSF12 pathway in NSCLC. Lu et
al (20) reported that hypoxia
is a driving force for acquired gefitinib resistance through the
regulation of the EMT process, and epigenetic regulators, including
LSD1 and PLU-1, thereby serving crucial roles in hypoxia-induced
gefitinib resistance. Li et al (21) investigated the role of DEAD-Box
Helicase 17 (DDX17) in acquired gefitinib resistance and observed
that exportin/importin-dependent nucleocytoplasmic shuttling of
DDX17 serves important roles in acquired gefitinib resistance
through the activation of β-catenin in NSCLC. Overexpression of
peptidylarginine deiminase IV (PAD4) has been demonstrated to
decrease the activity of EMT by inhibiting the expression of
ETS-domain containing protein (Elk1), which then suppresses the
resistance of NSCLC cell lines to gefitinib (22). Feng et al (23) indicated that Krüppel-like factor 4
(KLF4) is an important contributing factor for gefitinib resistance
in c-Met-overexpressed-NSCLC cells by repressing the expression of
apoptosis-related proteins. In addition, microRNAs have been
recently reported to be widely involved in acquired gefitinib
resistance in NSCLC (24,25). In conclusion, the mechanisms of
gefitinib resistance remain unclear. Further studies are urgently
required to elucidate the underlying mechanisms of gefitinib
resistance in NSCLC.
Identification of potential factors that regulate
the resistance of NSCLC cells to EGFR-TKIs, including gefitinib may
provide more precise information that facilitates effective
therapy. The present study identified that CTEN is upregulated in
gefitinib-resistant NSCLC cells, and that CTEN may lower the
sensitivity of gefitinib-resistant NSCLC cells to gefitinib.
Subsequently, it was observed that CTEN exerts a promoting effect
on the growth of gefitinib-resistant NSCLC cells in vitro or
in vivo. In addition, the mRNA expression of CTEN is
significantly increased in patients with NSCLC with resistance to
gefitinib, suggesting that CTEN may contribute toward gefitinib
resistance in NSCLC. In conclusion, CTEN may be a potential
therapeutic target for the treatment of patients with NSCLC with
acquired gefitinib resistance. However, the present study is
confined to a small number of enrolled patients with NSCLC.
Therefore, prospective studies with a larger number of NSCLC
patients are required to validate the findings of the present
study.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Wuxi Science
and Technology Project (2018) and the Youth Fund of Wuxi Health
Committee (grant no. 201904).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XL, YZ and YP conducted the experiments, analyzed
and interpreted the data. MC and XZ aided in collecting plasma
samples from patients with NSCLC. TZ conceived this study and wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics statement and consent to
participate
All patients provided written informed consent and
the present study was approved by the Ethics Committee of the
Jiangyin Hospital affiliated to Medical College of Southeast
University. All animal experimental protocols were approved by the
Animal Research Ethics Committee of Jiangyin Hospital affiliated to
Medical College of Southeast University.
Patient consent for publication
All patients provided written informed consent for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ansari J, Palmer DH, Rea DW and Hussain
SA: Role of tyrosine kinase inhibitors in lung cancer. Anticancer
Agents Med Chem. 9:569–575. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Muhsin M, Graham J and Kirkpatrick P:
Gefitinib. Nat Rev Drug Discov. 2:515–516. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lo HS and Lo TB: Cten, a COOH-terminal
tensin-like protein with prostate restricted expression, is
down-regulated in prostate cancer. Cancer Res. 62:4217–4221.
2002.PubMed/NCBI
|
|
5
|
Albasri A, Seth R, Jackson D, Benhasouna
A, Crook S, Nateri AS, Chapman R and Ilyas M: C-Terminal
Tensin-like (CTEN) is an oncogene which alters cell motility
possibly through repression of E-cadherin in colorectal cancer. J
Pathol. 218:57–65. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Albasri A, Aleskandarany M, Benhasouna A,
Powe DG, Ellis IO, Ilyas M and Green AR: CTEN (C-terminal
tensin-like), a novel oncogene overexpressed in invasive breast
carcinoma of poor prognosis. Breast Cancer Res Treat. 126:47–54.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li YQ, Mizokami A, Izumi K, Narimoto K,
Shima T, Zhang J, Dai J, Keller ET and Namiki M: CTEN/tensin 4
expression induces sensitivity to paclitaxel in prostate cancer.
Prostate. 70:48–60. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Al-Ghamdi S, Cachat J, Albasri A, Ahmed M,
Jackson D, Zaitoun A, Guppy N, Otto WR, Alison MR, Kindle KB and
Ilyas M: C-Terminal tensin-like gene functions as an oncogene and
promotes cell motility in pancreatic cancer. Pancreas. 42:135–140.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sasaki H, Moriyama S, Mizuno K, Yukiue H,
Konishi A, Yano M, Kaji M, Fukai I, Kiriyama M, Yamakawa Y and
Fujii Y: Cten mRNA expression was correlated with tumor progression
in lung cancers. Lung Cancer. 40:151–155. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sjoestroem C, Khosravi S, Zhang G,
Martinka M and Li G: C-Terminal tensin-like protein is a novel
prognostic marker for primary melanoma patients. PLoS One.
8:e804922013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lo SH: C-Terminal tensin-like (CTEN): A
promising biomarker and target for cancer. Int J Biochem Cell Biol.
51:150–154. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thorpe H, Asiri A, Akhlaq M and Ilyas M:
Cten promotes epithelial-mesenchymal transition through
post-transcriptional stabilization of snail. Mol Carcinog.
56:2601–2609. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Asiri A, Toss MS, Raposo TP, Akhlaq M,
Thorpe H, Alfahed A, Asiri A and Ilyas M: Cten promotes
epithelial-mesenchymal transition (EMT) in colorectal cancer
through stabilisation of src. Pathol Int. 69:381–391. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hong SY, Shih YP, Lo A and Lo SH:
Identification of subcellular targeting sequences of Cten reveals
its role in cell proliferation. Biochim Biophys Acta Mol Cell Res.
1866:450–458. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu WM and Liao YC: Downregulation of
C-terminal tensin-like protein (CTEN) suppresses prostate cell
proliferation and contributes to acinar morphogenesis. Int J Mol
Sci. 19:31902018. View Article : Google Scholar
|
|
17
|
Lu X, Gao J, Zhang Y, Zhao T, Cai H and
Zhang T: CTEN induces epithelial-mesenchymal transition (EMT) and
metastasis in non small cell lung cancer cells. PLoS One.
13:e01988232018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cho JH, You YM, Yeom YI, Lee DC, Kim BK,
Won M, Cho BC, Kang M, Park S, Yang SJ, et al: RNF25 promotes
gefitinib resistance in EGFR-mutant NSCLC cells by inducing
NF-κB-mediated ERK reactivation. Cell Death Dis. 9:5872018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li X, Xin Z, Chunlu Y, Su C, Qiming S and
Shun X: The lncRNA RHPN1-AS1 downregulation promotes gefitinib
resistance by targeting miR-299-3p/TNFSF12 pathway in NSCLC. Cell
Cycle. 17:1772–1783. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu Y, Liu YF, Sebastian O and Glazer PM:
Hypoxia promotes resistance to EGFR inhibition in NSCLC cells via
the histone demethylases, LSD1 and PLU-1. Mol Cancer Res.
16:1458–1469. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li K, Mo C, Gong D, Chen Y, Huang Z, Li Y,
Zhang J, Huang L, Li Y, Fuller-Pace FV, et al: DDX17
nucleocytoplasmic shuttling promotes acquired gefitinib resistance
in non-small cell lung cancer cells via activation of β-catenin.
Cancer Lett. 400:194–202. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Duan Q, Pang C, Chang N, Zhang J and Liu
W: Overexpression of PAD4 suppresses drug resistance of NSCLC cell
lines to gefitinib through inhibiting Elk1-mediated
epithelial-mesenchymal transition. Oncol Rep. 36:551–558. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feng W, Xie Q, Liu S, Ji Y and Jin L: KLF4
promotes c-met amplification-mediated gefitinib resistance in
NSCLC. Cancer Sci. 109:1775–1786. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiao D, Chen J, Li Y, Tang X, Wang J, Xu
W, Song J, Li Y, Tao H and Chen Q: miR-1-3p and miR-206 sensitizes
HGF-induced gefitinib-resistant human lung cancer cells through
inhibition of c-Met signalling and EMT. J Cell Mol Med.
22:3526–3536. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cao X, Lai S, Hu F, Li G, Wang G, Luo X,
Fu X and Hu J: miR-19a contributes to gefitinib resistance and
epithelial mesenchymal transition in non-small cell lung cancer
cells by targeting c-Met. Sci Rep. 7:29392017. View Article : Google Scholar : PubMed/NCBI
|