Introduction
Lung cancer is the leading cause of
cancer-associated deaths worldwide (1), with the 5-year survival rate of
patients with lung cancer being <20% (2). Non-small cell lung cancer accounts for
>85% of lung cancer cases and ~60% of these cases are classified
as lung adenocarcinoma (LUAD) (3).
At the time of diagnosis, ~70% of patients with lung cancer have
locally advanced or metastatic disease (4). Although the development of
anti-angiogenic drugs, EGFR inhibitors and other novel anticancer
agents has greatly improved the treatment of lung cancer, the
5-year survival rate remains <15% (5). Therefore, the present study aimed to
provide a valuable theoretical basis for the study of the
mechanisms underlying the development of LUAD and novel directions
for the further investigation of the pathogenesis of LUAD.
It has been reported that competing endogenous RNAs
(ceRNAs) serve an important role in the post-transcriptional
regulation of genes by competing with other RNA molecules to bind
to specific microRNAs (miRNAs/miRs) via common miRNA response
elements (6,7). Numerous studies have demonstrated that
the regulatory mechanisms of ceRNAs are critical in the development
and progression of several types of cancer, including breast
(8), bladder (9) and lung cancer (10). An increasing number of long
non-coding RNAs (lncRNAs) have been identified to serve vital roles
in the pathogenesis of LUAD through the mechanisms of ceRNAs
(11,12). For example, Dong et al
(13) demonstrated that the lncRNA
DiGeorge syndrome critical region gene 5 promoted LUAD progression
via inhibiting hsamiR-22-3p. Additionally, Xiong et al
(14) revealed that lncRNA nuclear
paraspeckle assembly transcript 1 (NEAT1) accelerated LUAD
deterioration by acting as a ceRNA to regulate miR-193a-3p
expression. Furthermore, it has been reported that exosomes derived
from chondrosarcoma cells carry the lncRNA receptor activity
modifying protein 2-antisense RNA 1 (RAMP2-AS1), which acts as a
ceRNA of miR-2355-5p to modulate the expression levels of vascular
endothelial growth factor receptor 2 (VEGFR2), thus actively
regulating the angiogenic ability of human umbilical vein
endothelial cells (HUVECs) (15).
Consequently, it has been speculated that exosomes carrying
RAMP2-AS1 may be a novel biomarker and therapeutic target for
chondrosarcoma (15). Therefore,
exploring lncRNA-associated ceRNA mechanisms in LUAD may lead to
the development of effective diagnostic and therapeutic
strategies.
Over the past few decades, with the development of
high-throughput technology, rapid progress has been made in the
identification of differentially expressed genes to further explore
the molecular mechanisms underlying cancer development. In the
present study, two microarray datasets (GSE113852 and GSE130779)
were downloaded from the Gene Expression Omnibus (GEO) to identify
the key lncRNAs and mRNAs. Furthermore, a functional enrichment
analysis was performed and an interaction network was constructed
to explore the functions of the key genes associated with LUAD. The
present study aimed to identify the key genes associated with the
development of LUAD and to provide available target genes for the
treatment and diagnosis of LUAD by performing bioinformatics
analyses.
Materials and methods
Identification of key lncRNAs and
mRNAs
GSE113852 and GSE130779 gene expression profiles
were downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). In the GSE113852
dataset, GSM3121285-GSM3121311 and GSM3121312-GSM3121338 included
27 paired normal lung and lung cancer samples, respectively. In the
GSE130779 dataset, GSM3753429-GSM3753432 and GSM3753437-GSM3753440
included 8 LUAD samples, whereas GSM3753433-GSM3753436 and
GSM3753441-GSM37534344 included 8 normal paired samples.
Subsequently, differential gene expression analysis was performed
using the GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) web tool with
a threshold of |log2 fold-change (FC)| >1 and P<0.05.
Subsequently, the differentially expressed lncRNAs and mRNAs in the
two datasets were identified using Venn diagrams
(Venny2.1https://bioinfogp.cnb.csic.es/tools/venny/index.html). The
gene expression profile of lung adenocarcinoma was obtained from
The Cancer Genome Atlas (TCGA) database (https://tcga-data.nci.nih.gov/tcga/). The Starbase
V2.0 (http://starbase.sysu.edu.cn/starbase2/) database was
used to screen for miRNAs that could bind to RAMP2-AS1 as ceRNAs.
Starbase V2.0 is a database used for the systematical
identification of RNA-RNA and protein-RNA interaction networks
(16). Furthermore, the target genes
of miR-296-5p were obtained via TargetScan (17) (http://www.targetscan.org/vert_72/) and miRDB
(18) (http://mirdb.org/) databases. Both these online
databases are used for miRNA target prediction and functional
annotations. The expression pattern of miR-296-5p in LUAD was
acquired from the database of differentially expressed miRNAs in
human Cancers (dbDEMC; (http://www.picb.ac.cn/dbDEMC/index.html) (19). Finally, the target genes were
intersected with differentially expressed mRNAs to obtain the key
mRNAs.
Expression and prognostic
analyses
The expression status of RAMP2-AS1 in tumors was
acquired using the Gene Expression Profiling Interactive Analysis
(GEPIA; http://gepia.cancer-pku.cn/detail.php) online tool.
GEPIA is a web server for gene expression profiling and interactive
analyses of normal and cancer samples (20). In addition, the UALCAN platform
(http://ualcan.path.uab.edu/) is an
interactive web-portal for facilitating tumor subgroup gene
expression and survival analyses (21). Therefore, this platform was used to
investigate the expression levels of key mRNAs in LUAD and adjacent
normal tissues, as well as their association with cancer stage
(22), nodal metastasis status
(23) and histological subtype. N0
represents no regional lymph node metastasis; N1 represents
metastases in 1–3 axillary lymph nodes; N2 represents metastases in
4–9 axillary lymph nodes; N3 represents metastases in ≥10 axillary
lymph nodes. The correlation between the expression of RAMP2-AS1
and the key mRNAs in LUAD was analyzed using Spearman's rank
correlation test in GEPIA. The overall survival (OS) analysis of
RAMP2-AS1 was also evaluated using the GEPIA online tool. Finally,
the OS and first progression (FP) analyses of key mRNAs were
performed using Kaplan-Meier plotter (24) (2014 version). The log-rank test was
used to determine differences in the survival rate between the high
and low expression groups.
LUAD samples
Tumor and adjacent normal tissue samples (>2 cm
from tumor) used in the present study were collected from surgery
from 40 patients with LUAD at Zhongnan Hospital of Wuhan University
(Wuhan, China) between January 2018 and January 2019. The patients
included 22 males and 18 females, and had a median age of 64 years
(range, 37–75 years). None of the patients had received any
anticancer therapy prior to surgery. Each patient provided written
informed consent, which was in accordance with the ethical
guidelines of Zhongnan Hospital of Wuhan University. Additionally,
the collection of human tumor tissues was approved by the Ethical
Committee of Zhongnan Hospital of Wuhan University.
Functional enrichment analysis
The cBio Cancer Genomics Portal (cBioPortal;
http://cbioportal.org) was used to investigate
the interactions between the key mRNAs and obtain the important
genes involved. cBioPortal provides a web resource for exploring,
visualizing and analyzing multidimensional cancer genomics data,
enabling researchers to interactively explore genetic alterations
across samples, genes and pathways (25). Furthermore, Gene Ontology (GO) and
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway function
enrichment analyses were performed using the FunRich software
(version 3.1.3) (26). This open
access functional enrichment and network analysis tool provides
graphical representation, such as Venn, pie charts and heatmaps, of
the data with customizable font, scale and color (26).
Interaction network construction
The Search Tool for the Retrieval of Interacting
Genes/Proteins (STRING; http://string-db.org/) database incorporates known and
predicted protein-protein association data for a large number of
organisms, including direct (physical), as well as indirect
(functional) interactions (27).
Therefore, based on the STRING database, the ceRNA network was
constructed using Cytoscape (version 3.6.1) (28), which is an open source software
project that integrates biomolecular interaction networks with
expression profiles, phenotypes and other molecular states into a
unified conceptual framework.
Reverse transcription-quantitative PCR
(RT-qPCR) assays
Total RNA was extracted from LUAD tissues using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. Following total
RNA isolation, RT was performed according to the protocol of the
UEIris II RT-PCR System for First-Strand cDNA Synthesis (US
Everbright® Inc.) kit. The SYBR Premix Ex Taq (US
Everbright® Inc.) kit was employed to perform qPCR
(denaturation, 30 sec at 95°C; annealing, 30 sec at 58°C;
extension, 30 sec at 72°C; 35 cycles) on the ABI 7900 system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) and GAPDH
served as the endogenous control. Comparative quantification was
performed using the 2−ΔΔCq method (29). The primers were purchased from Sangon
Biotech Co., Ltd., and their sequences are listed in Table I.
 | Table I.List of primers used in the present
study. |
Table I.
List of primers used in the present
study.
| Name | Sequences
(5′-3′) |
|---|
| CD44 F |
GACAACGCAGCAGAGTAA |
| CD44 R |
TGTGTGGGTAATGAGAGGTA |
| NCALD F |
TCATCGCCTTGAGTGTAA |
| NCALD R |
CCGTCTCTATTGGTGTCC |
| CCND3 F |
CACACCACATCTAAGCCTGAA |
| CCND3 R |
CCCAATCCAAATGCAATAAC |
| MACF1 F |
CTGTGCCTGTGTGTTGAG |
| MACF1 R |
TGGACTGCGTGGTTTTAG |
| RAMP2-AS1 F |
CTTGGATCATGGGCACGGAT |
| RAMP2-AS1 R |
GTCAAGTCACCTCTTGCCCT |
| GAPDH F |
GAAAGCCTGCCGGTGACTAA |
| GAPDH R |
GCATCACCCGGAGGAGAAAT |
Statistical analysis
Statistical analyses were performed using GraphPad
Prism (version 7.0; GraphPad Software, Inc.). Data are expressed as
the mean ± standard deviation. Comparisons between two groups
(normal vs. tumor tissues) were analyzed using a paired Student's
t-test. Comparisons among multiple groups were analyzed using
one-way ANOVA followed by Tukey's post hoc test. All experiments
were performed in triplicate. P<0.05 was considered to indicate
a statistically significant difference.
Results
Identification of key lncRNA and
mRNAs
A total of three differentially expressed lncRNAs
(Fig. 1A) and 368 differentially
expressed mRNAs (Fig. 1B) were
obtained from the GSE113852 and GSE130779 datasets using the GEO2R
and Venn diagrams. Among them, lncRNA RAMP2-AS1 was the most
significantly downregulated (Table
II) and was therefore chosen for subsequent studies. Samples
were divided according to the expression levels of all analyzed
RNAs from high to low, with values higher than the median value
considered as high expression and values lower than the median
value considered as low expression. TCGA results revealed that
RAMP2-AS1 expression was downregulated in the majority of tumor
types (Fig. 1C) and was
significantly downregulated in LUAD tissues compared with in normal
tissues (Fig. 1D). Furthermore, the
prognostic analysis in GEPIA revealed that increased expression
levels of RAMP2-AS1 were associated with an improved OS (Fig. 1E). Furthermore, three miRNAs binding
to RAMP2-AS1 were identified using Starbase, namely miR-296-5p,
miR-1301-3p and miR-654-5p. However, according to the dbDEMC, the
differential expression of miR-296-5p was the most marked in LUAD
(Fig. 1G). Subsequently, the target
genes of miR-296-5p were obtained using TargetScan and miRDB
databases. The intersection of these target genes with the 368
differentially expressed mRNAs identified in the aforementioned
datasets revealed five key mRNAs, namely CD44, cyclin D3 (CCND3),
neurocalcin δ (NCALD), microtubule actin crosslinking factor 1
(MACF1) and potassium channel tetramerization domain containing 15
(KCTD15) (Fig. 1F).
 | Figure 1.Identification of key lncRNAs and
mRNAs in LUAD. Venn diagrams of the intersection of differentially
expressed (A) lncRNAs and (B) mRNAs from the GSE113852 and
GSE130779 datasets. (C) Expression levels of RAMP2-AS1 in different
types of cancer as analyzed using TCGA. Green indicates that
RAMP2-AS1 expression was downregulated, while red indicates that it
was upregulated. (D) Expression levels of RAMP2-AS1 in LUAD and
normal tissues as analyzed using TCGA. (E) Prognostic analysis of
RAMP2-AS1 expression using Gene Expression Profiling Interactive
Analysis. (F) Venn diagrams of the intersection of 368
differentially expressed mRNAs and the target genes of miR-296-5p
identified using TargetScan and miRDB. (G) Differential expression
profile of miR-296-5p, miR-1301-3P and miR-654-5P. Green indicates
downregulation, while red indicates upregulation. T, tumor; N,
normal; lncRNA, long non-coding RNA; LUAD, lung adenocarcinoma;
RAMP2-AS1, receptor activity modifying protein 2-antisense RNA 1;
TCGA, The Cancer Genome Atlas; ACC, adrenocortical carcinoma; BLCA,
bladder urothelial carcinoma; BRCA, breast invasive carcinoma;
CESC, cervical squamous cell carcinoma and endocervical
adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon
adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell
lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme;
HNSC, head and neck squamous cell carcinoma; KICH, kidney
chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney
renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG,
brain lower grade glioma; LIHC, liver hepatocellular carcinoma;
LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian
serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG,
pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma;
READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous
melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell
tumors; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine
corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM,
uveal melanoma; TPM, transcripts per million. |
| Table II.Information of long non-coding RNAs
in the GSE113852 and GSE130779 datasets. |
Table II.
Information of long non-coding RNAs
in the GSE113852 and GSE130779 datasets.
| Name | GSE113852
LogFC | GSE130779
P-value | LogFC | P-value |
|---|
| RAMP2-AS1 | −2.65 |
2.06×10−18 | −3.16 |
7.52×10−3 |
| ADAMTS9-AS2 | −1.66 |
6.70×10−16 | −2.42 |
6.04×10−4 |
| Linc00312 | −1.58 |
1.45×10−11 | −2.97 |
3.73×10−4 |
Expression and prognostic
analyses
According to the expression profiles included in the
GSE113852 and GSE130779 datasets, the expression levels of
RAMP2-AS1, CD44, CCND3, NCALD, MACF1 and KCTD15 were significantly
downregulated in LUAD compared with in normal tissues (Fig. 2). As shown in Fig. 3, the expression levels of CD44,
CCND3, NCALD, MACF1 and KCTD15 were downregulated in the vast
majority of LUAD histological subtypes compared with in normal
samples; additionally, they were differentially expressed according
to tumor stage and nodal metastasis status.
 | Figure 3.Expression levels of the selected
genes in the UALCAN database. Expression levels of selected genes
in lung adenocarcinoma based on (A) sample type, (B) individual
cancer stage, (C) nodal metastasis status and (D) histological
subtype. *P<0.05; **P<0.01; ***P<0.001. N0, no regional
lymph node metastasis; N1, metastases in 1–3 axillary lymph nodes;
N2, metastases in 4–9 axillary lymph nodes; N3, metastases in ≥10
axillary lymph nodes; NOS, not otherwise specified; LBC, lung
bronchioloalveolar carcinoma; CCND3, cyclin D3; NCALD, neurocalcin
δ; MACF1, microtubule actin crosslinking factor 1; KCTD15,
potassium channel tetramerization domain containing 15. |
The correlation between the expression levels of
RAMP2-AS1 and CD44, CCND3, NCALD, MACF1 and KCTD15 in LUAD was
evaluated using Pearson's rank correlation test in GEPIA, revealing
that RAMP2-AS1 expression was significantly positively correlated
with the expression levels of CD44, CCND3, NCALD, MACF1 and KCTD15
(Fig. 4A; P<0.05; R>0).
Additionally, the prognostic value of each key mRNA was determined
using Kaplan-Meier plotter analysis. The analysis indicated that
high expression levels of CD44, CCND3, NCALD and MACF1 resulted in
an improved OS and FP in patients with LUAD, while KCTD15
expression exhibited the opposite effect (Fig. 4B and C). Therefore, KCTD15 was
excluded from subsequent analyses.
Functional enrichment analysis
The co-expression analysis of CD44, CCND3, NCALD and
MACF1 was assessed using cBioPortal. The analysis revealed 50 genes
that may be associated with CD44, CCND3, NCALD and MACF1 (Fig. 5A). Subsequently, these genes were
subjected to GO and KEGG enrichment analyses in FunRich to
determine their possible molecular functions. KEGG pathway analysis
revealed that these genes were enriched in the ‘cyclin D associated
events in G1’, ‘generic transcription pathway’, ‘CDC42
signaling events’, ‘stabilization and expansion of the E-cadherin
adherens junction’ and ‘N-cadherin signaling events’ (Fig. 5B). The genes involved in the enriched
pathways are shown in Table II.
Furthermore, GO analysis revealed that the predicted genes were
mainly enriched in biological processes such as ‘cell
communication’ and ‘signal transduction’, cellular components such
as ‘mediator complex’, ‘cytosol’ and ‘nucleus’, and molecular
functions such as ‘kinase regulator activity’, ‘TF regulator
activity’ and ‘kinase binding’ (Fig.
5C).
Construction of interaction
networks
Subsequently, the aforementioned 50 genes were
subjected to the STRING database analysis to construct the
predicted protein-protein interaction network. The interaction
network was then imported into the Cytoscape software. Using the
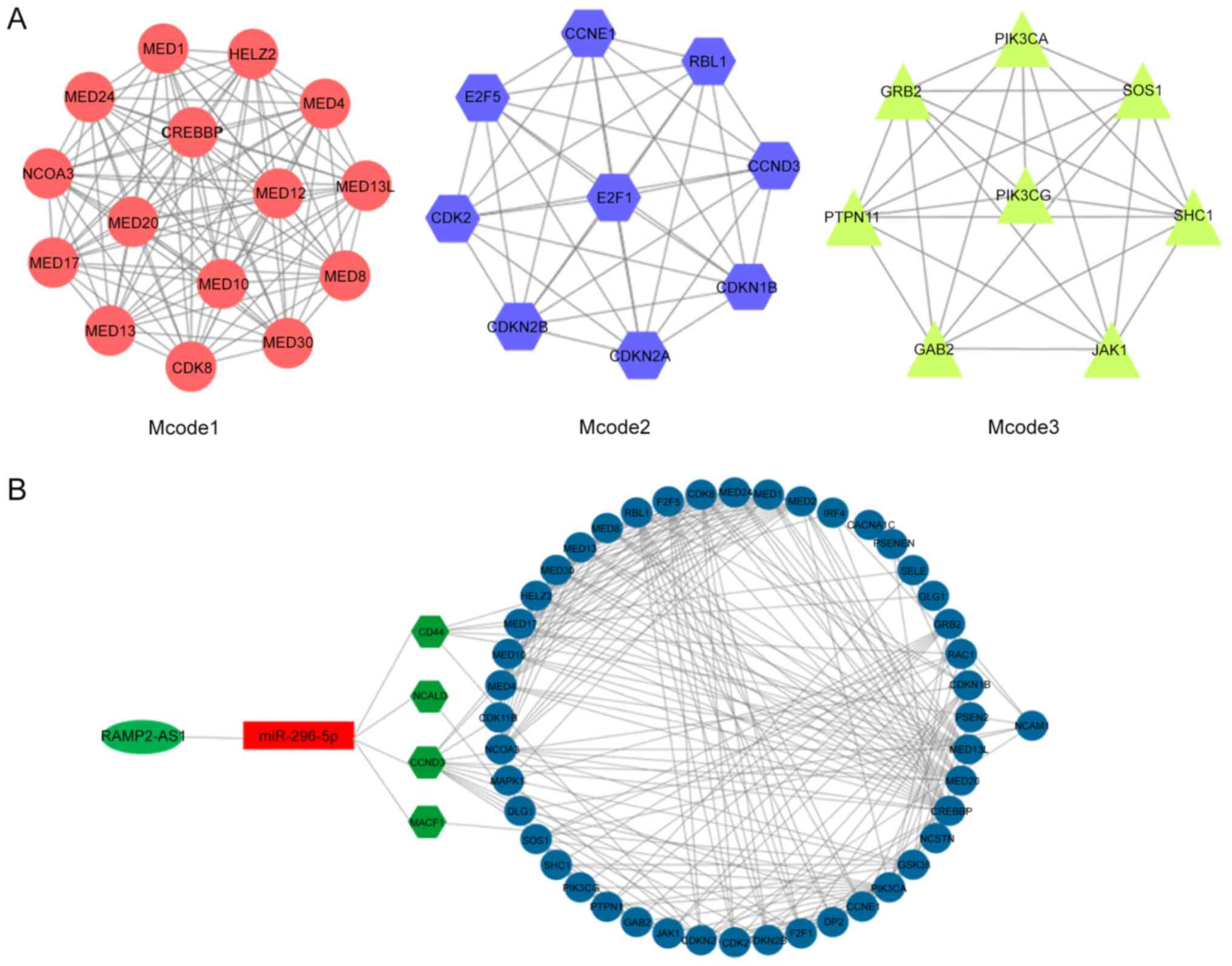
Mcode function of Cytoscape, three enriched modules were identified
that exhibited a marked overlap with the genes identified in the
previous enrichment analysis (Fig.
6A). The functions of these three modules were mainly enriched
in these processes, namely ‘generic transcription pathway’, ‘cyclin
D associated events in G1’, ‘N-cadherin signaling
events’, ‘stabilization and expansion of the E-cadherin adherens
junction’ and ‘CDC42 signaling events’ (Fig. 5B), reflecting the main functions of
the network composed of RAMP2-AS1 and the target genes CD44, CCND3,
NCALD and MACF1. Subsequently, Cytoscape was used for the
visualization of the protein-protein interaction network of
RAMP2-AS1, miR-296-5p, CD44, CCND3, NCALD and MACF1, shown in
Fig. 6B.
Expression levels of lncRNA RAMP2-AS1,
CD44, CCND3, NCALD, MACF1 and miR-296-5p in tumor tissues
The expression levels of RAMP-AS1, miR-296-5p, CD44,
CCND3, NCALD and MACF1 were detected in 40 tumor and adjacent
tissue samples via RT-qPCR. The results demonstrated that the
expression levels of RAMP2-AS1 (P<0.05), CD44 (P<0.05), CCND3
(P<0.001), NCALD (P<0.01) and MACF1 (P<0.001) were
significantly downregulated, while miR-296-5p expression
(P<0.0001) was significantly upregulated in tumor tissues
compared with in adjacent tissues (Fig.
7), which was consistent with the results obtained with the
bioinformatics analysis.
 | Figure 7.Expression levels of RAMP2-AS1, CD44,
CCND3, NCALD, MACF1 and miR-296-5p in tumor and adjacent tissues.
lncRNA RAMP2-AS1, CD44, CCND3, NCALD and MACF1 expression was
significantly downregulated, while miR-296-5p expression was
significantly upregulated in tumor tissues compared with in
adjacent normal tissues. *P<0.05; **P<0.01; ***P<0.001;
****P<0.0001. lncRNA RAMP2-AS1, long non-coding RNA receptor
activity modifying protein 2-antisense RNA 1; CCND3, cyclin D3;
NCALD, neurocalcin δ; MACF1, microtubule actin crosslinking factor
1; miR, microRNA. |
Correlation analysis
In addition, the correlation between the expression
levels of miR-296-5p and those of RAMP2-AS1, CD44, CCND3, NCALD and
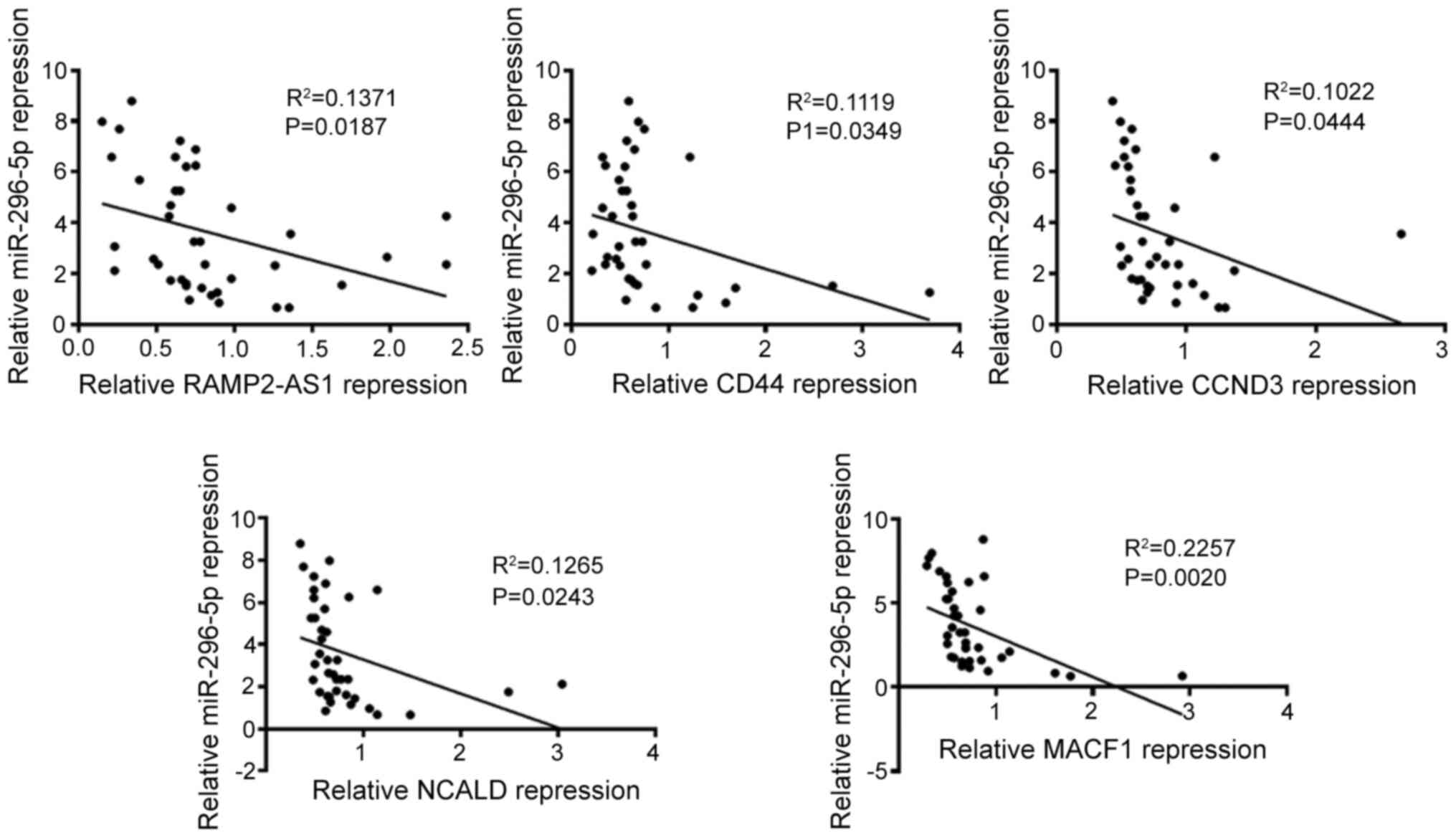
MACF1 was evaluated (Fig. 8). The
results revealed that the expression levels of miR-296-5p were
negatively correlated with that of RAMP2-AS1 (R2=0.1371;
P=0.0187), CD44 (R2=0.1119; P=0.0349), CCND3
(R2=0.1022; P=0.0444), NCALD (R2=0.1265;
P=0.0243) and MACF1 (R2=0.2257; P=0.0020), as analyzed
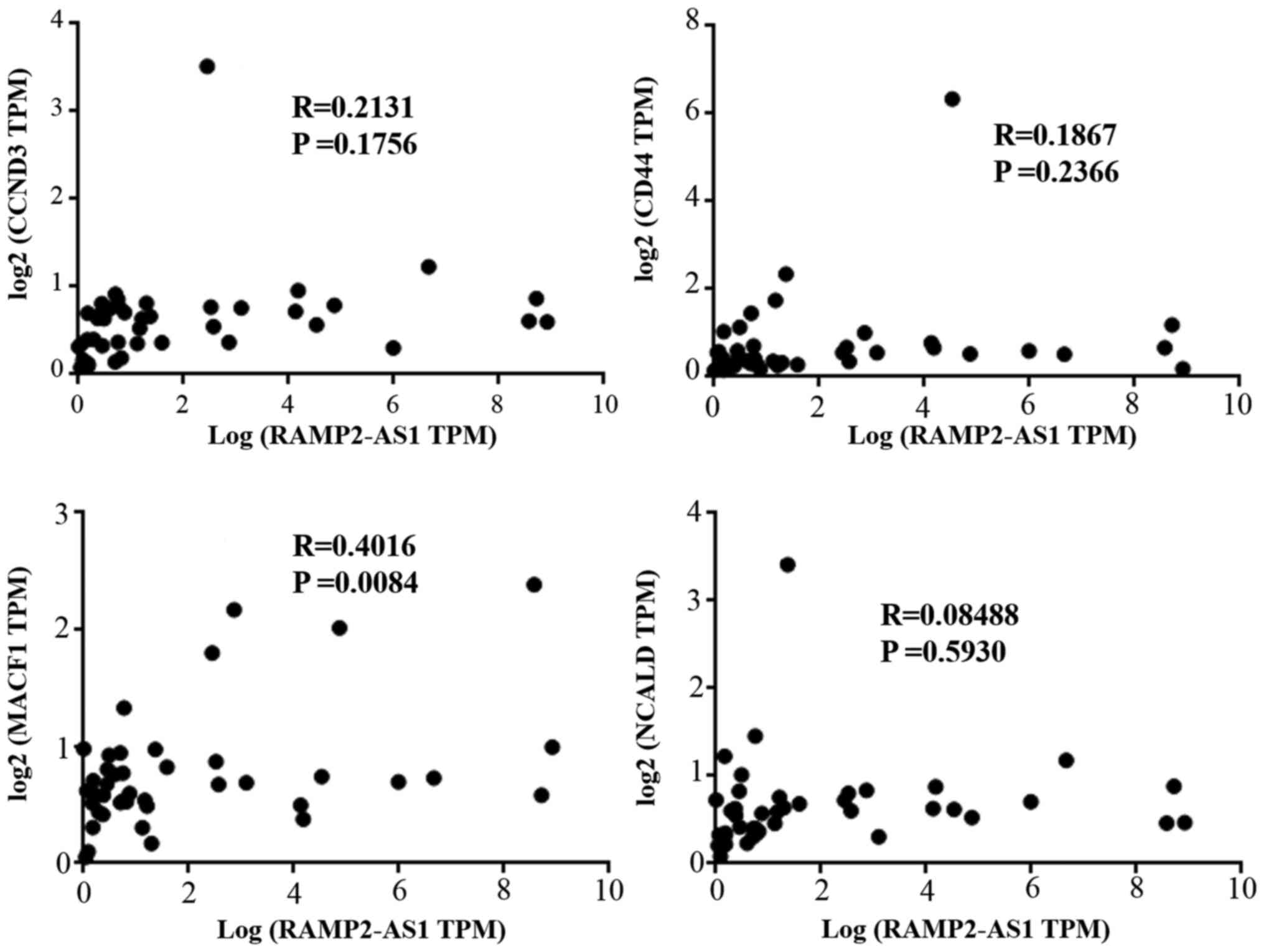
using Spearman's correlation. Subsequently, Pearson's correlation
analysis was performed between the expression levels of RAMP2-AS1
and those of CD44, CCND3, NCALD and MACF1 in the tumor tissues of
the aforementioned 40 patients. The results revealed that RAMP2-AS1
expression was significantly positively correlated with that of
MACF1 (Fig. 9; R=0.4016; P=0.0084),
which was consistent with the results obtained with the
bioinformatics analysis.
Discussion
In recent years, the microarray technology has been
considered as an effective method to identify differentially
expressed genes. An increasing number of studies have demonstrated
that dysregulated genes serve a key role in the occurrence and
development of LUAD (30–32). In the present study, three
differentially expressed lncRNAs and 368 mRNAs were identified from
the GSE113852 and GSE130779 datasets. RAMP2-AS1 was selected as a
key lncRNA for subsequent analyses, as it was significantly
downregulated in LUAD, as well as in most types of cancer, and its
upregulation was associated with improved OS. Consistent with these
findings, a previous study has demonstrated that RAMP2-AS1
expression is significantly decreased in primary glioblastoma
tissues compared with in normal brain tissues and that its
decreased expression levels are associated with poor OS in patients
with glioblastoma (33).
Additionally, RAMP2-AS1 expression in the serum of patients with
chondrosarcoma is closely associated with local invasiveness,
distant metastasis and poor prognosis in patients with
chondrosarcoma (15). Overexpression
of RAMP2-AS1 decreases the proliferation of glioblastoma cells
in vitro, as well as glioblastoma xenografts in vivo
(33). The aforementioned studies
suggest that RAMP2-AS1 may be used as a biomarker for the prognosis
of LUAD.
Furthermore, it has been reported that lncRNA
RAMP2-AS1 in exosomes derived from chondrosarcoma cells may act as
a ceRNA, which combined with miR-2355-5p may modulate VEGFR2
expression, thus positively regulating the angiogenic ability of
HUVECs (15). Therefore, the present
study hypothesized that RAMP2-AS1 may act as a ceRNA to bind to
miRNAs, regulate the expression of target genes and affect the
occurrence of LUAD. Therefore, miR-296-5p was identified to bind to
RAMP2-AS1 as a ceRNA via the Starbase online database, and high
miR-296-5p expression was detected in LUAD. Numerous studies have
demonstrated the impact of lncRNA-miRNA-mRNA functional networks on
the tumorigenesis of human carcinoma (34–36). For
example, it has been revealed that NEAT1 promotes the development
of hepatocellular carcinoma cells via regulating the
miR-296-5p/CNN2 axis (37). Chen
et al (38) indicated that
lncRNA Forkhead box D3 antisense RNA 1 exerted antitumor effects
via upregulating miR-296-5p expression in thyroid cancer. Based on
the ceRNA hypothesis, 5 mRNAs (CD44, CCND3, NCALD, MACF1 and
KCTD15) were identified as target genes for miR-296-5p. Notably,
the expression levels of RAMP2-AS1, CD44, CCND3, NCALD, KCTD15 and
MACF1 were all downregulated in LUAD tissues compared with in
normal tissues, and RAMP2-AS1 expression was positively correlated
with the expression levels of CD44, CCND3, NCALD and MACF1 through
database analysis. Additionally, the present study revealed that
the expression levels of these 5 mRNAs affected the prognosis of
patients with LUAD, suggesting that patients with high expression
levels of the 5 mRNAs survived longer than those with low
expression levels. The results of the Kaplan-Meier plotter analysis
determined that the high expression groups of CD44, CCND3, NCALD
and MACF1 had an improved prognosis compared with the low
expression groups, while KCTD15 exhibited the opposite trend. To
the best of our knowledge, KCTD15 upregulation has never been
reported to be associated with pathological states, although it has
been indirectly associated with several types of cancer, such as
pleomorphic adenoma and medulloblastoma (39,40). The
specific role of KCTD15 in LUAD should be investigated in future
studies. The present data indicated that RAMP2-AS1 may act as a
ceRNA to bind miR-2355-5p, regulate its target genes CD44, CCND3,
NCALD and MACF1, and then affect the development of LUAD.
Furthermore, the expression levels of CD44, CCND3, NCALD, MACF1 and
KCTD15 were downregulated in the vast majority of LUAD histological
subtypes compared with in normal samples, and they were
differentially expressed according to tumor stage and nodal
metastasis status. However, a limitation of the present study is
that no comparison was made between these expression levels and
patient clinicopathological characteristics, and prognosis data was
not collected for the 40 clinical samples.
Screening using the cBioPortal and FunRich tools
revealed that 50 genes were closely associated with CD44, CCND3,
NCALD and MACF1. These genes were enriched in the ‘cyclin D
associated events in G1’, ‘generic transcription
pathway’, ‘CDC42 signaling events’, ‘stabilization and expansion of
the E-cadherin adherens junction’ and ‘N-cadherin signaling
events’. In addition, using the STRING database and Cytoscape,
three enriched modules among these 50 genes were predicted. The
genes involved in the enriched modules overlapped with those
identified in the enrichment analysis, thus indicating that
‘generic transcription pathway’, ‘cyclin D associated events in
G1’, ‘N-cadherin signaling events’, ‘stabilization and
expansion of the E-cadherin adherens junction’ and ‘CDC 42
signaling events’ were the main functions of the network composed
of RAMP2-AS1, miR-296-5p, CD44, CCND3, NCALD and MACF1. The data
further determined that miR-296-5p expression was negatively
correlated with that of RAMP2-AS1, CD44, CCND3, NCALD and MACF1,
while RAMP2-AS1 expression was positively correlated with MACF1
expression.
MACF1 is a spectraplakin cytoskeletal crosslinking
protein that can decrease the toxicity to normal tissues while
improving the efficacy of radiation (41). It has been used as a targeted
diagnostic marker for glioblastoma (42). Notably, a previous study has
suggested that MACF1 mutations are associated with HPV-negative
vulvar cancer (43). It is well
known that EGFR mutations can influence the prognosis of cancer.
EGFR may be used as a promising therapeutic target and EGFR
mutations are associated with a poor prognosis in patients with
ovarian cancer (44). However, the
specific mechanism in LUAD remains unclear. It is well known that
miRNAs can target one or more genes to affect their role in tumors.
Due to the lack of stratification based on EGFR mutations, the
potential association between key genes and EGFR mutations has not
been clarified in the current study, which is another limitation
that should be further investigated in future research.
In the present study, various databases were used to
analyze genes, which may be different from the selected databases
of other studies. For example, the two datasets used by Li et
al (11) consisted of
non-metastatic and metastatic samples, while another study directly
derived data from TCGA database (12). In addition, the present study
performed differential gene expression, functional enrichment,
molecular network and prognostic analyses, as well as analyzing the
correlation between gene expression levels and different modules,
using the GEPIA database to achieve data visualization, which is an
advantage of this study. However, in vitro experiments to
corroborate the results of the present database analysis were not
performed, and the current results are not comprehensive.
Therefore, further investigations should be performed.
Overall, the present study suggested that
miR-296-5p, RAMP2-AS1, CD44, CCND3, NCALD and MACF1 may serve as
potential reliable biomarkers for the detection of LUAD, and
provided a possible theoretical basis for the pathogenesis of LUAD.
However, the possible molecular mechanisms associated with the
described ceRNA regulatory network were based on bioinformatics
analyses and basic in vitro experiments in LUAD tissues.
Therefore, the underlying regulatory mechanism should be further
investigated in future research.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific
Research Project of Hubei Provincial Health Committee (grant. no.
WJ2019M194), the Xisike-Hengrui Cancer Research Fund (grant no.
Y-HR2018-328) and the Xisike-BMS Cancer Immunotherapy Research Fund
(grant no. Y-BMS2019-003).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article. The datasets generated and/or
analyzed during the current study are available in the Gene
Expression Profiling Interactive Analysis (http://gepia.cancer-pku.cn/detail.php), the cBio
Cancer Genomics Portal (cBioPortal; http://cbioportal.org) and the UALCAN platform
(http://ualcan.path.uab.edu/).
Authors' contributions
ZS and BZ designed the study. YZ and ZC performed
the experiments. ZS, YZ and ZC analyzed the data. ZS wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All patients provided written informed consent, in
accordance with the ethical guidelines of Zhongnan Hospital of
Wuhan University. Additionally, the collection of human tumor
tissues was approved by the Ethical Committee of Zhongnan Hospital
of Wuhan University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ma D, Li S, Cui Y, Li L, Liu H, Chen Y and
Zhou X: Paclitaxel increases the sensitivity of lung cancer cells
to lobaplatin via PI3K/Akt pathway. Oncol Lett. 15:6211–6216.
2018.PubMed/NCBI
|
|
3
|
Warth A, Muley T, Meister M, Stenzinger A,
Thomas M, Schirmacher P, Schnabel PA, Budczies J, Hoffmann H and
Weichert W: The novel histologic International Association for the
Study of Lung Cancer/American Thoracic Society/European Respiratory
Society classification system of lung adenocarcinoma is a
stage-independent predictor of survival. J Clin Oncol.
30:1438–1446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tay Y, Kats L, Salmena L, Weiss D, Tan SM,
Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, et al:
Coding-independent regulation of the tumor suppressor PTEN by
competing endogenous mRNAs. Cell. 147:344–357. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang R, Xing L, Zheng X, Sun Y, Wang X and
Chen J: The circRNA circAGFG1 acts as a sponge of miR-195-5p to
promote triple-negative breast cancer progression through
regulating CCNE1 expression. Mol Cancer. 18:42019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Su H, Tao T, Yang Z, Kang X, Zhang X, Kang
D, Wu S and Li C: Circular RNA cTFRC acts as the sponge of
MicroRNA-107 to promote bladder carcinoma progression. Mol Cancer.
18:272019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tay Y, Karreth FA and Pandolfi PP:
Aberrant ceRNA activity drives lung cancer. Cell Res. 24:259–260.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li L, Peng M, Xue W, Fan Z, Wang T, Lian
J, Zhai Y, Lian W, Qin D and Zhao J: Integrated analysis of
dysregulated long non-coding RNAs/microRNAs/mRNAs in metastasis of
lung adenocarcinoma. J Transl Med. 16:3722018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li DS, Ainiwaer JL, Sheyhiding I, Zhang Z
and Zhang LW: Identification of key long non-coding RNAs as
competing endogenous RNAs for miRNA-mRNA in lung adenocarcinoma.
Eur Rev Med Pharmacol Sci. 20:2285–2295. 2016.PubMed/NCBI
|
|
13
|
Dong HX, Wang R, Jin XY, Zeng J and Pan J:
lncRNA DGCR5 promotes lung adenocarcinoma (LUAD) progression via
inhibiting hsa-mir-22-3p. J Cell Physiol. 233:4126–4136. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiong DD, Li ZY, Liang L, He RQ, Ma FC,
Luo DZ, Hu XH and Chen G: The LncRNA NEAT1 Accelerates Lung
Adenocarcinoma Deterioration and Binds to Mir-193a-3p as a
Competitive Endogenous RNA. Cell Physiol Biochem. 48:905–918. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheng C, Zhang Z, Cheng F and Shao Z:
Exosomal lncRNA RAMP2-AS1 Derived from Chondrosarcoma Cells
Promotes Angiogenesis Through miR-2355-5p/VEGFR2 Axis. OncoTargets
Ther. 13:3291–3301. 2020. View Article : Google Scholar
|
|
16
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42D:D92–D97. 2014. View Article : Google Scholar
|
|
17
|
Andrews MC, Cursons J, Hurley DG, Anaka M,
Cebon JS, Behren A and Crampin EJ: Systems analysis identifies
miR-29b regulation of invasiveness in melanoma. Mol Cancer.
15:722016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang H, Zhao Y, Chen T, Liu G, He N and Hu
H: miR-371 promotes proliferation and metastasis in hepatocellular
carcinoma by targeting PTEN. BMB Rep. 52:312–317. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang Z, Wu L, Wang A, Tang W, Zhao Y, Zhao
H and Teschendorff AE: dbDEMC 2.0: Updated database of
differentially expressed miRNAs in human cancers. Nucleic Acids
Res. 45D:D812–D818. 2017. View Article : Google Scholar
|
|
20
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res.
45W:W98–W102. 2017. View Article : Google Scholar
|
|
21
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A Portal for Facilitating Tumor Subgroup Gene
Expression and Survival Analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fouad TM, Barrera AMG, Reuben JM, Lucci A,
Woodward WA, Stauder MC, Lim B, DeSnyder SM, Arun B, Gildy B, et
al: Inflammatory breast cancer: A proposed conceptual shift in the
UICC-AJCC TNM staging system. Lancet Oncol. 18:e228–e232. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shin J, Shin S, Lee JH, Song KB, Hwang DW,
Kim HJ, Byun JH, Cho H, Kim SC and Hong SM: Lymph node size and its
association with nodal metastasis in ductal adenocarcinoma of the
pancreas. J Pathol Transl Med. 54:387–395. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nagy Á, Lánczky A, Menyhárt O and Győrffy
B: Validation of miRNA prognostic power in hepatocellular carcinoma
using expression data of independent datasets. Sci Rep. 8:92272018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pathan M, Keerthikumar S, Ang CS, Gangoda
L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim
A, et al: FunRich: An open access standalone functional enrichment
and interaction network analysis tool. Proteomics. 15:2597–2601.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Szklarczyk D, Morris JH, Cook H, Kuhn M,
Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al:
The STRING database in 2017: Quality-controlled protein-protein
association networks, made broadly accessible. Nucleic Acids Res.
45D:D362–D368. 2017. View Article : Google Scholar
|
|
28
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qiu M, Xu Y, Wang J, Zhang E, Sun M, Zheng
Y, Li M, Xia W, Feng D, Yin R, et al: A novel lncRNA, LUADT1,
promotes lung adenocarcinoma proliferation via the epigenetic
suppression of p27. Cell Death Dis. 6:e18582015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shang J, Wang Z, Chen W, Yang Z, Zheng L,
Wang S and Li S: Pseudogene CHIAP2 inhibits proliferation and
invasion of lung adenocarcinoma cells by means of the WNT pathway.
J Cell Physiol. 234:13735–13746. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mao S, Li Y, Lu Z, Che Y, Huang J, Lei Y,
Wang Y, Liu C, Wang X, Zheng S, et al: PHD finger protein 5A
promoted lung adenocarcinoma progression via alternative splicing.
Cancer Med. 8:2429–2441. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu S, Mitra R, Zhao MM, Fan W, Eischen
CM, Yin F and Zhao Z: The Potential Roles of Long Noncoding RNAs
(lncRNA) in Glioblastoma Development. Mol Cancer Ther.
15:2977–2986. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou SL, Tang QL, Zhou SX and Ren RZ:
miR-296-5p suppresses papillary thyroid carcinoma cell growth via
targeting PLK1. Eur Rev Med Pharmacol Sci. 23:2084–2091.
2019.PubMed/NCBI
|
|
35
|
Li S, Zheng H, Chen L, Xu C, Qu X, Qin Z,
Gao J, Li J and Liu J: Expression Profile and Potential Functions
of Circulating Long Noncoding RNAs in Acute Ischemic Stroke in the
Southern Chinese Han Population. Front Mol Neurosci. 12:2902019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang P, Ning S, Zhang Y, Li R, Ye J, Zhao
Z, Zhi H, Wang T, Guo Z and Li X: Identification of
lncRNA-associated competing triplets reveals global patterns and
prognostic markers for cancer. Nucleic Acids Res. 43:3478–3489.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li Y, Ding X, Xiu S, Du G and Liu Y:
LncRNA NEAT1 Promotes Proliferation, Migration And Invasion Via
Regulating miR-296-5p/CNN2 Axis In Hepatocellular Carcinoma Cells.
OncoTargets Ther. 12:9887–9897. 2019. View Article : Google Scholar
|
|
38
|
Chen Y, Gao H and Li Y: Inhibition of
LncRNA FOXD3-AS1 suppresses the aggressive biological behaviors of
thyroid cancer via elevating miR-296-5p and inactivating
TGF-β1/Smads signaling pathway. Mol Cell Endocrinol.
500:1106342020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Choi JS, Cho BH, Kim HJ, Kim YM and Jang
JH: Identification of new genes of pleomorphic adenoma. Medicine
(Baltimore). 98:e184682019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Spiombi E, Angrisani A, Fonte S, De Feudis
G, Fabretti F, Cucchi D, Izzo M, Infante P, Miele E, Po A, et al:
KCTD15 inhibits the Hedgehog pathway in Medulloblastoma cells by
increasing protein levels of the oncosuppressor KCASH2.
Oncogenesis. 8:642019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bonner K, Borlay D, Kutten O and Quick QA:
Inhibition of the Spectraplakin Protein Microtubule Actin
Crosslinking Factor 1 Sensitizes Glioblastomas to Radiation. Brain
Tumor Res Treat. 8:43–52. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Afghani N, Mehta T, Wang J, Tang N, Skalli
O and Quick QA: Microtubule actin cross-linking factor 1, a novel
target in glioblastoma. Int J Oncol. 50:310–316. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Prieske K, Alawi M, Oliveira-Ferrer L,
Jaeger A, Eylmann K, Burandt E, Schmalfeldt B, Joosse SA and
Woelber L: Genomic characterization of vulvar squamous cell
carcinoma. Gynecol Oncol. 158:547–554. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mallmann-Gottschalk N, Sax Y, Kimmig R,
Lang S and Brandau S: EGFR-Specific Tyrosine Kinase Inhibitor
Modifies NK Cell-Mediated Antitumoral Activity against Ovarian
Cancer Cells. Int J Mol Sci. 20:202019. View Article : Google Scholar
|