Introduction
Cancer cachectic muscle atrophy (CCMA) is a
multifactorial metabolic syndrome characterized by muscle atrophy
and a progressive loss of muscle function (1). It occurs in ~85% of terminal patients
with upper gastrointestinal cancer and was responsible for ~20% of
all cancer deaths worldwide in 2010 (2). Conventional nutritional support cannot
fully reverse CCMA, and no effective treatment has been reported
(3). Although the pathogenesis of
CCMA has been constantly researched in the past decade, it remains
not well established.
CCMA is associated with increased muscle proteolysis
and decreased protein synthesis, which involve the calpain system
(4), the ubiquitin-proteasome system
(5), the autophagy-lysosome system
(6) and the PI3K/AKT signaling
pathway (7). Since only the calpain
system can degrade intact myofilaments, calpain-dependent cleavage
of myofilaments is considered as the initial step in muscle
proteolysis, serving a critical role in CCMA (8).
Calpains are a family of 15 calcium-activated
cysteine proteases, including two ubiquitously expressed members,
calpain-1 and calpain-2, and one muscle-specific member, calpain-3
(9,10). Calpastatin (CAST) is the only known
ubiquitously expressed endogenous calpain inhibitor (9). Calpains and CAST are generally
considered to be localized in the cytoplasm and involved in
numerous physiological and disease processes, including muscular
dystrophy, diabetes, neurological disorders and hematonosis
(11,12). Calpain-1, calpain-2 and CAST have
been found in the mitochondria of cardiomyocytes and pulmonary
smooth muscle cells; in these cells, activation of mitochondrial
calpain induces mitochondrial injury and cell damage (13,14).
It has been reported that CCMA is also associated
with mitochondrial injury (15). To
the best of our knowledge, the role of mitochondrial calpain in
CCMA has not yet been investigated. In the present study, a
Transwell-plate system was used to develop a myotube-carcinoma cell
co-culture model to simulate the cancer cachexia environment in
vitro. The presence of calpains in the mitochondria of skeletal
muscle and their potential role in CCMA were investigated.
Materials and methods
Cell culture
Mouse C2C12 myoblasts and CT26 colon carcinoma cells
were obtained from the American Type Culture Collection and
maintained in DMEM (Invitrogen; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin-streptomycin. The cells were
cultured in an atmosphere of 5% CO2 at 37°C.
To establish the co-culture system, C2C12 myoblasts
were seeded in 6-well plates at a density of 30,000
cells/cm2 and cultured in growth medium for 24–48 h at
37°C to reach 90–100% confluence. Subsequently, the growth medium
was replaced by differentiation medium composed of DMEM and 2%
horse serum (Gibco; Thermo Fisher Scientific, Inc.) to induce
myoblast differentiation. The myoblasts were cultured for another 4
days at 37°C to allow their differentiation into myotubes, and the
differentiation medium was replaced by growth medium. At the same
time, CT26 cells were seeded (20,000 cells/cm2) into
Transwell inserts (Corning Life Sciences) on a different 6-well
plate containing growth medium. After a further 24 h of culture at
37°C, the inserts were placed into the wells containing myotubes,
and the medium was changed to growth medium with or without the
calpain inhibitors CAST (cat. no. 208902; 1 µM; EMD Millipore) and
calpeptin (CAPT; cat. no. C8999; 50 µM; Sigma-Aldrich; Merck KGaA).
After 24 h of co-culture at 37°C, the myotubes were fixed for
immunocytochemistry or harvested for further analysis.
The co-culture combinations consisted of 5 groups:
i) Sham myotubes (without CT26 cells in the insert) without calpain
inhibitors (NC group); ii) myotubes co-cultured with CT26 cells but
without calpain inhibitors (CO group); iii) myotubes with CT26
cells and CAST (CAST group); iv) myotubes with CT26 cells and CAPT
(CAPT group); and v) myotubes with CT26 cells and CAST plus CAPT
(CC group).
Immunocytochemical analysis
Myotubes were fixed, permeabilized, blocked and
incubated with anti-myosin heavy chain (MHC) primary antibody
(1:200; cat. no. ab91506; Abcam) followed by Alexa Fluor
488-conjugated goat anti-rabbit IgG secondary antibody (1:500; cat.
no. ab150077; Abcam) as previously described (16). The myotubes were then mounted with
Fluoroshield Mounting Medium containing DAPI (cat. no. ab104139;
Abcam). Images were acquired with a fluorescence microscope (Nikon
Corporation; magnification, ×100) and analyzed with ImageJ software
(version 1.46r; National Institutes of Health).
Myotube diameters were measured as previously
described (17). Briefly, 20
images/well were captured, and the diameters of the five largest
myotubes (those containing ≥3 nuclei when viewed at ×100
magnification) in each image were measured. Next, the mean diameter
of a single myotube was calculated based on three independent
measurements. The measurement points were separated by 200 µm. This
method was also used to calculate the mean diameter ± SD of the 100
largest myotubes in each well.
Isolation of myotube mitochondria
The Mitochondria Isolation kit for Cultured Cells
(cat. no. ab110170; Abcam) was used to isolate myotube cytoplasm
and mitochondria according to the manufacturer's protocol as
previously described (18). Both
cytoplasm and mitochondria were collected for further analysis.
Western blot analysis
Myotube cytosolic and mitochondrial proteins were
extracted and separated via SDS-PAGE, blocked and stained with
primary antibodies and HRP-conjugated secondary antibodies
(1:2,000; cat. nos. ab97051 and ab6789; Abcam), and visualized as
previously described (19). Images
of the membranes were recorded with ChemiDoc XRS+ system (Bio-Rad
Laboratories, Inc.) and analyzed using Quantity One software
(version 4.6.6; Bio-Rad Laboratories, Inc.). The following primary
antibodies were used: MHC (1:1,000; cat. no. ab124937), calpain-1
(1:1,000; cat. no. ab108400), calpain-2 (1:2,000; cat. no.
ab126600), CAST (1:1,000; cat. no. ab28252), NADH dehydrogenase
(ubiquinone) iron-sulfur protein 3, mitochondrial (NDUFS3; 1:2,000;
cat. no. ab177471), cyclophilin D (CYCD; 1:2,000; cat. no.
ab181983), phosphorylated- (p-)AKT S473 (1:1,000; cat. no.
ab81283), AKT (1:5,000; cat. no. ab179463), p-mTOR S2448 (1:1,000;
cat. no. ab109268), mTOR (1:1,000; cat. no. ab32028), p-eukaryotic
translation initiation factor 4E-binding protein 1 (p-4EBP1) T37
(1:1,000; cat. no. ab75767), 4EBP1 (1:2,000; cat. no. ab32024),
p-FoxO3a S253 (1:1,000; cat. no. ab31109), FoxO3a (1:1,000; cat.
no. ab70315), atrogin1 (1:2,000; cat. no. ab168372),
muscle-specific RING finger protein 1 (MuRF1; 1:2,000; cat. no.
ab172479), GAPDH (1:2,000; cat. no. ab181602) and voltage-dependent
anion-selective channel protein 1 (VDAC; 1:1,000; cat. no.
ab154856), all purchased from Abcam; calpain-3 (1:1,000; cat. no.
sc-365277) and spectrin [cat. no. sc-46696; detects both
full-length (F)-spectrin and cleaved (C)-spectrin] from Santa Cruz
Biotechnology, Inc.; and apoptosis-inducing factor (AIF; 1:1,000;
cat. no. 4642S) from Cell Signaling Technology, Inc.
Calpain activity assay
A calpain activity assay kit (cat. no. QIA120; EMD
Millipore) was used to measure calpain activity in samples of
myotube cytoplasm and mitochondria, according to the manufacturer's
protocol as previously described (20). The assay plates were read using a
SpectraMax M5 microplate reader with a wavelength of 380 nm
(Molecular Devices, LLC).
Complex I enzyme activity assay
The Complex I Enzyme Activity Microplate Assay kit
(cat. no. ab109721; Abcam) was used to measure mitochondrial
complex I enzyme activity in samples of myotube mitochondria
according to the manufacturer's protocol as previously described
(18). The assay plates were read
using a SpectraMax M5 microplate reader with a wavelength of 450
nm.
JC-1 staining assay
Myotubes from 2 wells were collected using trypsin
(cat. no. 25200072; Gibco; Thermo Fisher Scientific, Inc.) and
plated in 96-well dark plates. JC-1 dye (cat. no. C2006; Beyotime
Institute of Biotechnology) was used to detect the mitochondrial
membrane potential (Δψm) according to the manufacturer's
protocol. Δψm was measured by quantifying the
fluorescence emission shift from green (530 nm) monomers to red
(590 nm) aggregates (21). The assay
plates were read using a SpectraMax M5 microplate reader. Data were
expressed as fold increase in the red/green ratio.
Mitochondrial permeability transition
pore (MPTP) opening
Myotubes were collected and plated in 96-well dark
plates. MitoProbe Transition Pore Assay kit (cat. no. M34153;
Thermo Fisher Scientific, Inc.) was used to measure MPTP opening
according to the manufacturer's protocol as previously described
(21). Myotubes were incubated with
calcein AM dye (included in the kit) for 15 min and then incubated
with CoCl2 for 15 min at 37°C. Ionomycin was used as a positive
control. The assay plates were read using a SpectraMax M5
microplate reader with a wavelength of 480 nm.
Statistical analysis
Each experiment was repeated at least three times,
and all data were analyzed using SPSS version 19.0 (IBM Corp.).
Results are shown as the mean ± SD. Statistical comparisons between
groups were analyzed using one-way ANOVA followed by Tukey's
post-hoc test when equal variances were assumed or Dunnett's T3
test when equal variances were not assumed. P<0.05 was
considered to indicate a statistically significant difference.
Results
Calpain inhibitors decrease myotube
atrophy
Myotube diameter was significantly decreased
following 24 h of co-culture (CO group) compared with the NC group
(Fig. 1), suggesting that the cell
co-culture model can simulate muscle atrophy. Compared with
untreated myotubes (CO group), both CAST and CAPT treatment, as
well as the combination of both treatments (CC group),
significantly increased myotube diameter, indicating that
inhibition of calpain attenuated myotube atrophy during co-culture
of myoblasts and colon carcinoma cells.
 | Figure 1.Effect of calpain inhibitors in
myotube atrophy. Immunofluorescence staining for anti-MHC antibody
in mouse C2C12 myotubes. MHC staining outlines the myotubes
(green). DAPI was used to stain nuclei (blue). Magnification, ×100.
Scale bar, 100 µm. The myotube diameter was measured for each group
and is represented in the bar graph. Data are represented as mean ±
SD. ***P<0.001 vs. CO. MHC, myosin heavy chain; NC group, sham
myotubes without CT26 cells and without calpain inhibitors; CO
group, myotubes with CT26 cells without calpain inhibitors; CAST
group, myotubes with CT26 cells and CAST; CAPT group, myotubes with
CT26 cells and CAPT; CC group, myotubes with CT26 cells and CAST
plus CAPT; CAST, calpastatin; CAPT, calpeptin. |
Calpain inhibitors prevent calpain
activation during co-culture
Co-culture activated both cytosolic and
mitochondrial calpains in myotubes. Both cytosolic calpain-1
(cyt-CPN1) and mitochondrial calpain-1 (mit-CPN1) contents were
increased by co-culture compared with the NC group (Fig. 2A, B and I). Direct enzyme activity
assays revealed that cytosolic and mitochondrial calpain activities
were also increased by co-culture (Fig.
2L and M). Cytosolic calpain activation can also be measured by
calculating the C-spectrin/F-spectrin ratio in myotubes (22). The results indicated that this ratio
was significantly increased in co-cultured myotubes, indicating the
increased formation of C-spectrin (Fig.
2A and F-H). Compared with the CO group, CAST, CAPT and CC
treatment markedly decreased both CPN-1 expression in the western
blots (Fig. 2A and B) and cytosolic
calpain activity (Fig. 2L), the
formation of C-spectrin (Fig. 2F-H)
and the mit-CPN1 content (Fig. 2A and
I). Cyt-CPN1 content (Fig. 2A and
B) and mitochondrial calpain activity (Fig. 2M) were also decreased by CAST, CAPT
and CC treatment, but did not reach a statistically significant
difference. These data indicated that CAST, CAPT and CC treatment
attenuated cytosolic and mitochondrial calpain activation.
Cyt-CPN-2, cyt-CPN3, cyt-CAST, mit-CPN-2 and mit-CAST were also
detected in myotubes, but their protein expression levels were not
altered by co-culture, CAST, CAPT or CC treatment (Fig. 2A, C-E, J and K).
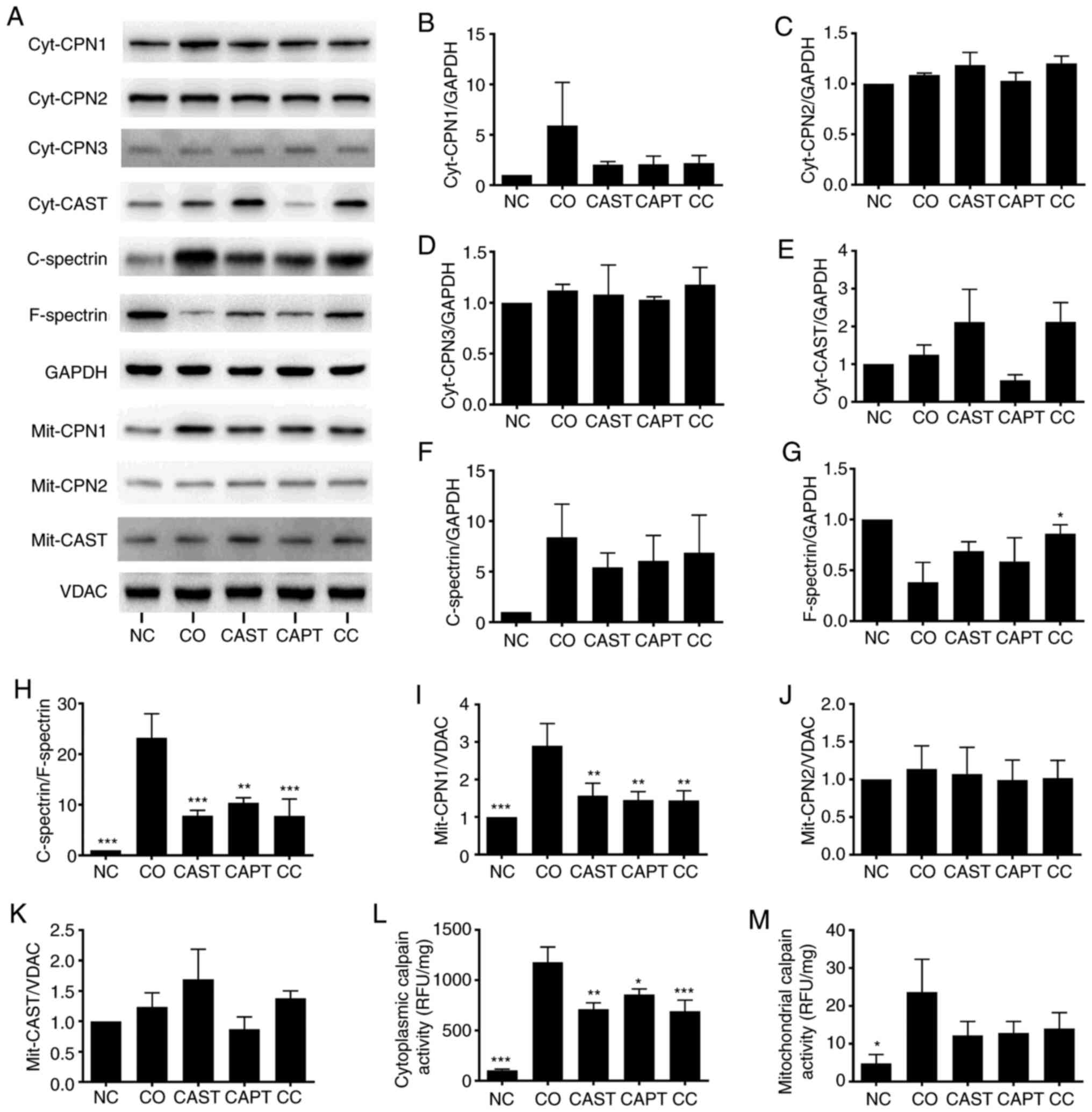 | Figure 2.Effect of calpain inhibitors in the
activation of calpain during co-culture. (A) Western blot results.
Quantification of (B) cyt-CPN1, (C) cyt-CPN2, (D) cyt-CPN3 and (E)
cyt-CAST normalized to GAPDH. (F) Quantification of cytosolic
C-spectrin normalized to GAPDH. (G) Quantification of cytosolic
F-spectrin normalized to GAPDH. (H) Quantification of
C-spectrin/F-spectrin ratio. (I) Quantification of mit-CPN1
normalized to VDAC. Quantification of (J) mit-CPN2 and (K) mit-CAST
normalized to VDAC. (L) Quantification of cytosolic calpain
activity. (M) Quantification of mitochondrial calpain activity.
Data are represented as the mean ± SD. *P<0.05, **P<0.01 and
***P<0.001 vs. CO. C-spectrin, cleaved spectrin; F-spectrin,
full-length spectrin; NC group, sham myotubes without CT26 cells
and without calpain inhibitors; CO group, myotubes with CT26 cells
without calpain inhibitors; CAST group, myotubes with CT26 cells
and CAST; CAPT group, myotubes with CT26 cells and CAPT; CC group,
myotubes with CT26 cells and CAST plus CAPT; CAST, calpastatin;
CAPT, calpeptin; cyt-, cytosolic; CPN, calpain; VDAC,
voltage-dependent anion-selective channel protein 1; mit-,
mitochondrial; RFU, relative fluorescence unit. |
Calpain inhibitors improve complex I
activity in myotube mitochondria following co-culture
Complex I is the first respiratory complex of the
mitochondrial electron transport chain (13). Activation of mitochondrial calpain
damages complex I activity, which impairs energy generation, leads
to MPTP opening and cardiac injury during reperfusion (13). Complex I activity was significantly
decreased in mitochondria following co-culture compared with the NC
control (Fig. 3A). CAST, CAPT and CC
treatment slightly improved complex I activity, but did not reach
statistical significance (Fig. 3A).
Additionally, NDUFS3 is a core subunit of complex I that is
essential for NADH oxidation and subsequent electron transfer
through the complex, maintaining the activity of complex I
(13). NDUFS3 content was not
altered by co-culture, CAST, CAPT or CC treatment (Fig. 3B and C).
 | Figure 3.Effect of calpain inhibitors on
complex I activity and MPTP opening in myotubes during co-culture
with colon carcinoma cells. (A) Quantification of mitochondrial
complex I activity. (B) Western blot results. (C) Quantification of
mitochondrial NDUFS3 normalized to VDAC. (D) Quantification of JC-1
red/green ratio. (E) Quantification of MPTP opening ratio. (F)
Quantification of mitochondrial CYCD normalized to VDAC. (G)
Quantification of mitochondrial AIF normalized to VDAC. Data are
represented as the mean ± SD. *P<0.05 and ***P<0.001 vs. CO.
NC group, sham myotubes without CT26 cells and without calpain
inhibitors; CO group, myotubes with CT26 cells without calpain
inhibitors; CAST group, myotubes with CT26 cells and CAST; CAPT
group, myotubes with CT26 cells and CAPT; CC group, myotubes with
CT26 cells and CAST plus CAPT; CAST, calpastatin; CAPT, calpeptin;
VDAC, voltage-dependent anion-selective channel protein 1; NDUFS3,
NADH dehydrogenase (ubiquinone) iron-sulfur protein 3,
mitochondrial; AIF, apoptosis-inducing factor; CYCD, cyclophilin D;
MPTP, mitochondrial permeability transition pore; mOD, mean optical
density. |
Calpain inhibitors increase the
Δψm in myotubes following co-culture
JC-1 was used to assess the Δψm in
myotubes (21). The Δψm
was reflected by the JC-1 red/green ratio. Compared with the NC
group, the JC-1 red/green ratio in the CO group was significantly
decreased by 70% (Fig. 3D),
indicating damage of the Δψm following co-culture.
However, treatment with CAST, CAPT or CC significantly improved
this ratio compared with the CO group (Fig. 3D), indicating that the activation of
calpain during co-culture is able to damage the Δψm in
myotubes.
Calpain inhibitors decrease the MPTP
opening in myotube following co-culture
The opening of the MPTP results in a permeation of
the mitochondrial membrane that leads to the loss of Δψm
(21). Thus, the MitoProbe assay was
used to assess the MPTP opening in myotubes (21). The MPTP opening was significantly
increased in the CO group compared with in the NC group (Fig. 3E), indicating the increased MPTP
opening following co-culture. However, CAST, CAPT and CC treatment
seemed to decrease the MPTP opening compared with the CO group,
although there were no significant differences (Fig. 3E).
CYCD is a key factor that regulates MPTP opening
(23). Compared with the NC group,
co-culture significantly decreased the CYCD content in myotube
mitochondria (Fig. 3B and F),
whereas CAST and CC treatment significantly increased the CYCD
content compared with co-culture alone. The present results
suggested that the activation of calpain during co-culture
decreased the CYCD content, which led to MPTP opening in
mitochondria.
Calpain inhibitors preserve AIF
content in myotube mitochondria
In addition to promoting the MPTP opening, calpain
can cleave AIF and induce its release from the mitochondria to the
cytoplasm (24,25). Similarly, the current results
demonstrated that co-culture significantly decreased the AIF
content in myotube mitochondria compared with the NC group, and
that this decrease was significantly attenuated by CAST and CC
treatment (Fig. 3B and G). However,
assays failed to detect the cleaved AIF in both cytoplasm and
mitochondria (data not shown).
Calpain inhibitors increase the
AKT/mTOR activity and decrease FoxO3a activity in myotubes
following co-culture
AKT/mTOR and FoxO3a serve a critical role in
regulating muscular protein metabolism (26). Foxo3a is a transcription factor that
activates the expression of ubiquitin ligase Atrogin-1 and MuRF1;
Akt can phosphorylate FoxO3a and decrease its transcriptional
activity (26). Co-culture
significantly decreased the ratios of p-AKT/AKT, p-mTOR/mTOR and
p-FoxO3a/FoxO3a in myotubes compared with the NC control group
(Fig. 4A-C and E). Treatment with
CAST, CAPT and CC significantly increased p-AKT/AKT and p-mTOR/mTOR
ratios compared with the CO group (Fig.
4A-C), but not the p-FoxO3a/FoxO3a ratio (Fig. 4E). The p-4EBP1/4EBP1 ratio was not
significantly affected by co-culture or treatment with the calpain
inhibitors CAST or CAPT (Fig. 4A and
D). The present results suggested that activation of calpain
during co-culture decreased AKT/mTOR activity and increased FoxO3a
activity in myotubes.
 | Figure 4.Effect of calpain inhibitors in
AKT/FoxO3a signaling pathway activity and atrogin-1 content in
myotubes during co-culture with colon carcinoma cells. (A) Western
blot results. (B) Quantification of p-AKT/AKT ratio. (C)
Quantification of p-mTOR/mTOR ratio. (D) Quantification of
p-4EBP1/4EBP1 ratio. (E) Quantification of p-FoxO3a/FoxO3a ratio.
(F) Quantification of atrogin-1 normalized to GAPDH. (G)
Quantification of MuRF1 normalized to GAPDH. Data are represented
as the mean ± SD. *P<0.05, **P<0.01 and ***P<0.001 vs. CO.
p-, phosphorylated; NC group, sham myotubes without CT26 cells and
without calpain inhibitors; CO group, myotubes with CT26 cells
without calpain inhibitors; CAST group, myotubes with CT26 cells
and CAST; CAPT group, myotubes with CT26 cells and CAPT; CC group,
myotubes with CT26 cells and CAST plus CAPT; CAST, calpastatin;
CAPT, calpeptin; 4EBP1, eukaryotic translation initiation factor
4E-binding protein 1; MuRF1, muscle-specific RING finger protein
1. |
Calpain inhibitors decrease atrogin-1
content in myotubes following co-culture
Atrogin-1 and MuRF1 are two muscle-specific
ubiquitin ligases that drive muscle protein degradation (8). As shown in Fig. 4A and F, co-culture significantly
increased atrogin-1 content in myotubes compared with the NC
control group, whereas CAST, CAPT and CC treatment significantly
ameliorated this change. However, MuRF1 content was not
significantly affected by co-culture or treatment with the calpain
inhibitors CAST or CAPT (Fig. 4A and
G).
Discussion
Cancer cachexia is a multifactorial syndrome that
affects ~50–80% of patients with cancer, depending on the tumor
type (27,28). In patients with gastric or pancreatic
cancer, the incidence is >80%, whereas ~50% of patients with
colon, lung or prostate cancer are affected, and ~40% of patients
with breast cancer or some leukemias develop the syndrome (27,28). In
the present study, calpain-1, calpain-2 and CAST were detected in
mouse myotube mitochondria. In addition, the results revealed that
co-culture of myoblasts with colon carcinoma cells activated
calpain in myotube mitochondria, caused MPTP opening and
Δψm damage, which led to mitochondrial injury. Moreover,
co-culture activated calpain in myotube cytoplasm, caused the
deactivation of AKT/mTOR and the activation of FoxO3a/atrogin-1.
Calpain inhibitors (CAST, CAPT or CC) prevented calpain activation
in both cytoplasm and mitochondria during co-culture, accompanied
by inhibition of Δψm damage and atrogin-1 expression.
Therefore, the present results indicated that calpain inhibitors
protected the myotube by inhibiting both cytosolic and
mitochondrial calpain activity.
CT26 colorectal adenocarcinoma cell conditioned
medium is often used to induce myotube atrophy to simulate CCMA
in vitro (29,30). In addition, CT26 colorectal
adenocarcinoma and BALB/c mice can be used to develop cancer
cachectic tumor-bearing mice, as previously described (4). Thus, in the present study, CT26 cells
were used to develop a cell co-culture model with myoblasts to
simulate CCMA.
Complex I is the first respiratory complex of the
mitochondrial electron transport chain (31). Activation of mitochondrial calpain
damages complex I activity, which impairs energy generation, leads
to MPTP opening and cardiac reperfusion injury (13,31). In
the present study, co-culture impaired complex I activity in
myotube mitochondria, whereas calpain inhibitors slightly improved
complex I activity, indicating that the activation of mitochondrial
calpain impairs complex I activity, which may lead to MPTP opening
and energy generation impairment.
The mechanisms of complex I damage involves complex
I subunit damage and post-translational modifications (32). NDUFS3 is a core subunit of complex I
that is essential for NADH oxidation and subsequent electron
transfer through the complex, maintaining the activity of complex I
(13). In the present study, NDUFS3
content in myotube mitochondria was not altered by co-culture or by
CAST, CAPT and CC treatment, indicating that the decreased complex
I activity was not due to altered NDUFS3 content. It has been
reported that complex I activity can be damaged by a conformational
change (induced by a sulfhydryl oxidation of the second cysteine
residue in complex I) (33).
Therefore, the decreased complex I activity upon co-culture in the
current study may be due to this post-translational modification,
although this requires further investigation.
The MPTP is a non-selective pore located on the
membrane of the mitochondria (34).
MPTP opening increases the permeability of the mitochondrial
membrane and leads to the loss of Δψm and to
mitochondrial injury, which in turn causes CCMA (27). In mouse hearts, calpain inhibitors
attenuate the ischemia-reperfusion and induce MPTP opening and
Δψm depolarization (20).
Additionally, calpain inhibitors ameliorate the
microcystin-LR-induced MPTP opening and Δψm
depolarization in cultured hepatocytes (35), suggesting that calpain activation
contributes to MPTP opening and Δψm depolarization.
Consistent with the aforementioned findings, the results of the
present study demonstrated that co-culture activated calpain in
myotube mitochondria, causing MPTP opening and Δψm
depolarization. By contrast, administration of CAST, CAPT or CC
ameliorated these changes, suggesting that the activation of
mitochondrial calpain induces MPTP opening and Δψm
depolarization. Furthermore, co-culture decreased the CYCD content
in myotube mitochondria, whereas treatment with calpain inhibitors
improved CYCD content, suggesting that the activation of
mitochondrial calpain may increase MPTP opening through the
cleavage of CYCD. However, no cleaved band of CYCD was detected in
myotubes.
AIF is a mitochondrial flavoprotein that functions
as an antioxidant in the mitochondrial intermembrane space
(11). Activation of mitochondrial
calpain cleaves AIF to truncated AIF (t-AIF) and releases t-AIF
from the mitochondria to the cytoplasm, allowing it to translocate
to the nucleus, inducing DNA degradation and apoptosis (13,36). In
the present study, co-culture decreased AIF content in myotube
mitochondria, which was then reversed by calpain inhibitors,
suggesting that activation of mitochondrial calpain induced the
loss of AIF in the mitochondria. However, t-AIF was not detected in
myotubes in the present study, suggesting that co-culture may
induce myotube apoptosis via other mechanisms, which should be
further investigated in future studies.
AKT/mTOR and FoxO3a serve a critical role in
regulating muscular protein synthesis and proteolysis (26). AKT phosphorylates mTOR to increase
its activity and promote protein synthesis (37). Activated mTOR in turn phosphorylates
4EBP1 (a negative regulator of the eukaryotic translation
initiation factor 4E) to inhibit its activity (38). Additionally, AKT phosphorylates
FoxO3a, inhibiting its activation and nuclear entrance, which
decreases the expression levels of atrogin-1 and MuRF1 (6,26). In
the present study, co-culture decreased AKT and mTOR activity and
increased FoxO3a activity and atrogin-1 content in myotubes,
whereas calpain inhibitors ameliorated these changes, suggesting
that the activation of cytosolic calpain acted through AKT/mTOR and
FoxO3a/atrogin-1 to disrupt muscular protein metabolism.
Activation of autophagy contributes to muscle
wasting (7,39). Therefore, future studies should
investigate whether the autophagy-lysosome system may be involved
in the atrophy of myotubes.
To the best of our knowledge, calpain-1, calpain-2
and CAST were for the first time demonstrated to be present in
mouse myotube mitochondria in the present study. Calpain inhibitors
protected the myotubes during co-culture by inhibiting both
cytosolic and mitochondrial calpain activity. CAST is an endogenous
calpain inhibitor, whereas CAPT is a commonly used non-selective
and reversible calpain inhibitor; however, they both have
off-target effects (9,40,41).
Therefore, a genetic approach is required to further clarify the
potential role of mitochondrial calpain in CCMA. Investigation of
mitochondrial calpain will provide new insights to understand the
mechanism of CCMA. The present results will further help to develop
focused approaches to attenuate CCMA by manipulating the
mitochondrial and cytosolic calpain activity.
Acknowledgements
Not applicable.
Funding
The present was supported by Joint Funds for The
Innovation of Science and Technology, Fujian province (grant no.
2018Y9083).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ drafted the main manuscript and performed the
main experiments. XL and SC conceived and designed the experiments.
LZ helped with cell culture, western blotting and
immunofluorescence staining experiments, and was responsible for
analyzing the data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
4EBP1
|
eukaryotic translation initiation
factor 4E-binding protein 1
|
|
AIF
|
apoptosis-inducing factor
|
|
CAPT
|
calpeptin
|
|
CAST
|
calpastatin
|
|
CCMA
|
cancer cachectic muscle atrophy
|
|
CYCD
|
cyclophilin D
|
|
MHC
|
myosin heavy chain
|
|
MPTP
|
mitochondrial permeability transition
pore
|
|
Δψm
|
mitochondrial membrane potential
|
References
|
1
|
Fearon K, Strasser F, Anker SD, Bosaeus I,
Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N,
Mantovani G, et al: Definition and classification of cancer
cachexia: An international consensus. Lancet Oncol. 12:489–495.
2011. View Article : Google Scholar
|
|
2
|
Donohoe CL, Ryan AM and Reynolds JV:
Cancer cachexia: Mechanisms and clinical implications.
Gastroenterol Res Pract. 2011:6014342011. View Article : Google Scholar
|
|
3
|
Yoshida T, Semprun-Prieto L, Sukhanov S
and Delafontaine P: IGF-1 prevents ANG II-induced skeletal muscle
atrophy via Akt- and Foxo-dependent inhibition of the ubiquitin
ligase atrogin-1 expression. Am J Physiol Heart Circ Physiol.
298:H1565–H1570. 2010. View Article : Google Scholar
|
|
4
|
Lin XY and Chen SZ: Calpain inhibitors
ameliorate muscle wasting in a cachectic mouse model bearing CT26
colorectal adenocarcinoma. Oncol Rep. 37:1601–1610. 2017.
View Article : Google Scholar
|
|
5
|
Johns N, Stephens NA and Fearon KC: Muscle
wasting in cancer. Int J Biochem Cell Biol. 45:2215–2229. 2013.
View Article : Google Scholar
|
|
6
|
Tsubouchi H, Yanagi S, Miura A, Matsumoto
N, Kangawa K and Nakazato M: Ghrelin relieves cancer cachexia
associated with the development of lung adenocarcinoma in mice. Eur
J Pharmacol. 743:1–10. 2014. View Article : Google Scholar
|
|
7
|
Penna F, Costamagna D, Pin F, Camperi A,
Fanzani A, Chiarpotto EM, Cavallini G, Bonelli G, Baccino FM and
Costelli P: Autophagic degradation contributes to muscle wasting in
cancer cachexia. Am J Pathol. 182:1367–1378. 2013. View Article : Google Scholar
|
|
8
|
Goll DE, Neti G, Mares SW and Thompson VF:
Myofibrillar protein turnover: The proteasome and the calpains. J
Anim Sci. 86 (Suppl):E19–E35. 2008. View Article : Google Scholar
|
|
9
|
Dókus LE, Yousef M and Bánóczi Z:
Modulators of calpain activity: Inhibitors and activators as
potential drugs. Expert Opin Drug Discov. 15:471–486. 2020.
View Article : Google Scholar
|
|
10
|
Huang J and Zhu X: The molecular
mechanisms of calpains action on skeletal muscle atrophy. Physiol
Res. 65:547–560. 2016. View Article : Google Scholar
|
|
11
|
Ozaki T, Tomita H, Tamai M and Ishiguro S:
Characteristics of mitochondrial calpains. J Biochem. 142:365–376.
2007. View Article : Google Scholar
|
|
12
|
Goll DE, Thompson VF, Li H, Wei W and Cong
J: The calpain system. Physiol Rev. 83:731–801. 2003. View Article : Google Scholar
|
|
13
|
Chen Q, Thompson J, Hu Y, Dean J and
Lesnefsky EJ: Inhibition of the ubiquitous calpains protects
complex I activity and enables improved mitophagy in the heart
following ischemia-reperfusion. Am J Physiol Cell Physiol.
317:C910–C921. 2019. View Article : Google Scholar
|
|
14
|
Kar P, Chakraborti T, Roy S, Choudhury R
and Chakraborti S: Identification of calpastatin and mu-calpain and
studies of their association in pulmonary smooth muscle
mitochondria. Arch Biochem Biophys. 466:290–299. 2007. View Article : Google Scholar
|
|
15
|
Tisdale MJ: Mechanisms of cancer cachexia.
Physiol Rev. 89:381–410. 2009. View Article : Google Scholar
|
|
16
|
Zeng X, Chen S, Yang Y and Ke Z: Acylated
and unacylated ghrelin inhibit atrophy in myotubes co-cultured with
colon carcinoma cells. Oncotarget. 8:72872–72885. 2017. View Article : Google Scholar
|
|
17
|
Trendelenburg AU, Meyer A, Rohner D, Boyle
J, Hatakeyama S and Glass DJ: Myostatin reduces Akt/TORC1/p70S6K
signaling, inhibiting myoblast differentiation and myotube size. Am
J Physiol Cell Physiol. 296:C1258–C1270. 2009. View Article : Google Scholar
|
|
18
|
Zhang XL, Wang ZZ, Shao QH, Zhang Z, Li L,
Guo ZY, Sun HM, Zhang Y and Chen NH: RNAi-mediated knockdown of
DJ-1 leads to mitochondrial dysfunction via Akt/GSK-3β and JNK
signaling pathways in dopaminergic neuron-like cells. Brain Res
Bull. 146:228–236. 2019. View Article : Google Scholar
|
|
19
|
Zeng X, Chen S, Lin Y and Ke Z: Acylated
and unacylated ghrelin inhibit apoptosis in myoblasts cocultured
with colon carcinoma cells. Oncol Rep. 39:1387–1395. 2018.
|
|
20
|
Thompson J, Hu Y, Lesnefsky EJ and Chen Q:
Activation of mitochondrial calpain and increased cardiac injury:
Beyond AIF release. Am J Physiol Heart Circ Physiol. 310:H376–H384.
2016. View Article : Google Scholar
|
|
21
|
Hendriks KDW, Joschko CP, Hoogstra-Berends
F, Heegsma J, Faber KN and Henning RH: Hibernator-derived cells
show superior protection and survival in hypothermia compared to
non-hibernator cells. Int J Mol Sci. 9:18642020. View Article : Google Scholar
|
|
22
|
Kudo-Sakamoto Y, Akazawa H, Ito K, Takano
J, Yano M, Yabumoto C, Naito AT, Oka T, Lee JK, Sakata Y, et al:
Calpain-dependent cleavage of N-cadherin is involved in the
progression of post-myocardial infarction remodeling. J Biol Chem.
289:19408–19419. 2014. View Article : Google Scholar
|
|
23
|
Fakharnia F, Khodagholi F, Dargahi L and
Ahmadiani A: Prevention of cyclophilin D-mediated mPTP opening
using cyclosporine-A alleviates the elevation of necroptosis,
autophagy and apoptosis-related markers following global cerebral
ischemia-reperfusion. J Mol Neurosci. 61:52–60. 2017. View Article : Google Scholar
|
|
24
|
Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin
XM, Clark RS, Graham SH and Chen J: Critical role of calpain I in
mitochondrial release of apoptosis-inducing factor in ischemic
neuronal injury. J Neurosci. 27:9278–9293. 2007. View Article : Google Scholar
|
|
25
|
Polster BM, Basañez G, Etxebarria A,
Hardwick JM and Nicholls DG: Calpain I induces cleavage and release
of apoptosis-inducing factor from isolated mitochondria. J Biol
Chem. 280:6447–6454. 2005. View Article : Google Scholar
|
|
26
|
Clavel S, Siffroi-Fernandez S, Coldefy AS,
Boulukos K, Pisani DF and Dérijard B: Regulation of the
intracellular localization of Foxo3a by stress-activated protein
kinase signaling pathways in skeletal muscle cells. Mol Cell Biol.
30:470–480. 2010. View Article : Google Scholar
|
|
27
|
Argilés JM, Busquets S, Stemmler B and
López-Soriano FJ: Cancer cachexia: Understanding the molecular
basis. Nat Rev Cancer. 14:754–762. 2014. View Article : Google Scholar
|
|
28
|
Fearon KC, Glass DJ and Guttridge DC:
Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell
Metab. 16:153–166. 2012. View Article : Google Scholar
|
|
29
|
Lokireddy S, Wijesoma I, Bonala S, Wei M,
Sze S, McFarlane C, Kambadur R and Sharma M: Retraction: Myostatin
is a novel tumoral factor that induces cancer cachexia. Biochem J.
473:11112016. View Article : Google Scholar
|
|
30
|
Sun R, Zhang S, Hu W, Lu X, Lou N, Yang Z,
Chen S, Zhang X and Yang H: Valproic acid attenuates skeletal
muscle wasting by inhibiting C/EBPβ-regulated atrogin1 expression
in cancer cachexia. Am J Physiol Cell Physiol. 311:C101–C115. 2016.
View Article : Google Scholar
|
|
31
|
Karamanlidis G, Lee CF, Garcia-Menendez L,
Kolwicz SC Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang
W and Tian R: Mitochondrial complex I deficiency increases protein
acetylation and accelerates heart failure. Cell Metab. 18:239–250.
2013. View Article : Google Scholar
|
|
32
|
Hollander JM, Thapa D and Shepherd DL:
Physiological and structural differences in spatially distinct
subpopulations of cardiac mitochondria: Influence of cardiac
pathologies. Am J Physiol Heart Circ Physiol. 307:H1–H14. 2014.
View Article : Google Scholar
|
|
33
|
Galkin A, Abramov AY, Frakich N, Duchen MR
and Moncada S: Lack of oxygen deactivates mitochondrial complex I:
Implications for ischemic injury? J Biol Chem. 284:36055–36061.
2009. View Article : Google Scholar
|
|
34
|
Halestrap AP: What is the mitochondrial
permeability transition pore? J Mol Cell Cardiol. 46:821–831. 2009.
View Article : Google Scholar
|
|
35
|
Ding WX, Shen HM and Ong CN: Calpain
activation after mitochondrial permeability transition in
microcystin-induced cell death in rat hepatocytes. Biochem Biophys
Res Commun. 291:321–331. 2002. View Article : Google Scholar
|
|
36
|
Ye H, Cande C, Stephanou NC, Jiang S,
Gurbuxani S, Larochette N, Daugas E, Garrido C, Kroemer G and Wu H:
DNA binding is required for the apoptogenic action of apoptosis
inducing factor. Nat Struct Biol. 9:680–684. 2002. View Article : Google Scholar
|
|
37
|
Glass DJ: Skeletal muscle hypertrophy and
atrophy signaling pathways. Int J Biochem Cell Biol. 37:1974–1984.
2005. View Article : Google Scholar
|
|
38
|
Sandri M: Signaling in muscle atrophy and
hypertrophy. Physiology (Bethesda). 23:160–170. 2008.
|
|
39
|
Tardif N, Klaude M, Lundell L, Thorell A
and Rooyackers O: Autophagic-lysosomal pathway is the main
proteolytic system modified in the skeletal muscle of esophageal
cancer patients. Am J Clin Nutr. 98:1485–1492. 2013. View Article : Google Scholar
|
|
40
|
Yoshida M, Miyasaka Y, Ohuchida K, Okumura
T, Zheng B, Torata N, Fujita H, Nabae T, Manabe T, Shimamoto M, et
al: Calpain inhibitor calpeptin suppresses pancreatic cancer by
disrupting cancer-stromal interactions in a mouse xenograft model.
Cancer Sci. 107:1443–1452. 2016. View Article : Google Scholar
|
|
41
|
Tsujinaka T, Kajiwara Y, Kambayashi J,
Sakon M, Higuchi N, Tanaka T and Mori T: Synthesis of a new cell
penetrating calpain inhibitor (calpeptin). Biochem Biophys Res
Commun. 153:1201–1208. 1988. View Article : Google Scholar
|














