Introduction
According to the International Agency for Research
on Cancer, hepatocellular carcinoma (HCC) is the second leading
cause of tumor-associated death worldwide (1). HCC commonly develops in patients with
chronic hepatitis, such as viral hepatitis (2). Various treatments for HCC, including
resection, transplantation and interventional therapy, have
undergone immense progress over the last decades, but the prognosis
of HCC remains poor in patients at the late stage (3). Additionally, the high rate of
postsurgical recurrence and metastasis (50–70% at 5 years)
represents a major challenge, as this disease is highly refractory
to conventional chemotherapy and radiation (4). Currently, the Barcelona Clinic Liver
Cancer (BCLC) staging classification is the most extensively used
classification system for HCC, which can be applied for the
assessment of patient prognosis and the selection of appropriate
therapies (5). However, it has been
reported that patients with HCC with the same BCLC stage may
include various tumor subtypes, such as nodular or infiltrating
tumors, thus resulting in differences in treatment responses and
survival (6). Therefore, it is
critical to identify novel and reliable prognostic molecular
signatures in HCC from basic and clinical research.
N6-methyladenosine (m6A), methylated at the N6
position of adenosine, is the most abundant epigenetic and
evolutionarily conserved modification of mRNAs and non-coding RNAs
in mammals (7–10). In total RNA, 0.1–0.4% of adenosines
are modified by m6A methylation (11). m6A methylation affects almost every
aspect of RNA metabolism, including abundance, alternative
splicing, stability, nuclear export, decay and translation
(12–14), thus negatively regulating protein
expression in a post-translational manner. The identification of
m6A adenosine methyltransferases (‘writers’), demethylases
(‘erasers’) and binding proteins (‘readers’) revealed that m6A
modification is reversible (15).
Increasing evidence has indicated that the m6A modification may be
involved in various physiological processes and diseases, including
circadian rhythms, stem cell differentiation and
maternal-to-zygotic transition (16), as well as the carcinogenesis of
several types of tumor, including cervical cancer (17), prostate cancer (18), breast cancer (19), pancreatic cancer (20) and HCC (21). The characterization for m6A sparked a
renewed interest in this particular RNA modification. However, its
expression pattern, as well as its prognostic value, has not been
fully elucidated in HCC.
In the present study, the m6A patterns were
estimated based on the 20 widely reported m6A RNA regulators and
were systematically characterized for the potential subtypes in a
multiomics view, including somatic mutations and DNA
methylation.
Materials and methods
Public data source
TCGA- Liver hepatocellular carcinoma (LIHC) cohort
data, including RNA sequencing, mutation and clinical data, were
downloaded from The National Cancer Institute Genomic Data Commons
(https://portal.gdc.cancer.gov/).
Maftools v2.4.12 (22) was utilized
to infer significant cancer mutated genes using default parameters.
Illumina Human Methylation 450 Beadchip (450K array; Illumina,
Inc.) was used to measure the DNA methylation data. For a gene with
>1 probe mapping to its promoter, the median β value was
considered. MethylMix v2.18.0 (23)
was used to identify the expression of genes associated with
methylation events. The International Cancer Genome Consortium
(ICGC; http://icgc.org) Japan cohort with 203 patients
with HCC and the meta-Gene Expression Omnibus (GEO) cohorts
[GSE14520 (24) and GSE76427
(25); www.ncbi.nlm.nih.gov/geo] with 336 patients with HCC
were utilized as the validation cohorts. In addition, validation of
the translation of m6A-associated genes was performed using the
Human Protein Atlas (HPA) database (version 19.2; http://www.proteinatlas.org).
Protein-protein interaction (PPI)
network construction and correlation analysis
The PPI network among m6A RNA methylation regulators
was constructed using Cytoscape v3.6.1 (www.cytoscape.org). Spearman correlation analysis was
employed to reveal the correlation among different m6A RNA
methylation regulators.
Consensus clustering analysis
To identify m6A patterns and classify patients for
further analysis, patients with HCC in TCGA cohort were grouped
using the ConsensusClusterPlus v1.52.0 (26) package, which was repeated 1,000 times
to ensure the stability of classification. The optimal K value was
determined according to consensus matrices, Consensus Cumulative
Distribution Function and Delta Area. Kaplan-Meier analysis
(27) with log-rank test was
performed to compare the survival of patients between clusters.
Screening of prognostic signatures and
key prognostic genes
Univariate Cox proportional hazards regression was
used to assess the independent m6A RNA methylation regulators,
whose expression levels were significantly associated with the
survival of patients. Hazard ratios (HRs) were used to identify
protective (HR<1) or risk-associated genes (HR>1).
LASSO-penalized Cox regression analysis performed using the glmnet
v3.0-2 package (https://cran.r-project.org/package=glmnet) was used to
achieve variable shrinkage and selection of key independent m6A RNA
methylation regulators (28). An
optimal model was determined based on a linear combination of the
expression profiles of independent prognostic m6A RNA methylation
regulators, weighted by the estimated regression coefficient
derived from the LASSO Cox regression model coefficients multiplied
with its mRNA expression level. The following formula was used to
calculate the risk score of each patient: Risk score= ∑ X J × coef
J, where coef J is the coefficient, and X J is the relative
expression level of each gene standardized by Z-score.
Subsequently, patients with HCC from TCGA were divided into high-
and low-risk groups, according to the optimal cut-off value of risk
scores obtained from the survminer v0.4.6 package (https://cran.r-project.org/package=survminer). Cox
regression analysis was used to evaluate the association between
risk score and disease-free survival (DFS) or overall survival
(OS), in which age, sex, Tumor-Node-Metastasis (TNM) stage
(29), Neoplasm_cancer_status and
grade were used as covariates. Neoplasm_cancer_status is a
statement about the progression (or not) of the original disease,
whereas ‘normal’ is a statement that there was no disease to begin
with. The Kaplan-Meier survival analysis with log-rank test and
receiver operating characteristic (ROC) curve analysis were used to
validate the multigene prognostic signature.
Gene Set Enrichment Analysis
(GSEA)
GSEA was performed to detect the significantly
different signaling pathways in the set of expressed genes between
the high- and low-risk groups in the enrichment of the MSigDB
Collection (c2.all.v7. 0. Symbols; http://www.gsea-msigdb.org/gsea/msigdb/collections.jsp#C2).
Gene set permutations were performed 1,000 times for each analysis.
The Benjamini-Hochberg method was used to correct the P-values for
multiple testing. The normalized enrichment score (NES), nominal
P-value and false discovery rate (FDR) q-value indicated the
significance of the association between gene sets and signaling
pathways. |NES|>1, FDR≤0.25 and P<0.05 were considered to
indicate a statistically significant difference.
Statistical analysis
Data analysis was performed using R v3.6 software
(https://cran.r-project.org/src/base/R-3/). All
statistical tests were two-sided. The Wilcoxon signed-rank test and
Mann-Whitney U test were used for paired and unpaired samples,
respectively, when the population with non-normal distribution or
uneven variance. Cox regression analysis was performed to identify
whether the final prognostic model was independent of traditional
clinical features (including age, sex, TNM stage and histological
grade). The Kaplan-Meier survival analysis with log-rank test and
ROC curve analysis were used to assess the predictive ability of
the prognostic model. P<0.05 was considered to indicate a
statistically significant difference.
Results
Landscape of m6A RNA methylation
regulators in HCC
The mRNA expression levels of the known
m6A-associated regulators were analyzed in TCGA cohort, including
m6A ‘writers’, such as METTL3, METTL4, WTAP, ZC3H13, RBM15, RBM15B
and VIRMA, m6A ‘readers’, such as YTHDF1, YTHDF2, YTHDF3, YTHDC1,
YTHDC2, HNRNPC, HNRNPA2B1, IGF2BP1, IGF2BP2, IGF2BP3 and RBMX, and
m6A ‘erasers’, such as FTO and ALKBH5. Compared with normal liver
tissues, patients with HCC generally exhibited a higher proportion
of m6A genes, except ZC3H13 (Fig. 1A and
B). Furthermore, the expression levels of these aberrant
m6A-associated genes were validated in the HPA database (Fig. S1). Notably, METTL3, WTAP, RBM15B,
YTHDF1, YTHDF2, YTHDF3, HNRNPA2B1, IGF2BP1, IGF2BP2 and IGF2BP3
were absent from the HPA database. Overall, these data confirmed
the highly significant dysregulation of several m6A-associated
regulators in human HCC. The associations between each individual
m6A RNA methylation regulator and the pathological features of HCC
were further investigated. Patients with G1/G2 pathological grade
were divided into the low (L) grade group, and patients with G3/G4
stage were divided into the high (H) grade group. The expression
levels of HNRNPA2B1, HNRNPC, IGF2BP1, IGF2BP2, IGF2BP3, RBM15B,
RBM15, RBMX, METTL4, VIRMA, YTHDC1, METTL3, YTHDF1 and YTHDF2 were
higher in the H group compared with in the L group. Patients with
tumor (T)1/T2 stage were divided into the L stage group, and
patients with T3/T4 stage were divided into the H stage group. The
expression levels of HNRNPA2B1, METTL4, RBM15B, RBMX, YTHDC1,
YTHDF3, METTL3, YTHDF1 and YTHDF2 were higher in the H group
compared with in the L group. In summary, the expression levels of
HNRNPA2B1, METTL3, METTL4, RBMX, YTHDF1 and YTHDF2 were
significantly increased as the pathological grade and T-stage
increased (Figs. 2 and S2).
Fig. 3 shows that the
majority of m6A RNA methylation regulators was positively
correlated with each other, and the correlation between HNRNPC and
RBMX was the most significant. Moreover, the PPI network depicted a
comprehensive landscape of the interactions of m6A RNA methylation
regulators and the writers, including WTAP, VIRMA and METTL4,
ranked first according to the degree of connectivity (Fig. S3). The correlation coefficients
between WTAP and IGF2BP3, YTHDF2, METTL3 and YTHDF1 were 0.47,
0.65, 0.50 and 0.58, respectively. The correlation coefficients
between VIRMA and IGF2BP3, YTHDF2, METTL3 and YTHDF1 were 0.37,
0.47, 0.52 and 0.44, respectively. The correlation coefficients
between METTL14 and IGF2BP3, YTHDF2, METTL3 and YTHDF1 were 0.41,
0.57, 0.60 and 0.63, respectively (Fig.
3).
Association between m6A regulators and
HCC prognosis
Consensus Clustering is a method that provides
quantitative evidence for determining the number and membership of
possible clusters within a dataset, and to assess the stability of
the discovered clusters (30). The
expression similarity of m6A regulators and clustering stability
was assessed using the ConsensusClusterPlus package. The cohort of
patients with HCC was divided into three clusters, namely cluster
1, cluster 2 and cluster 3 (Fig.
4A-C). Fig. 4A shows the
heatmaps of the consensus matrices for k=3. Fig. 4B shows the cumulative distribution
functions of the consensus matrix for each k, estimated by a
histogram of 100 bins. Fig. 4C shows
the relative change in area under the CDF curve comparing k and
k-1. For k=2, there is no k-1, so the total area under the curve
rather than the relative increase is plotted. The patients were
divided into three clusters and there was no significant difference
in the number of samples in each cluster. Notably, survival
analysis revealed that cluster 1 was significantly associated with
an improved DFS and cluster 2 with a poor DFS, while cluster 3 was
characterized by an intermediate prognosis (Fig. 4D). Additionally, a favorable
prognostic trend for OS was observed, although not statistically
significant (data not shown), partly due to the limitation of the
cohort size.
Construction and validation of the
m6A-based HCC prognosis signature
Subsequently, the possible prognostic power of m6A
RNA methylation regulators in HCC was analyzed by performing
univariate Cox regression analysis. The results demonstrated that
HNRNPA2B1, HNRNPC, IGF2BP3, METTL3, METTL4, RBM15, RBM15B, RBMX,
VIRMA, WTAP, YTHDC1, YTHDC2, YTHDF1 and YTHDF2 were significantly
associated with DFS, and HNRNPA2B1, IGF2BP3, METTL3, WTAP, YTHDF1
and YTHDF2 were significantly associated with OS. Increased
expression levels of HNRNPA2B1, IGF2BP3, METTL3, WTAP, YTHDF1 and
YTHDF2 indicated poorer OS and DFS rates in patients with HCC
(Fig. S4). Applying the LASSO
analysis, in which the selected m6A RNA methylation regulators were
required to appear 900 times out of 1,000 repetitions, four m6A RNA
methylation regulators, namely IGF2BP3, YTHDF1, YTHDF2 and METTL3,
were selected. The distribution of the risk score was different
among different clusters (Mann-Whitney U test; P=0.0013; data not
shown), and cluster 2, which was associated with a poorer DFS
(Fig. 4D), had the highest risk
score. To further investigate the prognostic role of the four-gene
risk signature, patients with HCC were assigned into groups based
on high- or low-risk scores using the cut-off value obtained using
the survminer package, and it was observed that the high-risk group
had a shorter DFS rate compared with the low-risk group (Fig. 5A). The AUC values of 1, 2 and 3 years
were 0.783, 0.720 and 0.701, respectively (Fig. 5C). The efficacy of the classifier was
further evaluated using OS, and the results were similar to those
for DFS (Fig. 5B). The AUC values of
1, 2 and 3 years were 0.745, 0.785 and 0.794, respectively
(Fig. 5D). Similarly, patients with
HCC in the meta-GEO and ICGC cohorts were divided into high- and
low-risk groups, the high-risk group of the meta-GEO cohort
exhibited worse OS and DFS rates compared with the low-risk group
(Fig. S5A-D), and the high-risk
group of the ICGC cohort (recurrence time was not collected)
exhibited worse OS rates compared with the low-risk group (Fig. S5E and F).
m6A-based prognosis classifier and
clinicopathological characteristics in patients with HCC
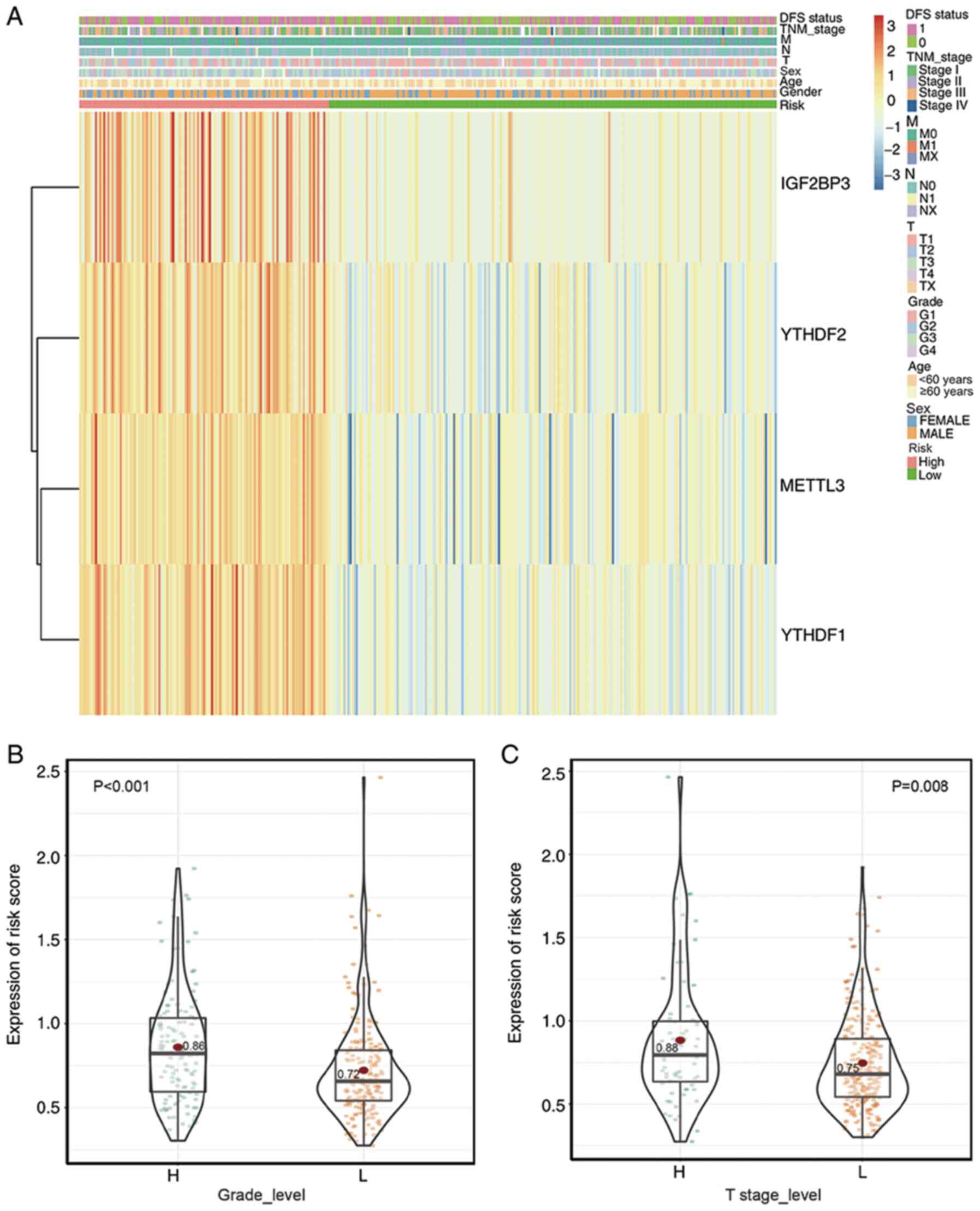
The heat map in Fig.
6A shows the expression levels of the four selected m6A RNA
methylation regulators and the clinicopathological variables in the
high- and low-risk groups. There was no marked difference between
the two groups regarding sex and age. Similar to their association
with prognosis, the low-risk group, with lower expression levels of
the four genes, was significantly associated with lower T stage and
pathological grade (Fig. 6B and C).
Univariate and multivariate Cox regression analyses were performed
to evaluate whether the prognostic signature-based risk score was
an independent factor for prognosis. When the m6A signature was
evaluated as a continuous variable with the Cox regression model,
the univariate and multivariate analyses revealed that the stage
and risk score were significantly associated with OS (Table I). These results suggested that the
risk signature may be a risk factor for HCC and may independently
predict the prognosis of patients with HCC.
 | Table I.Univariate and multivariate
regression analysis of the association between the risk score and
clinicopathological features with overall survival. |
Table I.
Univariate and multivariate
regression analysis of the association between the risk score and
clinicopathological features with overall survival.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Factor | HR | P-value | 95% CI | HR | P-value | 95% CI |
|---|
| Age (≥60 vs. <60
years) | 1.11 | 0.492 | 0.83–1.48 | 1.24 | 0.177 | 0.91–1.68 |
| Sex (male vs.
female) | 0.91 | 0.531 | 0.67–1.23 | 0.98 | 0.920 | 0.71–1.37 |
| Pathological grade
(G3/G4 vs. G1/G2) | 1.10 | 0.543 | 0.81–1.48 | 0.95 | 0.767 | 0.69–1.32 |
| Risk score (high
vs. low) | 3.00 | <0.001 | 2.01–4.47 | 2.51 | <0.001 | 1.59–3.97 |
| Tumor stage (III/IV
vs. I/II) | 2.08 | <0.001 | 1.49–2.89 | 1.79 | 0.001 | 1.26–2.54 |
Differential biological signaling
pathways, somatic mutation landscape and methylation-driven genes
between the HCC risk score subtypes
To further identify the potential biological
processes of the risk score subtypes, GSEA comparing the high- and
low-risk groups was performed. The results revealed that the
samples in the high-risk group were enriched in ‘regulation of
transcription’ and ‘immune system development’, while the samples
in the low-risk group were enriched in ‘lipid biosynthetic process’
(Fig. 7).
To identify the associations between the
distributions of somatic alterations and the HCC risk score
subtypes, 526 genes were identified with shared mutations between
the high- and low-risk groups. Specifically, a missense mutation in
TP53 and FLG was predominantly observed in the high-risk group,
while the mutation frequency of ALB was higher in the low-risk
group compared with in the high-risk group (Fig. 8).
Methylation-driven genes are genes with different
degree of methylation and expression between different groups.
After downloading and processing the methylation data, 569
methylation-driven genes associated with risk score subtypes were
screened via the MethylMix R package (Table II). Among these genes, 461 genes
(81.02%) were hypomethylated and the remaining 108 genes (18.98%)
were hypermethylated.
| Table II.Methylation-driven genes associated
with risk score subtypes of hepatocellular carcinoma. |
Table II.
Methylation-driven genes associated
with risk score subtypes of hepatocellular carcinoma.
| A, Top 10
hypermethylated genes |
|---|
|
|---|
| Gene | logFC |
|---|
| FMO3 | 0.558359143 |
| RBP5 | 0.550688961 |
| AKR7L | 0.546025322 |
| SLC2A2 | 0.527909372 |
| SLC27A2 | 0.451295029 |
| CFHR5 | 0.428486584 |
| CD14 | 0.416272718 |
| ACADL | 0.411132983 |
| SERPINC1 | 0.410346926 |
| APOC3 | 0.401360659 |
|
| B, Top 10
hypomethylated genes |
|
| Gene | logFC |
|
| SP5 | −0.684538772 |
| BMP4 | −0.677013955 |
| FOXD3 | −0.641820658 |
| EVI2A | −0.600077451 |
| ZNF702P | −0.570013961 |
| FOXE1 | −0.560776731 |
| LRFN4 | −0.545665424 |
| AIM2 | −0.545136870 |
| LTC4S | −0.499335824 |
| TMEFF1 | −0.495389988 |
Discussion
The occurrence and development of HCC is a
multi-step complex process involved with genetic or epigenetic
factors (31,32). Therefore, elucidating the underlying
molecular events accounting for the tumorigenesis, diagnosis and
precise individual therapy of HCC remain the greatest challenges.
Previous studies have demonstrated that m6A affects the epigenetic
regulation of RNA, including mRNA stability (33), alternative splicing (34) and microRNA biogenesis (35), which in turn regulate gene
expression. The dysregulation of m6A genes affects the pathogenesis
of a variety of human diseases, including obesity, neuronal
disorders and immunological diseases, as well as promoting the
initiation, expansion and progression of malignancies, including
HCC (21,36,37).
Previous studies (38–40) on mRNA m6A modification have
associated the methylation levels of m6A with the intracellular
writing and erasing genes, while the regulatory functions of
methylation sites in biological processes is performed by protein
molecules that read gene expression (41). Therefore, in tumors, both
m6A-associated genes and protein expression levels may become
potential diagnostic markers for tumor molecular diagnosis and
potential targets for molecular targeted therapies.
The abnormal methylation of m6A mRNA has exhibited
prognostic value in multiple types of tumor, such as cervical
cancer (42), acute myeloid leukemia
(43) and pancreatic cancer
(44). Considering the importance of
m6A modification in HCC, it can be reasonably speculated that
m6A-associated genes may have broad prospects in the prognostic
evaluation of HCC, and that using a multigene signature generated
using various algorithms may improve the prognostic prediction in
patients with HCC compared with using a single molecule.
The risk model created in the present study
consisted of four m6A-associated genes, and the risk score was an
independent prognostic marker according to the multivariate
analysis. In terms of validity and reliability, the four-gene
signature performed well even in the external validation datasets.
Furthermore, the high- and low-risk HCC groups presented with
different significantly mutated genes, of which TP53 was markedly
mutated in the high-risk HCC group compared with in the low-risk
group. The association between the TP53 signaling pathway and
YTHDF2 expression has been previously reported (45), suggesting that the high-risk HCC
group with increased TP53 mutations may be more likely to be
involved in the activation of cancer signaling pathways. In
addition, the identified METTL3/RDM1/TP53/ERK signaling pathway
(46) may help to clarify the
potential association of TP53 mutation and the risk signature in
the present study. In the present study, integrative analysis by
mRNA expression and promoter CpG islands methylation manifested a
broad spectrum of gene silencing in the high-risk HCC group
compared with that in the low-risk HCC group. These data may
provide a new perspective to study the mechanism of m6A
modulation.
Signaling pathways involved in the regulation of
transcription, immune system development and lipid biosynthesis
were differentially enriched in the phenotypes of the risk score in
the present study. IGF2BPs (including IGF2BP1/2/3) promote the
stability and storage of their target mRNAs (such as MYC) in an
m6A-dependent manner under normal and stress conditions, and
therefore affect gene expression output (47). Moreover, the K homology domains of
IGF2BPs are required for their recognition of m6A and are critical
for their oncogenic functions (48).
YTHDF2 mainly regulates mRNA stability (49), and acts as a tumor-inhibiting factor
in HCC (50,51). YTHDF2 deficiency promotes HCC growth,
vasculature remodeling and metastasis via a potential mechanism
that involves the reprogramming of the epi-transcriptome under
hypoxia (50,52). Although the four genes in the current
gene signature have not been reported to be associated with
dyslipidemia, YTHDC2 may bind to the mRNA of lipogenic genes,
including sterol regulatory element-binding protein 1c, fatty acid
synthase, stearoyl-CoA desaturase 1 and acetyl-CoA carboxylase 1,
to decrease their mRNA stability and inhibit gene expression
(53). RNA m6A modification
regulates anti-tumor immunity response via YTHDF1, which regulates
tumorigenicity and cancer stem cell-like activity in HCC (48). Transcripts encoding lysosomal
proteases are marked by m6A and recognized by YTHDF1; binding of
YTHDF1 to these transcripts increases the translation of lysosomal
cathepsins in dendritic cells, and inhibition of cathepsins
markedly enhances cross-presentation of wild-type dendritic cells
(54). Furthermore, the therapeutic
efficacy of programmed death-ligand 1 checkpoint blockade is
enhanced in Ythdf1−/− mice, implicating YTHDF1 as a
potential therapeutic target in anticancer immunotherapy (54).
The present study presents some limitations.
Firstly, although several independent external validations were
performed in the present study, it was difficult to consider all
variations among patients from different geographical regions,
since tissues and data were retrospectively collected from publicly
available databases. In addition, other key clinical pathological
features, such as surgical procedures, the number of lymph nodes
and α-fetoprotein levels, were not included. Secondly, the
mechanism and association between the risk score subtypes and
single nucleotide polymorphisms and DNA methylation require further
study. Thirdly, IGF2BP3, YTHDF2, METTL3 and YTHDF1 were indicated
to be associated with HCC. However, the mechanism underlying the
higher prognostic efficiency of the combination of these molecules,
as determined from three independent cohorts, remains unclear.
Finally, the expression levels and prognostic role of these four
genes require further validation by well-designed, prospective,
multicenter studies.
Overall, the present study comprehensively analyzed
the associations between the mRNA expression levels of m6A
regulators with the initiation, development and prognosis of HCC.
Notably, a robust four-gene prognostic signature that was
significantly associated with the OS of patients with HCC was
constructed and validated in independent HCC cohorts, suggesting
that the present prognostic signature may act as a promising
biomarker for predicting the prognosis of patients with HCC.
Additionally, it may serve as a prognostic classifier for clinical
decision-making for the accurate prognosis prediction, treatment
and follow-up scheduling.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81301301 and
81601579), the Chen Xiao-ping Foundation for the Development of
Science and Technology of Hubei Province (grant no.
CXPJJH11800001-2018203) and the Natural Science Foundation of Hubei
province (grant no. 2018CFB553).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the National Cancer Institute
Genomic Data Commons (https://gdc.cancer.gov/), Gene Expression Omnibus
(https://www.ncbi.nlm.nih.gov/geo/),
International Cancer Genome Consortium (https://icgc.org/) and Human Protein Atlas databases
(http://www.proteinatlas.org/).
Authors' contributions
QJ and ZG designed the study. NH and CZ performed
data acquisition and collected the literature. PZ and QR performed
data analysis and interpretation, and drafted the manuscript. QJ
critically revised the manuscript for important intellectual
content. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
m6A
|
N6-methyladenosine
|
|
TCGA
|
The Cancer Genome Atlas
|
|
GEO
|
Gene Expression Omnibus
|
|
HPA
|
Human Protein Atlas
|
|
LASSO
|
least absolute shrinkage and selection
operator
|
|
DFS
|
disease-free survival
|
|
OS
|
overall survival
|
|
BCLC
|
Barcelona Clinic Liver Cancer
|
|
PPI
|
protein-protein interaction
|
|
HR
|
hazard ratio
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thorgeirsson SS and Grisham JW: Molecular
pathogenesis of human hepatocellular carcinoma. Nat Genet.
31:339–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jung HI, Jeong D, Ji S, Ahn TS, Bae SH,
Chin S, Chung JC, Kim HC, Lee MS and Baek MJ: Overexpression of
PD-L1 and PD-L2 is associated with poor prognosis in patients with
hepatocellular carcinoma. Cancer Res Treat. 49:246–254. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gish RG, Porta C, Lazar L, Ruff P, Feld R,
Croitoru A, Feun L, Jeziorski K, Leighton J, Gallo J and Kennealey
GT: Phase III randomized controlled trial comparing the survival of
patients with unresectable hepatocellular carcinoma treated with
nolatrexed or doxorubicin. J Clin Oncol. 25:3069–3075. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Llovet JM, Brú C and Bruix J: Prognosis of
hepatocellular carcinoma: The BCLC staging classification. Seminars
Liver Disease. 19:329–338. 1999. View Article : Google Scholar
|
|
6
|
Yoo JJ, Chung GE, Lee JH, Nam JY, Chang Y,
Lee JM, Lee DH, Kim HY, Cho EJ, Yu SJ, et al: Sub-classification of
advanced-stage hepatocellular carcinoma: A cohort study including
612 patients treated with sorafenib. Cancer Res Treat. 50:366–373.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoon KJ, Ringeling FR, Vissers C, Jacob F,
Pokrass M, Jimenez-Cyrus D, Su Y, Kim NS, Zhu Y, Zheng L, et al:
Temporal control of mammalian cortical neurogenesis by m(6)A
methylation. Cell. 171:877–889.e17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schibler U, Kelley DE and Perry RP:
Comparison of methylated sequences in messenger RNA and
heterogeneous nuclear RNA from mouse L cells. J Mol Biol.
115:695–714. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wei CM and Moss B: Nucleotide sequences at
the N6-methyladenosine sites of HeLa cell messenger ribonucleic
acid. Biochemistry. 16:1672–1676. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Desrosiers R, Friderici K and Rottman F:
Identification of methylated nucleosides in messenger RNA from
Novikoff hepatoma cells. Proc Natl Acad Sci USA. 71:3971–3975.
1974. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei CM, Gershowitz A and Moss B:
Methylated nucleotides block 5′ terminus of HeLa cell messenger
RNA. Cell. 4:379–386. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao BS, Roundtree IA and He C:
Post-transcriptional gene regulation by mRNA modifications. Nat Rev
Mol Cell Biol. 18:31–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meyer KD, Saletore Y, Zumbo P, Elemento O,
Mason CE and Jaffrey SR: Comprehensive analysis of mRNA methylation
reveals enrichment in 3′ UTRs and near stop codons. Cell.
149:1635–1646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Helm M and Motorin Y: Detecting RNA
modifications in the epitranscriptome: Predict and validate. Nat
Rev Genet. 18:275–291. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pinello N, Sun S and Wong JJ: Aberrant
expression of enzymes regulating m6A mRNA methylation: Implication
in cancer. Cancer Biol Med. 15:323–334. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Batista PJ: The RNA modification
N(6)-methyladenosine and its implications in human disease.
Genomics Proteomics Bioinformatics. 15:154–163. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou S, Bai ZL, Xia D, Zhao ZJ, Zhao R,
Wang YY and Zhe H: FTO regulates the chemo-radiotherapy resistance
of cervical squamous cell carcinoma (CSCC) by targeting β-catenin
through mRNA demethylation. Mol Carcinog. 57:590–597. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu ZX, Li LM, Sun HL and Liu SM: Link
between m6A modification and cancers. Front Bioeng Biotechnol.
6:892018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang
Z, Liu Y, Zhang X, Zhang W and Ye L: HBXIP-elevated
methyltransferase METTL3 promotes the progression of breast cancer
via inhibiting tumor suppressor let-7g. Cancer Lett. 415:11–19.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Taketo K, Konno M, Asai A, Koseki J,
Toratani M, Satoh T, Doki Y, Mori M, Ishii H and Ogawa K: The
epitranscriptome m6A writer METTL3 promotes chemo-and
radioresistance in pancreatic cancer cells. Int J Oncol.
52:621–629. 2018.PubMed/NCBI
|
|
21
|
Cheng X, Li M, Rao X, Zhang W, Li X, Wang
L and Huang G: KIAA1429 regulates the migration and invasion of
hepatocellular carcinoma by altering m6A modification of ID2 mRNA.
Onco Targets Ther. 12:3421–3428. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mayakonda A, Lin DC, Assenov Y, Plass C
and Koeffler HP: Maftools: Efficient and comprehensive analysis of
somatic variants in cancer. Genome Res. 28:1747–1756. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gevaert O: MethylMix: An R package for
identifying DNA methylation-driven genes. Bioinformatics.
31:1839–1841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Roessler S, Jia HL, Budhu A, Forgues M, Ye
QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX and Wang XW: A
unique metastasis gene signature enables prediction of tumor
relapse in early-stage hepatocellular carcinoma patients. Cancer
Res. 70:10202–10212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grinchuk OV, Yenamandra SP, Iyer R, Singh
M, Lee HK, Lim KH, Chow PK and Kuznetsov VA: Tumor-adjacent tissue
co-expression profile analysis reveals pro-oncogenic ribosomal gene
signature for prognosis of resectable hepatocellular carcinoma. Mol
Oncol. 12:89–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wilkerson D M, Hayes and Neil D:
ConsensusClusterPlus: a class discovery tool with confidence
assessments and item tracking. Bioinformatics. 26:1572–1573. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lacny S, Wilson T, Clement F, Roberts DJ,
Faris PD, Ghali WA and Marshall DA: Kaplan-Meier survival analysis
overestimates the risk of revision arthroplasty: A meta-analysis.
Clin Orthop Relat Res. 473:3431–3442. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tibshirani R: The lasso method for
variable selection in the Cox model. Stat Med. 16:385–395. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
American Joint Committee On Cancer, . AJCC
7th edition Cancer Staging Manual, 2009. https://cancerstaging.org/references-tools/deskreferences/Documents/AJCC%207th%20Ed%20Cancer%20Staging%20Manual.pdfDecember
3–2019
|
|
30
|
Monti S, Tamayo P, Mesirov J and Golub T:
Consensus clustering: A resampling-based method for class discovery
and visualization of gene expression microarray data. Machine
Learn. 52:91–118. 2003. View Article : Google Scholar
|
|
31
|
Sun LY, Li XY and Sun ZW: Progress of
epigenetics and its therapeutic application in hepatocellular
carcinoma. Yi Chuan. 37:517–527. 2015.(In Chinese). PubMed/NCBI
|
|
32
|
Feo F, Frau M, Tomasi ML, Brozzetti S and
Pascale RM: Genetic and epigenetic control of molecular alterations
in hepatocellular carcinoma. Exp Biol Med (Maywood). 234:726–736.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Geula S, Moshitch-Moshkovitz S,
Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V,
Peer E, Mor N, Manor YS, et al: m6A mRNA methylation facilitates
resolution of naïve pluripotency toward differentiation. Science.
347:1002–1006. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lence T, Akhtar J, Bayer M, Schmid K,
Spindler L, Ho CH, Kreim N, Andrade-Navarro MA, Poeck B, Helm M and
Roignant JY: m6A modulates neuronal functions and sex
determination in Drosophila. Nature. 540:242–247. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Alarcón CR, Lee H, Goodarzi H, Halberg N
and Tavazoie SF: N 6-methyladenosine marks primary microRNAs for
processing. Nature. 519:482–485. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
He L, Li H, Wu A, Peng Y, Shu G and Yin G:
Functions of N6-methyladenosine and its role in cancer. Mol Cancer.
18:1762019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen Y, Peng C, Chen J, Chen D, Yang B, He
B, Hu W, Zhang Y, Liu H, Dai L, et al: WTAP facilitates progression
of hepatocellular carcinoma via m6A-HuR-dependent epigenetic
silencing of ETS1. Mol Cancer. 18:1272019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Meyer KD and Jaffrey SR: The dynamic
epitranscriptome: N6-methyladenosine and gene expression control.
Nat Rev Mol Cell Biol. 15:313–326. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shi H, Wei J and He C: Where, when, and
how: Context-dependent functions of RNA methylation writers,
readers, and erasers. Mol Cell. 74:640–650. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zaccara S, Ries RJ and Jaffrey SR:
Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell
Biol. 20:608–624. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang Y, Hsu PJ, Chen YS and Yang YG:
Dynamic transcriptomic m6A decoration: Writers, erasers,
readers and functions in RNA metabolism. Cell Res. 28:616–624.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang X, Li Z, Kong B, Song C, Cong J, Hou
J and Wang S: Reduced m6A mRNA methylation is correlated
with the progression of human cervical cancer. Oncotarget.
8:98918–98930. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kwok CT, Marshall AD, Rasko JE and Wong
JJ: Genetic alterations of m(6)A regulators predict poorer survival
in acute myeloid leukemia. J Hematol Oncol. 10:392017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cho SH, Ha M, Cho YH, Ryu JH, Yang K, Lee
KH, Han ME, Oh SO and Kim YH: ALKBH5 gene is a novel biomarker that
predicts the prognosis of pancreatic cancer: A retrospective
multicohort study. Ann Hepatobiliary Pancreat Surg. 22:305–309.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li Y, Xiao J, Bai J, Tian Y, Qu Y, Chen X,
Wang Q, Li X, Zhang Y and Xu J: Molecular characterization and
clinical relevance of m6A regulators across 33 cancer
types. Mol Cancer. 18:1372019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen SL, Liu LL, Wang CH, Lu SX, Yang X,
He YF, Zhang CZ and Yun JP: Loss of RDM1 enhances hepatocellular
carcinoma progression via p53 and Ras/Raf/ERK pathways. Mol Oncol.
14:373–386. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang H, Weng H, Sun W, Qin X, Shi H, Wu
H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al: Recognition of RNA
N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and
translation. Nat Cell Biol. 20:285–295. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhao X, Chen Y, Mao Q, Jiang X, Jiang W,
Chen J, Xu W, Zhong L and Sun X: Overexpression of YTHDF1 is
associated with poor prognosis in patients with hepatocellular
carcinoma. Cancer Biomark. 21:859–868. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han
D, Fu Y, Parisien M, Dai Q, Jia G, et al:
N6-methyladenosine-dependent regulation of messenger RNA stability.
Nature. 505:117–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hou J, Zhang H, Liu J, Zhao Z, Wang J, Lu
Z, Hu B, Zhou J, Zhao Z, Feng M, et al: YTHDF2 reduction fuels
inflammation and vascular abnormalization in hepatocellular
carcinoma. Mol Cancer. 18:1632019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhong L, Liao D, Zhang M, Zeng C, Li X,
Zhang R, Ma H and Kang T: YTHDF2 suppresses cell proliferation and
growth via destabilizing the EGFR mRNA in hepatocellular carcinoma.
Cancer Lett. 442:252–261. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chen M, Wei L, Law CT, Tsang FH, Shen J,
Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, et al: RNA
N6-methyladenosine methyltransferase-like 3 promotes liver cancer
progression through YTHDF2-dependent posttranscriptional silencing
of SOCS2. Hepatology. 67:2254–2270. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhou B, Liu C, Xu L, Yuan Y, Zhao J, Zhao
W, Chen Y, Qiu J, Meng M, Zheng Y, et al:
N6-methyladenosine reader protein Ythdc2 suppresses
liver steatosis via regulation of mRNA stability of lipogenic
genes. Hepatology. Mar 9–2020.(Epub ahead of print). doi:
10.1002/hep.31220.
|
|
54
|
Han D, Liu J, Chen C, Dong L, Liu Y, Chang
R, Huang X, Liu Y, Wang J, Dougherty U, et al: Anti-tumour immunity
controlled through mRNA m(6)A methylation and YTHDF1 in dendritic
cells. Nature. 566:270–274. 2019. View Article : Google Scholar : PubMed/NCBI
|