Introduction
Pancreatic cancer poses a major threat to human
health, and its incidence is rapidly rising. Cancer data in 2020
revealed that the death toll from pancreatic cancer reached 470,000
worldwide, making it the seventh tumor in terms of
cancer-associated deaths; additionally, pancreatic cancer is the
tumor with the lowest 5-year survival rate among all types of tumor
(1). Aggressive metastasis is the
most important cause of the high mortality rate observed among
patients with pancreatic cancer (2).
However, the mechanism of metastasis remains unknown. The target
molecules that underline metastasis are of great importance in the
treatment of pancreatic cancer. Pancreatic ductal adenocarcinoma
(PDAC) is the most common type of pancreatic cancer and one of the
most challenging malignant tumors to treat (3). The median survival time of patients
with PDAC after diagnosis is 2–8 months, and the 5-year overall
survival rate is <7% (1,4). The poor prognosis in patients with
pancreatic cancer is mainly due to the aggressiveness of cancer
cells, early metastasis and non-responsiveness to the majority of
chemotherapy regimens (5). At
present, surgical resection is the only feasible treatment for
PDAC. However, <20% of tumors are resectable at the time of
diagnosis (6). Furthermore, patients
who undergo surgery may relapse, and the average survival time of
patients undergoing resection is 12–20 months (7). The majority of patients with pancreatic
cancer have early metastatic or locally advanced cancer at the time
of diagnosis, and the only effective treatment for these patients
is chemoradiation (8). However,
pancreatic cancer cells respond poorly to both chemotherapy and
radiation (9,10). Although gemcitabine-based
chemotherapy, as the standard treatment for advanced pancreatic
cancer, can improve prognosis, its effectiveness is limited since
cancer cells often become resistant (11). Therefore, further research on
pancreatic cancer is urgently needed, and novel diagnostic and
therapeutic approaches are required to improve the prognosis of
this disease.
Non-coding RNAs (ncRNAs) serve important roles in
tumor development. MicroRNAs (miRNAs/miRs) are highly conserved
single-stranded ncRNA molecules with a length of 18–25 nucleotides.
miRNAs can regulate gene expression by base-pairing with
3′-untranslated regions (3′-UTRs), thereby enhancing mRNA
degradation or inhibiting post-transcriptional translation
(12). To date, >2,500 miRNAs
have been identified in plants, animals and viruses (13). After miRNAs are produced in the cell
nucleus, they are delivered into the cytoplasm by nuclear
transporters and then guided into the RNA-induced silencing complex
(RISC), where they facilitate target gene mRNA degradation or
inhibit translation through complementary pairing with target gene
mRNA bases (14). miRNAs serve
important roles in tumor development (15,16). For
example, previous studies have reported the involvement of various
miRNAs (such as miR-21, miR-155 and miR-210) in the development and
progression of pancreatic cancer (17,18).
Fibroblast growth factor 2 (FGF2) is the main factor
leading to tissue fibrosis, and pancreatic fibrosis plays a key
role in the progression of pancreatic cancer (19). Sakai et al (20) found that, when mice were
subcutaneously inoculated with BxPC-3 pancreatic cancer cells and
simultaneously administered FGF2, the area of interstitial fibrosis
increased compared with that of mice administered with BxPC-3
alone. The fibrosis was characterized by increased accumulation of
murine collagen, which was associated with an increase in
monocyte/macrophage content in tumor tissue, and promoted the
progression of pancreatic cancer (20).
miR-203 is located on chromosome 14q32.33.
Compared with that in normal tissues, miR-203 exhibits
downregulated expression in diverse malignancies, including
bladder, non-small cell lung and endometrial cancer (21–23).
Numerous studies have demonstrated that miR-203 serves an important
role in tumor cell proliferation, migration and invasion (24,25). For
example, miR-203 expression is decreased in pancreatic cancer
compared with that in normal pancreatic tissue and chronic
pancreatitis, suggesting that miR-203 may be associated with
specific characteristics of tumors and their behavior (26). Other studies have suggested that FGF2
expression may be affected by miR-203 (27,28).
However, the relevant mechanisms of action and signaling pathways
in pancreatic cancer remain unknown. The present study established
cell models of miR-203 knockdown and overexpression to explore the
regulatory effects of miR-203 on FGF2 expression, as well as on the
proliferation, invasion and migration of pancreatic cancer
cells.
Materials and methods
Reagents and materials
Pancreatic cancer cell lines [PANC-1 (CVCL_0480),
AsPC-1 (CVCL_0152), BxPC-3 (CVCL_0186) and HPAC] were purchased
from The Cell Bank of Type Culture Collection of the Chinese
Academy of Sciences. Normal human pancreatic epithelial cells
[hTERT-HPNE E6/E7 (CRL-4036)] and 293T cells were also purchased
from The Cell Bank of Type Culture Collection of the Chinese
Academy of Sciences (293T cells were used for transfection and
luciferase reporter gene experiments). Dulbecco's modified Eagle's
medium (DMEM), fetal bovine serum (FBS) and Opti-MEM were procured
from Gibco (Thermo Fisher Scientific, Inc.).
Lipofectamine® 2000 was obtained from Invitrogen (Thermo
Fisher Scientific, Inc.). PrimeScript™ RT kit and SYBR Green dye
were purchased from Takara Bio, Inc. Mimic-miR-203-3p,
mimic-negative control (NC; scrambled), inhibitor-miR-203-3p and
inhibitor-NC (non-targeting) were obtained from Guangzhou RiboBio
Co., Ltd. The pGL3 reporter vector and Dual-Glo Luciferase Activity
Assay kit were purchased from Promega Corporation. The Annexin
V-FITC/PI Double-Staining Cell Apoptosis Assay kit was purchased
from Nanjing KeyGen Biotech Co., Ltd.
Cell culture and transfection
PANC-1, AsPC-1, BxPC-3, HPAC and HPNE cells were
cultured in DMEM containing 10% FBS and 1% streptomycin at 37°C in
an atmosphere of 5% CO2. The cells were passaged at 1:4
or 1:5 ratio and then used for experiments at the logarithmic
growth phase. The cells were digested with 0.25% trypsin, and the
digested cells were counted using a hemocytometer and diluted with
culture medium to a concentration of 5×104 cells/ml.
Subsequently, 100 µl cell suspension was added into each well of a
96-well culture plate, and incubated at 37°C in an atmosphere of 5%
CO2 for 24 h. According to the experimental results,
AsPC-1 cells were selected as the experimental cell line. For cell
transfection, 0.25 µg miRNA (mimic-miR-203, miR-NC, inhibitor-NC,
inhibitor-miR-203) was diluted with 25 µl serum-free Opti-MEM,
mixed gently and incubated at room temperature for 5 min.
Lipofectamine® 2000 (0.5 µl) was diluted with 25 µl
serum-free Opti-MEM, mixed gently and incubated at room temperature
for 5 min. Both components were subsequently combined, mixed gently
and incubated for 20 min at room temperature. The
miRNA-Lipofectamine 2000 mixture was then added to the wells, which
contained 50 µl Opti-MEM, and gently mixed. The sequences of the
miRs transfected into the cells are as follows: Mimic-miR-203,
5′-GUGAAAUGUUUAGGACCACUAG-3′; miR-NC, 5′-UUCUCCGAACGUGUCACGUTT-3′;
inhibitor-NC, 5′-CAGUACUUUUGUGUAGUACAA-3′; and inhibitor-miR-203,
5′-CUAGUGGUCCUAAACAUUUCAC-3′. After 4–6 h of incubation at 37°C,
the medium containing the siRNA-Lipofectamine 2000 mixture was
carefully aspirated and replaced with DMEM with 5% FBS and no
antibiotics. The culture plate was incubated at 37°C for 24 h
before detection of cell proliferation, apoptosis, invasion and
migration. In addition, transfection of FGF2 recombinant plasmid
(Guangzhou RiboBio Co., Ltd.; 25 nM/ml; control plasmid was an
empty vector) was performed. The specific method was the same as
aforementioned for the transfection of mimic/inhibitor-miR-203.
Regarding the cell culture with gemcitabine, 0.04 µg/ml gemcitabine
(LY 188011; MedChemExpress) was added as previously described
(29).
Detection of cell proliferation by the
Cell Counting Kit (CCK)-8 assay
After 24 h of transfection as aforementioned, 10 µl
CCK-8 reagent (Wuhan Boster Biological Technology Co., Ltd.) was
added to each well. The culture plate was incubated for 3 h at 37°C
and mixed gently on a shaker for 10 min. A microplate reader was
used to determine the optical density (OD) value of each well at a
wavelength of 450 nm. The inhibition rate = (NC group OD values -
experimental group OD values)/NC group OD values × 100.
Detection of apoptosis by Annexin
V-APC/7-AAD double-staining
AsPC-1 cells were washed twice with PBS, digested
with 0.25% trypsin for 5 min and centrifuged at 1,000 × g for 5 min
at 4°C to collect 5×105 cells, which were then
resuspended in 500 µl binding buffer (Wuhan Boster Biological
Technology Co., Ltd.). Subsequently, 5 µl Annexin V-APC was added
to the cell suspension, followed by mixing, and then 5 µl 7-AAD was
added, followed by mixing. The samples were incubated at room
temperature in the dark for 5–15 min, and apoptosis was detected
using a BD LSRII flow cytometer (BD Biosciences). The detection
data were analyzed and graphed using CellQuest Pro (BD
Biosciences).
Detection of migration and invasion by
wound healing and Transwell assays
For wound healing assays, cells at the logarithmic
growth phase were cultured to reached 100% confluence in 6-well
plates. The next day, the AsPC-1 cell layer was scratched with a
10-µl micropipette tip in the center of the well (denoted as 0 h).
DMEM with 1% FBS was used to avoid cell proliferation. The AsPC-1
cells were gently rinsed with PBS and incubated in 1%
FBS-containing DMEM. After 24 h of incubation at 37°C, the cells
were removed from the incubator, photographed using an inverted
light microscope (magnification, ×200; Leica Microsystems, Inc.),
and the cell migration distance was measured. The width of the
wound healing site was quantified and compared with baseline
values. All experiments were repeated independently in
triplicate.
Cancer cell invasion was tested using Transwell
assays. Cells were removed from serum-containing medium and
serum-starved for 24 h using serum-free medium. Matrigel (BD
Biosciences) was thawed at 4°C overnight, and 2×104
cells from each group in 200 µl serum-free medium were seeded in
the upper chamber (pore size, 8.0 µm; Corning, Inc.) precoated with
90 µl Matrigel at 37°C for 8 h. Subsequently, 600 µl RPMI-1640
medium (Wuhan Boster Biological Technology Co., Ltd.) containing
10% FBS was added to the lower chamber. After 24 h of incubation at
37°C, the upper chambers were fixed with 4% polymethanol (Wuhan
Boster Biological Technology Co., Ltd.) for 30 min at room
temperature, and then stained with 0.1% crystal violet for 30 min
at room temperature. The cells that migrated through the membrane
and invaded the underside of the upper chamber were photographed
using an inverted light microscope (magnification, ×200; Leica
Microsystems, Inc.). Five random fields were selected to calculate
the number of migrating or invading cells.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and reverse transcribed into cDNA according to the
manufacturer's protocol using PrimeScript™ RT kit (Thermo Fisher
Scientific, Inc.). RT-qPCR was performed on a CFX96 System (Bio-Rad
Laboratories, Inc.). The reactions consisted of 5.0 µl 2X SYBR
Green Master Mix, 0.5 µl each forward and reverse primers (2.5 µM),
1 µl cDNA and RNase-free double-distilled H2O. The
thermocycling conditions were as follows: 95°C for 5 min, followed
by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The relative
expression of target mRNA and miRNA was quantified using the
2−ΔΔCq method (30).
GAPDH was used as the reference gene to normalize mRNA and U6 was
used as the reference gene to normalize miRNA expression. The
primer sequences are shown in Table
SI.
Western blot analysis
Cells were washed with PBS to extract the total
protein using RIPA Lysis and Extraction Buffer (Wuhan Boster
Biological Technology Co., Ltd.). The concentration of the
extracted protein was measured with a BCA protein quantification
kit (Wuhan Boster Biological Technology Co., Ltd.) to prepare
samples for electrophoresis. Subsequently, samples were mixed with
a loading buffer (Wuhan Boster Biological Technology Co., Ltd.) at
a ratio of 5:1 and subjected to heat at 100°C for 10 min. A total
of 50 µg protein/lane from each sample was separated by 10%
SDS-PAGE and then transferred to a PVDF membrane. The membrane was
blocked with 5% skimmed milk powder in TBS-0.5% Tween (TBST) at
room temperature for 2 h and then incubated with the following
anti-human primary antibodies at 4°C for 12 h: FGF2 (cat. no.
BM4959; 1:1,000), FGF receptor 3 (FGFR3; cat. no. BM5016; 1:500),
FGFR2 (cat. no. BM4991; 1:1,000) and GAPDH (cat. no. BM3896;
1:10,000) (all Wuhan Boster Biological Technology Co., Ltd.).
Subsequently, a secondary antibody (HRP-conjugated affiniPure goat
anti-rabbit IgG; cat. no. BA1055, 1:5,000; Wuhan Boster Biological
Technology Co., Ltd.) was incubated with the membrane at 37°C for 2
h, and an ECL™ reagent (Wuhan Boster Biological Technology Co.,
Ltd.) was used for visualizing the protein bands after washing with
TBST 3 times for 19 min each. GADPH was used as a loading control
for normalization. The results were analyzed using ImageJ software
(v1.8.0; National Institutes of Health).
Bioinformatics analysis
TargetScan (http://www.targetscan.org/vert_72/) is a web server
that predicts biological targets of miRNAs by searching for the
presence of sites that match the seed region of each miRNA. FGF2
and miR-203-3p were inserted to analyze whether they could bind to
each other and to identify the binding sequence online.
Dual-luciferase reporter assay
The wild-type (WT) FGF2 fragments containing the
putative miR-203-3p binding site were prepared and cloned into the
pGL3 reporter vectors (Promega Corporation) to yield the FGF2-WT
reporter constructs. The mutated (Mut) versions of the fragments
were additionally prepared according to the manufacturer's protocol
with the GeneTailor™ Site-Directed Mutagenesis System (Invitrogen;
Thermo Fisher Scientific, Inc.), after which FGF2-Mut reporter
constructs were generated as aforementioned. After growing until
70–80% confluence, cotransfection of WT or Mut reporter vectors
with miR-203-3p mimic or NC mimic was implemented using
Lipofectamine 2000, as aforementioned. After 36 h of incubation at
37°C, firefly luciferase activity was detected according to the
manufacturer's instructions of the Dual-Glo Luciferase Activity
Assay kit and compared with Renilla luciferase activity. All
experiments were repeated independently in triplicate.
Statistical analysis
The experimental results are presented as the mean ±
SD and all experiments were repeated ≥3 times. SPSS 26.0 Software
(IBM Corp.) was used for statistical analysis. According to whether
the data conformed to the normal distribution, The data of CCK-8,
apoptosis, migration, invasion and RT-qPCR assays were analyzed
according to data types and comparison methods. Kruskal-Wallis test
was used for comparison between two groups (e.g. mimic-NC vs.
mimic-miR, inhibitor-NC vs. inhibitor-miR, AsPC1-NC vs.
AsPC1-FGF2), and the Bonferroni method was used for multiple
comparisons. Statistical comparison of mean values in two groups
were compared by unpaired Student's t-test. Multiple groups were
compared by one-way ANOVA with Dunnett's post hoc test. All graphs
were produced using GraphPad Prism (version 8.0; GraphPad Software,
Inc.) and ImageJ (v1.8.0; National Institutes of Health). P<0.05
was considered to indicate a statistically significant
difference.
Results
miR-203-3p expression in different
cell lines
RT-qPCR was used to quantify miR-203-3p expression
in the HPNE, BxPC-3, HPAC, PANC-1 and AsPC-1 cell lines, and it was
found that miR-203-3p expression was significantly lower in
pancreatic cancer cells than that in HPNE cells (Fig. 1A). Furthermore, miR-203-3p expression
was the lowest in AsPC-1 cells (Fig.
1A), indicating that the AsPC-1 cell line was suitable for
further experiments.
miR-203-3p inhibits the proliferation
of AsPC-1 cells
To investigate the role of miR-203-3p in pancreatic
cancer cells, inhibitor-miR-203-3p was used to knockdown
miR-203-3p, which targeted the junction sites of miR-203-3p.
Overexpression of miR-203-3p was achieved using mimic-miR-203-3p
for transfection into AsPC-1 cells. The results of the CCK-8 assay
revealed that miR-203-3p overexpression significantly inhibited the
proliferation of AsPC-1 cells (Fig.
1B). As shown in Table SII,
compared with that of the mimic-NC group, the inhibition rate of
the mimic-miR-203-3p group was increased. However, compared with
that of the inhibitor-NC group, the inhibition rate of the
inhibitor-miR-203-3p group did not markedly change (Fig. 1B and Table SII).
miR-203-3p increases the apoptosis of
pancreatic cancer cells
The AsPC-1 cell apoptosis rate was significantly
different among all groups. Compared with that of the mimic-NC
group, the apoptosis rate of the mimic-miR-203-3p group was
significantly increased, while compared with that of the
inhibitor-NC group, the apoptosis rate of the inhibitor-miR-203-3p
group was significantly decreased (Fig.
1C and D, and Table SIII).
miR-203-3p decreases cell migration
and invasion, and increases the sensitivity of pancreatic cancer
cells to gemcitabine
The present study examined the ability of cells
transfected with mimic- or inhibitor-miR-203-3p to invade and
migrate. Compared with that of the mimic-NC group, the
mimic-miR-203-3p group exhibited significantly decreased migration
and invasion at 24 h, while knockdown of miR-203-3p expression
significantly promoted cell migration and invasion (Fig. 2A-D). Compared with the gemcitabine
group and the mimic-NC + gemcitabine group, the proliferation curve
of the AsPC-1 cells in the mimic-miR-203-3p + gemcitabine group was
significantly decreased; however, compared with the gemcitabine
group and the inhibitor-NC + gemcitabine group, the
inhibitor-miR-203-3p + gemcitabine group exhibited higher
proliferation rates at 96 h (Fig.
2E). Overall, the current findings suggested that miR-203-3p
restrained the progression of pancreatic cancer and increased the
sensitivity of pancreatic cancer cells to gemcitabine in
vitro.
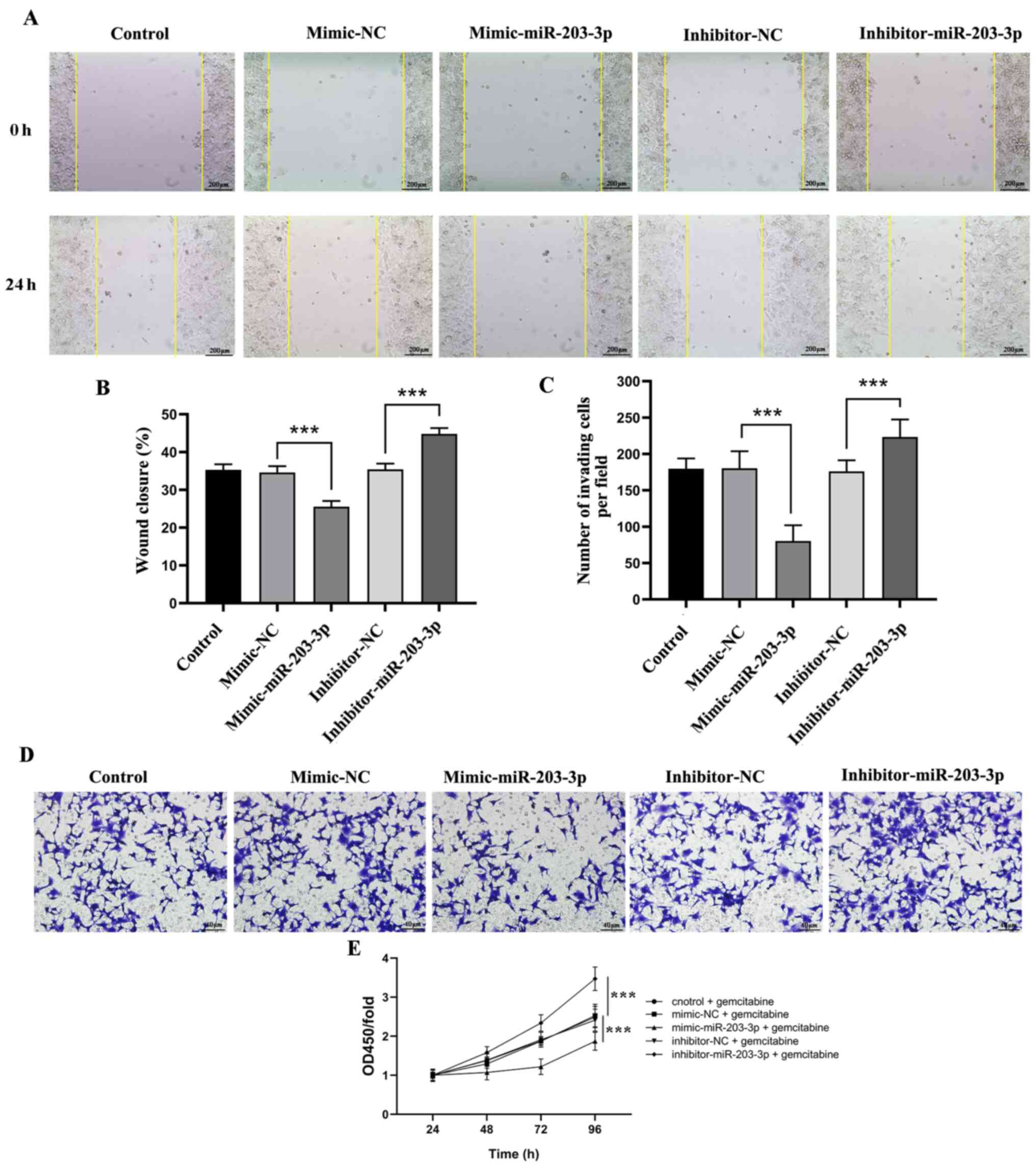 | Figure 2.miR-203-3p overexpression decreases
the migration and invasion of pancreatic cancer cells. (A) Effects
of miR-203-3p overexpression or knockdown expression on cell
migration were detected by wound healing assay (scale bar, 200 µm).
(B) Wound closure percentage of AsPC-1 cells treated with mimic-NC,
mimic-miR-203-3p, inhibitor-NC or inhibitor-miR-203-3p. (C) Number
of invading AsPC-1 cells treated with mimic-NC, mimic-miR-203-3p,
inhibitor-NC or inhibitor-miR-203-3p group. (D) Effects of
miR-203-3p overexpression or knockdown on cell invasion were
detected by Transwell assay (scale bar, 40 µm). (E) Proliferation
curve of AsPC-1 cells treated with mimic-NC, mimic-miR-203-3p,
inhibitor-NC or inhibitor-miR-203-3p group. Control represents
AsPC-1 cells without transfection. All data are presented as the
mean ± SD of three experiments. ***P<0.001. OD, optical density;
miR, microRNA; NC, negative control. |
miR-203-3p inhibits FGF2 expression in
pancreatic cancer cells
miR-203-3p expression was significantly decreased by
inhibitor-miR-203-3p, whereas it was significantly enhanced by
mimic-miR-203-3p (Fig. 3A).
Additionally, it was found that miR-203-3p overexpression in AsPC-1
cells significantly decreased FGF2 mRNA expression, while
miRNA-203-knockdown in AsPC-1 cells significantly increased FGF2
mRNA expression, as determined by RT-qPCR (Fig. 3A). Furthermore, the protein
expression levels of FGFR2 and FGFR3 were detected by western
blotting, revealing that miR-203-3p overexpression decreased FGFR2
and FGFR3 expression, while miR-203-3p-knockdown increased FGFR2
and FGFR3 expression (Fig. 3B).
These results indicated that upregulated miR-203-3p expression
elicited inhibitory effects on FGF2 expression.
To verify the role of FGF2 in AsPC-1 cells, the
control plasmid and the FGF2 overexpression plasmid were
simultaneously transfected, and RT-qPCR was performed. It was found
that FGF2 mRNA expression in the AsPC-1-FGF2 group increased
significantly compared with that in the AsPC-1-NC group (Fig. 3C). In addition, miR-203 was
significantly decreased after FGF2 overexpression (Fig. 3C).
FGF2 significantly affects the
proliferation of AsPC-1 cells
FGF2 overexpression in pancreatic cancer cells
significantly affected cell proliferation. Compared with that of
the AsPC-1 and AsPC-1-NC groups, the cell viability of the
AsPC-1-FGF2 group was significantly increased, indicating that FGF2
expression promoted the proliferation of AsPC-1 cells (Fig. 3D and Table SIV).
FGF2 affects AsPC-1 cell
apoptosis
The flow cytometry results indicated that the
apoptosis rate was significantly decreased in the AsPC-1-FGF2 group
compared with that in the AsPC-1-NC group, indicating that the
change in FGF2 expression significantly affected apoptosis
(Fig. 3E and F).
Overexpression of FGF2 increases cell
migration and invasion, and the resistance of pancreatic cancer
cell lines to gemcitabine
Compared with the AsPC-1NC group, the
wound closure ratio was significantly increased in the
AsPC-1FGF2 group (Fig. 4A and
B). Similarly, compared with the AsPC-1NC group, the
invading cell number was significantly increased in the
AsPC-1FGF2 group (Fig. 4C and
D). These results indicated that FGF2 promoted the migratory
and invasive abilities of the AsPC-1 pancreatic cancer cell line.
Compared with the gemcitabine group and the NC + gemcitabine group,
the AsPC-1 cells of the FGF2 + gemcitabine group exhibited
significantly higher proliferation efficiency (Fig. 4E).
 | Figure 4.Overexpression of FGF2 increases the
migration and invasion of pancreatic cancer cells. (A) Wound
healing assays were used to detect the migration of AsPC-1,
AsPC-1-NC and AsPC-1-FGF2 cell lines (scale bar, 200 µm). (B) Wound
closure percentage of AsPC-1, AsPC-1-NC and AsPC-1-FGF2 cells. (C)
Transwell assays were used to detect the invasion of AsPC-1,
AsPC-1-NC and AsPC-1-FGF2 cells (scale bar, 40 µm). (D) Number of
invading AsPC-1, AsPC-1-NC and AsPC-1-FGF2 cells. (E) Proliferation
curve of AsPC-1, AsPC-1-NC and AsPC-1-FGF2 group. ***P<0.001.
FGF2, fibroblast growth factor 2; NC, negative control; OD, optical
density. |
miR-203-3p directly interacts with the
3′-UTR region of FGF2 in pancreatic cancer cells
According to bioinformatics analysis using
TargetScan, a binding site to the 3′-UTR of FGF2 was predicted in
miR-203-3p (Fig. 5A). A
dual-luciferase reporter system was used to verify whether
miR-203-3p interacted with the binding site of the 3′-UTR of FGF2.
A dual-luciferase reporter gene vector for the target gene site
region (including WT and Mut) was successfully constructed, and was
co-transfected with mimic-miR-203a-3p and mimic-NC. The luciferase
activity in transfected cells was determined. The results revealed
that luciferase activity in AsPC-1 cells co-transfected with
mimic-miR-203-3p and WT vectors was significantly decreased
compared with that of cells transfected with MUT vectors (Fig. 5B). These results indicated that
miR-203-3p interacted directly with the 3′-UTR of FGF2. This
mechanism of direct binding indicated that miR-203-3p directly
regulated its target gene FGF2.
Discussion
Since no significant progress has been made lately
in the treatment of pancreatic cancer, research has focused on the
tumor cells per se, as well as on the tumor immune
microenvironment of pancreatic cancer (31). Previous studies have found that
overexpression of FGF2 and its receptor in pancreatic cancer cells
in the tumor microenvironment promotes the progression of the tumor
itself, and is associated with a poor prognosis (32–34).
Clinically resected pancreatic cancer specimens, particularly PDAC
specimens, have a hard consistency, and large quantities of
collagen can be observed by hematoxylin and eosin staining under an
optical microscope (35). Kostas
et al (36) described the
anti-apoptotic effects of FGF1 and FGF2 in cells, which were
independent of FGFR activation and downstream signaling.
Shirakihara et al (37) found
that FGF2 can promote epithelial-mesenchymal transition (EMT) in
injured tissues. Our previous study has revealed that the
heparanase/syndecan-1 axis can upregulate the FGF2 level and
increase the expression levels of downstream Palladin by activating
the PI3K/Akt signaling pathway, thereby leading to the activation
of EMT (38). It has been also
reported that EMT promotes the migration and invasion of pancreatic
cancer cells (38). Therefore, it is
necessary to identify a method that can interfere with the
expression of target proteins.
miRNA is a type of post-transcriptional regulatory
factor that does not directly act on genes, but downregulates the
expression levels of target genes by acting on the mRNA transcribed
from genes (39). The present study
detected miR-203-3p expression in pancreatic cancer cell lines
(PANC-1, AsPC-1, BxPC-3 and HPAC) and in normal pancreatic cells
(HPNE). Previous studies have investigated the role of miR-203 in
tumors (24,25,39). The
present study found that miR-203-3p expression was significantly
lower in pancreatic cancer cell lines compared with in normal
pancreatic cells, which is consistent with the results of Du et
al (24). Lin et al
(25) reported that miR-203-3p
expression is increased in pancreatic cancer tissues, but the miRNA
detection of tissues may be affected by the presence of
lymphocytes, fibroblasts, other interstitial cells and non-cellular
components, resulting in inaccurate results (40). Using in vitro experiments, the
present study indicated that miR-203-3p expression affected the
proliferation, apoptosis, invasion and migration of pancreatic
cancer cells. Overexpression of miR-203-3p significantly inhibited
the proliferation, invasion and migration of AsPC-1 cells, and
promoted their apoptosis. According to our preliminary experiments
(38) and the TargetScan sequence
prediction results, subsequent FGF2 detection was performed.
Our group has previously demonstrated that FGF2
overexpression can contribute to the resistance of pancreatic
cancer cells to chemotherapy (29,35).
Patients with pancreatic cancer and high FGF2 expression are not
responsive to postoperative chemotherapy with gemcitabine, and the
overall prognosis is poor (41). The
present study revealed that increased miR-203 expression inhibited
the transcription of FGF2 mRNA in AsPC-1 cells, thereby inhibiting
the progression of pancreatic cancer cells. In recent years, the
association between cytokines and tumorigenesis has gained
increasing attention. Cytokine-receptor interactions activate
various signaling pathways in cells and serve important roles in
regulating cell proliferation, apoptosis and angiogenesis (42). FGF2, a growth-promoting factor, is
highly expressed in breast, gastric and thyroid cancer, as well as
in other normal tissues, and aberrant FGF2/FGFR1 signaling may
promote tumor development (43).
miRNAs regulate gene expression after transcription. Therefore,
miRNAs are key regulators of gene expression and promising
candidates for biomarker development. Additionally, miRNAs guide
RISC to degrade mRNAs or prevent translation by base-pairing with
target gene mRNAs (12). The present
study demonstrated that miR-203-3p, combined with the 3′-UTR of
FGF2, exerted a direct regulatory effect. The current results are
consistent with those of previous studies (28,44), and
may explain why miR-203 affected the proliferation, invasion and
migration of pancreatic cancer cells. However, it was also revealed
that downregulation of miR-203 inhibited AsPC-1 cell apoptosis,
while overexpression of FGF2 promoted the viability of pancreatic
cancer cells. Therefore, miR-203 may also affect the viability of
AsPC-1 cells through other signaling pathways, and further studies
are required to explore this hypothesis.
Li et al (45)
has found that FGF2 prevents cancer cells from endoplasmic
reticulum (ER) stress-mediated apoptosis via enhancing
proteasome-mediated non-catalytic region of tyrosine kinase
degradation. The present study indicated that overexpression of
FGF2 in pancreatic cancer AsPC-1 cells promoted cell viability and
inhibited apoptosis. The specific mechanism may be through FGF2
binding and activating the tumor cell surface tyrosine kinase
receptor FGFR. Intracellular signaling (including Ras/MAPK,
PI3K/Akt and PLCγ/PKC) leads to the proliferation, migration or
differentiation of multiple types of cells and exerts significant
anti-apoptotic effects (46,47). In addition to this classic mode of
action, Kostas et al (36)
revealed that under ER stress conditions, FGF1 and FGF2 can be
transported to the cytoplasm and nucleus through endosomal
membranes in a receptor-independent manner to exert their
anti-apoptotic effects. FGF2 has significantly stronger
anti-apoptotic effects than FGF1 (48). Thus, the present study hypothesized
that, when tumor cells are deficient in FGF2, the signaling
pathways in tumor cells may be altered, the ER stress may be
unbalanced, and apoptosis may increase. This theory is consistent
with the present results, since downregulating intracellular FGF2
significantly induced apoptosis.
The regulation of miR-203-3p expression in
pancreatic cancer should also be further investigated. Li et
al (48) has revealed that
epidermal growth factor in esophageal squamous cells acts on the
upstream regulatory elements of miR-203 through CCAAT-enhancer
binding protein β LIP to downregulate miR-203-3p expression in the
cells. Zhang et al (49) has
demonstrated that cisplatin induces the production of E2F
transcription factor 1 (E2F1) in esophageal cancer cells, and then
acts on the E2F1 promoter to regulate miR-203 expression. The
abnormal expression levels of miR-203-3p in pancreatic cancer cells
require to be further studied.
The present study has some limitations. At present,
gemcitabine has become the main part of pancreatic cancer drug
treatment. In the current study, the joint effect of miR-203-3p and
gemcitabine was investigated, but the mechanism was not explored in
depth. Moreover, whether miR-203-3p can reverse the gemcitabine
resistance of pancreatic cancer cell lines should be further
investigated. Second, although miR-203-3p downregulated FGF2
expression, whether it changes the sensitivity of pancreatic cancer
cell lines to gemcitabine through FGF2 should also be further
studied.
In summary, the present study provided evidence that
miRNA-203-3p expression was abnormally low in pancreatic cancer
cells, and that miRNA-203-3p overexpression inhibited the
proliferation, invasion and migration, and increased the apoptosis
of pancreatic cancer cells. Furthermore, the present study has
strengthened the evidence that miR-203-3p may directly act on FGF2,
thus suggesting the value of miR-203 as an important biomarker for
pancreatic cancer progression.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by grants from the
Shanxi Science and Technology Department (grant nos. 201901D11412
and 201901D1111404) and Shanxi ‘136’ Leading Clinical Key Specialty
(grant nos. 2019XY002 and 2019XY012).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XFF and HCZ conceived the project. XFF, HCZ, CLY,
CZC and KW performed most of the experiments. HCZ wrote the
manuscript and collected the data. CLY and CZC collected and
analyzed the data. YZT, KW and FG contributed to the interpretation
of the data and the critical review of the manuscript. HLZ made
contributions to conception and design, acquisition of data and
revision and correction of the manuscript. HLZ, YZT and XFF
provided the funding for the study. The authenticity of all the raw
data was assessed by YZT and HLZ. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rawla P, Sunkara T and Gaduputi V:
Epidemiology of pancreatic cancer: Global trends, etiology and risk
factors. World J Oncol. 10:10–27. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Adamska A, Domenichini A and Falasca M:
Pancreatic ductal adenocarcinoma: Current and evolving therapies.
Int J Mol Sci. 18:13382017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gillen S, Schuster T, Meyer Zum
Büschenfelde C, Friess H and Kleeff J: Preoperative/neoadjuvant
therapy in pancreatic cancer: A systematic review and meta-analysis
of response and resection percentages. PLoS Med. 7:e10002672010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maitra A and Hruban RH: Pancreatic cancer.
Annu Rev Pathol. 3:157–188. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Siegel RL, Fedewa SA, Miller KD,
Goding-Sauer A, Pinheiro PS, Martinez-Tyson D and Jemal A: Cancer
statistics for hispanics/latinos, 2015. CA Cancer J Clin.
65:457–480. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
McGuigan A, Kelly P, Turkington RC, Jones
C, Coleman HG and McCain RS: Pancreatic cancer: A review of
clinical diagnosis, epidemiology, treatment and outcomes. World J
Gastroenterol. 24:4846–4861. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Garrido-Laguna I and Hidalgo M: Pancreatic
cancer: From state-of-the-art treatments to promising novel
therapies. Nat Rev Clin Oncol. 12:319–324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Neoptolemos JP, Stocken DD, Friess H,
Bassi C, Dunn JA, Hickey H, Beger H, Fernandez-Cruz L, Dervenis C,
Lacaine F, et al: A randomized trial of chemoradiotherapy and
chemotherapy after resection of pancreatic cancer. N Engl J Med.
350:1200–1210. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Amrutkar M and Gladhaug IP: Pancreatic
cancer chemoresistance to gemcitabine. Cancers (Basel). 9:1572017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li H, Jiang JD and Peng ZG:
MicroRNA-mediated interactions between host and hepatitis C virus.
World J Gastroenterol. 22:1487–1496. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Macfarlane LA and Murphy PR: MicroRNA:
Biogenesis, function and role in cancer. Curr Genomics. 11:537–561.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen X, Cheng JY and Yin J: Predicting
microRNA-disease associations using bipartite local models and
hubness-aware regression. RNA Biol. 15:1192–1205. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hatziapostolou M, Polytarchou C and
Iliopoulos D: miRNAs link metabolic reprogramming to oncogenesis.
Trends Endocrinol Metab. 24:361–373. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dillhoff M, Liu J, Frankel W, Croce C and
Bloomston M: MicroRNA-21 is overexpressed in pancreatic cancer and
a potential predictor of survival. J Gastrointest Surg.
12:2171–2176. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Szafranska AE, Davison TS, John J, Cannon
T, Sipos B, Maghnouj A, Labourier E and Hahn SA: MicroRNA
expression alterations are linked to tumorigenesis and
non-neoplastic processes in pancreatic ductal adenocarcinoma.
Oncogene. 26:4442–4452. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marzoq AJ, Giese N, Hoheisel JD and
Alhamdani MSS: Proteome variations in pancreatic stellate cells
upon stimulation with proinflammatory factors. J Biol Chem.
288:32517–32527. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sakai S, Iwata C, Tanaka HY, Cabral H,
Morishita Y, Miyazono K and Kano MR: Increased fibrosis and
impaired intratumoral accumulation of macromolecules in a murine
model of pancreatic cancer co-administered with FGF-2. J Control
Release. 230:109–115. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shen J, Zhang J, Xiao M, Yang J and Zhang
N: miR-203 suppresses bladder cancer cell growth and targets
twist1. Oncol Res. 26:1155–1165. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chi Y, Jin Q, Liu X, Xu L, He X, Shen Y,
Zhou Q, Zhang J and Jin M: miR-203 inhibits cell proliferation,
invasion, and migration of non-small-cell lung cancer by
downregulating RGS17. Cancer Sci. 108:2366–2372. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zierau O, Helle J, Schadyew S, Morgenroth
Y, Bentler M, Hennig A, Chittur S, Tenniswood M and Kretzschmar G:
Role of miR-203 in estrogen receptor-mediated signaling in the rat
uterus and endometrial carcinoma. J Cell Biochem. 119:5359–5372.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Du SL, Xu LY, Gao P, Liu QS, Lu FF, Mo ZH,
Fan ZZ, Cheng XL and Dong ZH: miR-203 regulates DJ-1 expression and
affects proliferation, apoptosis and DDP resistance of pancreatic
cancer cells. Eur Rev Med Pharmacol Sci. 23:8833–8840.
2019.PubMed/NCBI
|
|
25
|
Lin XM, Chen H and Zhan XL: miR-203
regulates JAK-STAT pathway in affecting pancreatic cancer cells
proliferation and apoptosis by targeting SOCS3. Eur Rev Med
Pharmacol Sci. 23:6906–6913. 2019.PubMed/NCBI
|
|
26
|
Greither T, Grochola LF, Udelnow A,
Lautenschläger C, Würl P and Taubert H: Elevated expression of
microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated
with poorer survival. Int J Cancer. 126:73–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
He S, Zhang G, Dong H, Ma M and Sun Q:
miR-203 facilitates tumor growth and metastasis by targeting
fibroblast growth factor 2 in breast cancer. Onco Targets Ther.
9:6203–6210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu M, Gu M, Zhang K, Zhou J, Wang Z and Da
J: miR-203 inhibition of renal cancer cell proliferation, migration
and invasion by targeting of FGF2. Diagn Pathol. 10:242015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fu XF, Tian YZ and Dong XS: Effects of
gemcitabine combined with heparanase inhibitor on invasion and
migration of pancreatic cancer PANC-1 cells. Chin Med Clin.
15:20–22. 2015.
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dąbkowski K, Bogacka B, Tarnowski M and
Starzyńska T: Pancreatic cancer microenvironment. Pol Merkur
Lekarski. 41:296–302. 2016.(In Polish).
|
|
32
|
Coleman SJ, Chioni AM, Ghallab M, Anderson
RK, Lemoine NR, Kocher HM and Grose RP: Nuclear translocation of
FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic
cancer cell invasion. EMBO Mol Med. 6:467–481. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mardhian DF, Vrynas A, Storm G, Bansal R
and Prakash J: FGF2 engineered SPIONs attenuate tumor stroma and
potentiate the effect of chemotherapy in 3D heterospheroidal model
of pancreatic tumor. Nanotheranostics. 4:26–39. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ren B, Cui M, Yang G, Wang H, Feng M, You
L and Zhao Y: Tumor microenvironment participates in metastasis of
pancreatic cancer. Mol Cancer. 17:1082018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fu XF, Dong XS, Gao F and Zhao HC: The
expression of fibroblast grouth factor 2 in pancreatic cancer and
its effect and mechanism on the invasion and metastasis of
pancreatic cancer cells. J Shanxi Med Univ. 87:26–31. 2017.
|
|
36
|
Kostas M, Lampart A, Bober J, Wiedlocha A,
Tomala J, Krowarsch D, Otlewski J and Zakrzewska M: Translocation
of exogenous FGF1 and FGF2 protects the cell against apoptosis
independently of receptor activation. J Mol Biol. 430:4087–4101.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shirakihara T, Horiguchi K, Miyazawa K,
Ehata S, Shibata T, Morita I, Miyazono K and Saitoh M: TGF-β
regulates isoform switching of FGF receptors and
epithelial-mesenchymal transition. EMBO J. 30:783–795. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen X and Zhao H, Chen C, Li J, He J, Fu
X and Zhao H: The HPA/SDC1 axis promotes invasion and metastasis of
pancreatic cancer cells by activating EMT via FGF2 upregulation.
Oncol Lett. 19:211–220. 2020.PubMed/NCBI
|
|
39
|
Wang N, Zheng J, Chen Z, Liu Y, Dura B,
Kwak M, Xavier-Ferrucio J, Lu YC, Zhang M, Roden C, et al:
Single-cell microRNA-mRNA co-sequencing reveals non-genetic
heterogeneity and mechanisms of microRNA regulation. Nat Commun.
10:952019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tan S, Xia L, Yi P, Han Y, Tang L, Pan Q,
Tian Y, Rao S, Oyang L, Liang J, et al: Exosomal miRNAs in tumor
microenvironment. J Exp Clin Cancer Res. 39:672020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Parente P, Parcesepe P, Covelli C,
Olivieri N, Remo A, Pancione M, Latiano TP, Graziano P, Maiello E
and Giordano G: Crosstalk between the tumor microenvironment and
immune system in pancreatic ductal adenocarcinoma: Potential
targets for new therapeutic approaches. Gastroenterol Res Pract.
2018:75306192018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee M and Rhee I: Cytokine signaling in
tumor progression. Immune Netw. 17:214–227. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Akl MR, Nagpal P, Ayoub NM, Tai B, Prabhu
SA, Capac CM, Gliksman M, Goy A and Suh KS: Molecular and clinical
significance of fibroblast growth factor 2 (FGF2 /bFGF) in
malignancies of solid and hematological cancers for personalized
therapies. Oncotarget. 7:44735–44762. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shi K, Qiu X, Zheng W, Yan D and Peng W:
miR-203 regulates keloid fibroblast proliferation, invasion, and
extracellular matrix expression by targeting EGR1 and FGF2. Biomed
Pharmacother. 108:1282–1288. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li B, Pi Z, Liu L, Zhang B, Huang X, Hu P,
Chevet E, Yi P and Liu J: FGF-2 prevents cancer cells from ER
stress-mediated apoptosis via enhancing proteasome-mediated Nck
degradation. Biochem J. 452:139–145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Turner N and Grose R: Fibroblast growth
factor signalling: From development to cancer. Nat Rev Cancer.
10:116–129. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mohammadi M, Olsen SK and Ibrahimi OA:
Structural basis for fibroblast growth factor receptor activation.
Cytokine Growth Factor Rev. 16:107–137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li J, Shan F, Xiong G, Chen X, Guan X,
Wang JM, Wang WL, Xu X and Bai Y: EGF-induced C/EBPβ participates
in EMT by decreasing the expression of miR-203 in esophageal
squamous cell carcinoma cells. J Cell Sci. 127:3735–3744.
2014.PubMed/NCBI
|
|
49
|
Zhang K, Dai L, Zhang B, Xu X, Shi J, Fu
L, Chen X, Li J and Bai Y: miR-203 is a direct transcriptional
target of E2F1 and causes G1 arrest in esophageal cancer cells. J
Cell Physiol. 230:903–910. 2015. View Article : Google Scholar : PubMed/NCBI
|

















