Introduction
Proto-oncogenic RAS mutations are found in the most
lethal cancers, as their gain-of-function phenotype underlies the
pathogenesis of up to 30% of all human cancers, and these tumors
are associated with the worst prognoses (1,2).
Such mutations have been implicated in the pathogenesis of numerous
cancers, including approximately 97% of pancreatic ductal
adenocarcinoma, 52% of colorectal adenocarcinomas, 32% of lung
adenocarcinomas (1,3,4), and
lower percentages of other cancers (5,6). As
an important oncogenic driver in cancer malignancy, KRAS is the
most frequently mutated isoform among the three human RAS genes
that encode highly homologous RAS proteins-namely, KRAS, NRAS, and
HRAS (7). In pancreatic cancer,
oncogenic mutations of the KRAS gene are major events that produce
a permanent, active KRAS protein which triggers various
intracellular pathways involved in malignancy (8). Under normal physiological conditions,
the KRAS gene encodes the GTPase transductor protein, which plays a
key role in the signal transduction cascades that regulate cell
growth, proliferation, migration, differentiation, survival, and
apoptosis (7–9). Throughout the regular KRAS GTPase
cycle, the KRAS protein switches between its inactive and active
forms by binding to guanosine diphosphate (GDP) and guanosine
triphosphate (GTP), respectively (10,11).
The KRAS protein predominantly exists in a GDP-bound inactive form,
but upon stimulation by growth factors it undergoes conformational
changes, binds to GTP, and becomes active. Active KRAS then
activates a range of molecules that mediate the transmission of
signals from the cell surface to the nucleus, leading eventually to
cellular processes essential for survival and proliferation
(7). Mutations in the KRAS
oncogene, which play a pivotal role in driving the progression of
pancreatic cancer, occur most frequently at codons 12, 13, and 61.
The most common mutation occurs at codon 12 (G12D); it results in
the amino acid substitution of aspartate for glycine, interfering
with GTP hydrolysis and thereby increasing the proportion of
active, GTP-bound KRAS, rendering it constitutively active
(8). Despite tremendous effort and
decades of intensive studies of KRAS activation, the KRAS mutant
has remained difficult to treat with drugs; as a result, a targeted
therapy exists currently for only one KRAS mutation (G12C), which
is found primarily in non-small-cell lung cancer (12). Much attention has been focused on
targeting signaling cascades downstream from KRAS, in particular
the PI3K, MAPK, and RAL-GEF pathways (7,13,14).
Direct targeting of the GTP-binding pocket of the mutant G12D KRAS
protein has proved unsuccessful (7). Moreover, there are no well-defined
druggable sites in the surface topology of this protein that are
vulnerable to high-affinity, small-sized antagonists, and thus, to
date, efforts to inhibit the functioning of mutant G12D KRAS have
not translated into clinical benefits (15). Of note, a study that used genetic
engineering techniques to shut down KRAS expression in cancer cells
showed that PI3K may compensate for KRAS loss and activate
MAP-kinase signaling (9,16). Some progress has been made with the
treatment of pancreatic cancer using small interfering RNA (siRNA),
directed against G12D-mutated KRAS; siRNA has been assessed in
vitro and in vivo and shown to induce a significant
decrease in KRAS levels, leading to an inhibition of cell
proliferation (17). In another
study, antisense specific to the KRAS gene sequence has been shown
to selectively deplete KRAS mRNA, as well as its protein product,
leading eventually to the inhibition of mutant KRAS cell
proliferation (18).
In light of the current interest in and success with
therapeutic antisense inhibitors of KRAS, we report here a
potential candidate treatment for pancreatic cancer that uses
PNA-based antisense. In general, PNA applications for developing
specific therapeutic agents that can target and inhibit the
expression of certain genes (19,20)
may prove to be useful. PNAs are synthetic, single-stranded
oligonucleotide analogues containing normal nucleobases, covalently
attached to backbone of a polyamide structure that consists of
repeating N-(2-aminoethyl) glycine units (21–23).
They are easily and quickly synthesized and have a simple chemical
structure that is resistant to degradation by nucleases and
proteases. In addition, they exhibit improved hybridization
characteristics, with high specificity and an affinity to
complementary sequences of RNA and DNA (24), and they can even differentiate
between similar sequences at the level of single-base mismatches
(25,26). Moreover, PNA binding to
single-stranded RNA or DNA is much stronger than that of
complementary DNA sequences (27,28).
Due to these advantageous characteristics, for this study, various
PNA molecules were hypothesized to be promising therapeutic or
diagnostic antisense or antigene molecules and were utilized to
regulate gene expression (29–34).
When applied as antisense agents, PNAs have been shown to bind to
the messenger RNA (mRNA) of the targeted gene and inhibit
translation activity (35).
PNA-based antisense sequences used as antibacterial agents against
key genes (i.e., encoding enzymes involved in the synthesis of DNA,
RNA, the cell envelope, fatty acids, and proteins) have also
induced inhibition of bacterial growth (22). Other PNA-based antisense sequences
have been shown to inhibit the expression of specific genes in
primary cells and in the cell lines of mice and humans (36). In contrast to the small interfering
RNAs (siRNAs) involved in the endogenous mechanism that regulates
gene expression by triggering the degradation of complementary mRNA
molecules (37–39), PNA-based antisense sequences can
inhibit specific mRNA molecules by means of splicing modulation,
blocking, and transcription arrest (36).
Although they may have many advantages in terms of
possible applications, the therapeutic use of PNAs remains limited,
due to their aggregation, low water solubility, and weak
intracellular penetration. To overcome these problems, PNAs can be
chemically modified to produce a charged form, or they can be
linked to a positively charged peptide such poly-L-lysine. If they
are positively charged, however, they are attracted to the
negatively charged cell membrane, whereas uncharged PNA molecules
might be able to penetrate cells through endocytosis (28,29).
For the current study, we designed and synthetized
PNA-based antisense molecules and investigated their ability to
pass through the cellular membrane of AsPC-1 cells and their
effects on cell growth, proliferation, and cell-cycle checkpoints.
The results strongly indicate that the penetration of PNA-based
antisense into cells caused apoptosis and arrested the cell cycle,
and thus it should be considered for further development as a
specific drug candidate for pancreatic cancer treatment.
Materials and methods
RNA preparation, RT-PCR, and the
sequencing of the mutated KRAS gene from the AsPC-1 cell line
To ascertain that the G12D-mutated KRAS gene
sequence within the AsPC-1 cell line was identical to the known
sequence in the GenBank (Accession No. AF493917), we amplified and
sequenced a fragment that harbored the G12D mutation. First,
forward (KRAS2-F: 5′-TGACTGAATATAAACTTGTGGT-3′) and reverse
(KRAS2-R: 5′-CTCATTGCACTGTACTCCTCTTG-3′) primers were designed and
utilized to amplify the size of the amplicon (202 bp) that spans
the KRAS G12D mutation, using the RT-PCR technique. To this
end, total RNA was prepared from 80–90% confluent-trypsinized
AsPC-1 cell culture, using GENZOL Tri RNA Pure Kit (Geneaid Biotech
Ltd., Taiwan) in keeping with the manufacturer's instructions. The
quantity and purity of the total RNA samples were assessed by
ultraviolet spectroscopy with a DS-11 spectrophotometer (DeNovix,
Inc.). One microgram of total RNA was subjected to a one-step
RT-PCR using a Maxime RT-PCR premix kit (INtRON Biotechnology)
containing a pellet of 10× RT-PCR buffer, dNTPs, an OptiScript
RT-system, hot start i-StarTaq DNA Polymerase, and 0.5 nM KRAS-2
forward and reverse primers. The RT-PCR cycling parameters were as
follows: reverse transcription at 45°C for 30 min, denaturation at
95°C for 5 min, 40 cycles of denaturation at 95°C for 30 sec,
annealing at 55°C for 1 min, elongation at 72°C for 1 min, and
terminal elongation at 72°C for 5 min. The PCR products were
electrophoresed in 1% agarose gel, and a DNA band of approximately
200 bp was excised from the gel and sent for sequencing to Hy-Labs
Laboratories Ltd. The DNA was sequenced in two orientations. A
202-bp sequence was BLASTed against the NCBI source and found to be
identical to the KRAS2 mRNA sequence AF493917.
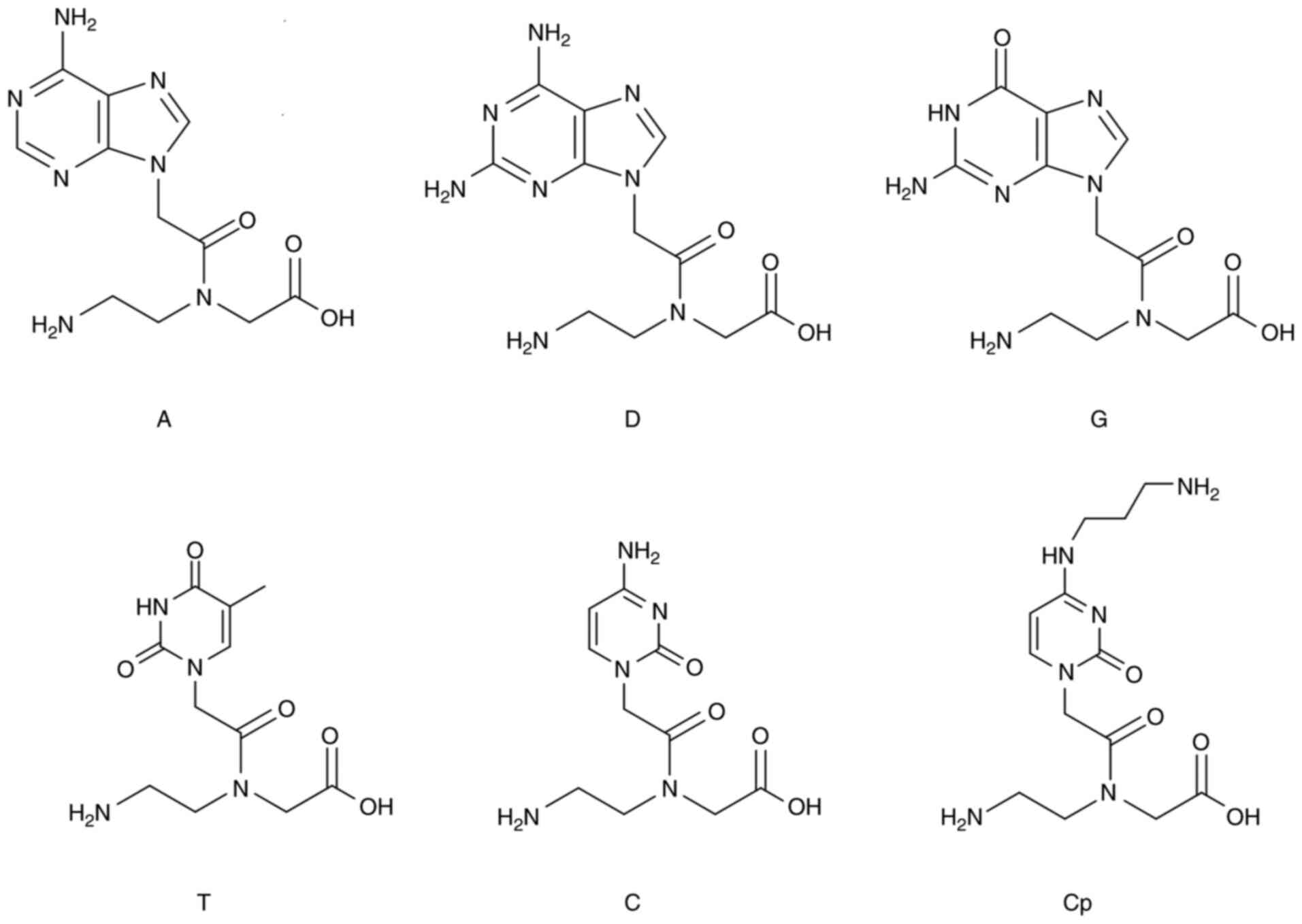
PNA design, synthesis, and
characterization
The three PNA-based molecules were designed to
target the mutated gene fragment 5′-TGGAGCTGATGGCGTAG-3′. They were
synthesized using solid-phase technology, as has been previously
described (40–42). The MALDI-TOF MS technique was used
to confirm that the synthesis was successful. Reverse-phase HPLC
was used for purification and analysis, the wavelength of the UV
detector was set to 260 nm, and a linear gradient of 10–25%
ACN/water was applied. The chemical structures of the
monomers/residues are shown in Fig.
1. The sequences of the three PNA-based molecules are as
follows:
PNA1: Composed of PNA-based monomers (A, C, T, G).
Three lysine residues are attached at the N-terminal edge. The
sequence is CTACGCCATCAGCTCCA-KKK.
PNA3: Monomer D replaces monomer A in targeting the
U nucleotide. Three lysine residues are attached at the N-terminal
edge. The sequence is CTDCGCCDTCDGCTCCD-KKK.
PNA14: Monomer Cp, a positively charged monomer,
replaces monomer C. No lysine residues are linked to this PNA-based
molecule. The sequence is CTDCpGCCDTCpDGCTCCpD.
Cell culture and viability tests
AsPC-1 cells were seeded in non-coated tissue
culture flasks or well-plates in RPMI1640 medium, supplemented with
10% fetal calf serum and 1% penicillin-streptomycin (Biological
Industries), at 37°C in a humidified atmosphere of 5%
CO2 in air. The medium was changed three times a week,
and the cells were split when they reached 80–90% confluency, using
Trypsin EDTA according to the manufacturer's instructions
(Biological Industries).
AsPC-1 cell permeability to PNA
molecules
To evaluate the permeability of cells to the PNAs,
we adapted the protocol recommended by the manufacturer with slight
modifications, which are outlined below. The transfection of the
PNAs was conducted using Lipofectamine 2000 (Thermo Fisher
Scientific, Inc.). Initially, AsPC-1 cells were seeded overnight in
a 24-well plate at a density of 5×105 per well, until
they reached confluency between 90 and 95%. The cells were then
treated with 0.5 ml of medium containing 0.5 or 2 µM of
fluorescently labeled PNA-1 that was first prepared in 100 µl of
Opti-MEM I medium. According to the manufacturer's instructions,
each 1 µM of PNA was mixed with 2.5 µl of lipofectamine 2000
(Thermo Fisher Scientific, Inc.) and incubated for 20 min at room
temperature before being dripped onto the cells. For the
permeability tests, 100 µl of PNA1/lipofectamine mixture were mixed
with 400 µl of RPMI1640 medium (without FCS), and the treated cells
were incubated for 24 h at 37°C. The cells were washed three times
in PBS buffer and fixed with 4% paraformaldehyde solution. They
were then stained with DAPI stain for 5 min at 37°C, washed with
PBS, and viewed through a Nikon Eclipse Ti2 fluorescent microscope
(Nikon Corporation, Tokyo, Japan). The intracellular fluorescence
was also measured using a plate reader with an excitation maximum
of 493 nm and an emission maximum of 528 nm.
Dissociation curves and melting
temperature (Tm) tests
Hybridization reactions were monitored using an
Exicycler™ 96 PCR system designed for real-time qPCR
(Bioneer, Korea), with qPCRBIO SyGreen Mix (PCR Biosystems Ltd.)
and the following hybridization-dissociation protocol: 95°C for 30
sec, cooling to 20°C, incubation at 20°C for 5 min, and gradual
reheating to 94°C, then a 1.0°C increase for 30 sec, and finally,
incubation for 1 min at 25°C. The SyGreen signal was recorded
incrementally (approximately every 20 sec) during the reheating
phase, and the average fluorescence measurement during this time
was reported. All of the hybridization reactions involved
combinations of equal molars of (0.5 µM) PNA, complementary target
oligonucleotides, and free PNAs (1.0 µM) in the presence of SyGreen
Mix. Dissociation curves and Tm evaluations were obtained using
Exicycler™ 96 PCR system analysis software. For
controls, we used oligonucleotides without mutations. Three
replications of each combination were run in two separate
experiments.
Cytotoxicity of the PNA molecules
To evaluate PNA cytotoxicity, AsPC-1 cells were
seeded overnight in a 24-well plate at a density of
5×105 per well and treated with 1 to 4 µM of PNAs or
10–20 µM of gemcitabine. The 100 µl PNA/lipofectamine mixture was
mixed with 400 µl of RPMI1640 medium (without FCS), and the treated
cells were incubated for 48 h at 37°C. The cells were then washed
three times in PBS buffer, and viability was determined with a XTT
Cell Proliferation Kit (Biological Industries) according to the
manufacturer's instructions, using a Thermo Scientific Varioskan
LUX Multimode Microplate Reader set at 450 nm and subtracted from
the reference absorbance at 620 nm. The experiments were repeated
independently three times.
Cell-cycle analysis
AsPC-1 cells were seeded in a 6-well plate at a
density of 5×105 cells per well and incubated overnight
at 37°C. The medium was changed and the cells were transfected with
2-µM PNAs. At 24 and 48 h after transfection, the cells were
trypsinized and collected with the growth media, centrifuged,
washed with PBS, and fixed with 70% ethanol for 1 h. Gemcitabine
(20 µM) and siRNA (20 µM) served as positive controls; the siRNA
was specific to the mRNA sequence of the mutant KRAS gene
(5′-GUUGGAGCUGAUGGCGUAGdTdT and 5′-CUACGCCAUCAGCUCCAACdTdT). This
was followed by incubation with 0.1% NP-40 for 5 min at 4°C and
then with ice and 100 µg/ml of RNase for 30 min. Finally, a 50
µg/ml solution of propidium iodide (PI) was added for 20 min.
Cell-cycle phase distributions were determined by flow cytometry,
using a Navios flow cytometer (Beckman Coulter, Miami, FL).
Apoptosis assay using Annexin V/PI
double staining
Apoptotic cell death was evaluated and quantified by
flow cytometry with an Annexin V FITC Detection kit
(Mebcyto® Apoptosis kit, MBL) (used according to the
manufacturer's recommended procedure) and a PI Double Staining kit.
To differentiate between apoptosis and necrosis, the cells were
stained with FITC-labeled Annexin V and PI.
The AsPC-1 cells (5×105) were seeded in a
six-well plate. The next day, the cells were transfected with PNAs
and incubated for 48 h. Adherent and floating cells were both
collected in order to detect early and late apoptosis events.
Treated and untreated cells were harvested by trypsinization,
washed, and suspended in ice-cold PBS. The washed cell pellet was
resuspended in ice-cold binding buffer containing FITC-conjugated
Annexin V and PI. The samples were then incubated in the dark at
room temperature for 15 min before being analyzed with a flow
cytometer [Navios flow cytometer (Beckman Coulter)] and Kaluza
software.
KRAS expression profile by
quantitative real-time PCR
RNA was extracted from the PNA-treated AsPC-1 cells,
as well as from non-treated cultured cells, using a GeneJET RNA
Purification kit (Thermo Fisher Scientific, Inc.). The RNA was
quantified with Nanodrop OneC (Thermo Fisher Scientific, Inc.).
First-strand cDNA was synthesized with a qScript cDNA Synthesis kit
(Quanta Bio.). The expression level of the mature KRAS was
quantified separately, using PerfeCTa SYBR Green FastMix (Quanta
Bio.) in accordance with the manufacturer's instructions, and
normalized with the use of GAPDH for internal expression control. A
real-time PCR was carried out with StepONePlus, and the results
were analysed with the comparative delta Ct method, using the
StepONePlus analysis software.
Western blot analysis
Following the treatment of the AsPC-1 cells for 48
h, the control and treated cells were collected and lysed with RIPA
buffer (Merck) and Complete Protease Inhibitor (Sigma) on ice for
20 min. Protein fractions were separated by centrifugation for 10
min at 13,000 rpm at 4°C. The proteins, in 20-µg aliquots, were
separated by SDS-PAGE and then transferred by semi-dry transfer to
0.45-micrometer-pore-size nitrocellulose membranes. The membranes
were blocked with 5% milk TBST solution for 1 h at room
temperature, followed by overnight incubation with rabbit
monoclonal Anti-Ras Antibody (Abcam). They were then washed three
times with TBST, each for 10 min, incubated with secondary
antibody, peroxidase-conjugated, AffiniPure Goat Anti-Rabbit IgG
(Jackson Immune Research Laboratories and Dako) for 1 h at room
temperature, and washed three more times. An antigen/antibody
complex was detected with an ECL kit Western Bright™ ECL
detection reagent (Advansta). The results were analyzed with an
Amersham Imager 600 (GE Healthcare).
For reference, and after detection with the specific
antibodies, all of the membranes were exposed to mouse
Anti-β-actin monoclonal antibody (MP Biomedicals). Donkey
polyclonal anti-mouse HRP secondary antibody and an ECL kit were
used to detect actin.
Statistics
All analyses were performed using SPSS 19.0 software
(IBM, Corp.). All data were expressed as mean value ± Standard
Error (SE). Statistical analyses were performed, using one-way
analysis of variance (ANOVA) test for comparison among multiple
groups followed by Bonferroni test for significancy. The SPSS
software served for calculation of differences. P<0.05 was
considered to indicate a statistically significant difference.
Results
PNA permeability
Cell membrane and nuclear envelope permeability to
PNA were determined by monitoring fluorescently labeled PNA1
(fluorescein isothiocyanate (FITC)-conjugated PNA1) transfected
into AsPC-1 cells. The fluorescence intensity was determined using
an fmax fluorescence microplate reader (see Fig. 2A). The cells were then stained with
DAPI and examined with a fluorescence microscope (Fig. 2B and C).
The results above demonstrate that the fluorescence
signal was positively correlated with the concentration of
FITC-PNA1. The fluorescence was observed in the cell cytoplasm.
Effect of PNAs on cell viability
Because the mutated KRAS gene plays a key role in
mediating the growth and proliferation of pancreatic cancer cells,
the PNAs specific to this gene were expected to affect cell
viability and growth. To assess this projected effect, AsPC-1 cells
were transfected with different concentrations of PNAs for 48 h,
after which the number of viable cells was quantitated using the
XTT assay (Fig. 3). Gemcitabine
was used as a positive control because it is one of the main
chemotherapy drugs used to treat pancreatic cancer. It is not
specific to the mutated KRAS gene but acts as a competitive
substrate of deoxycytidine triphosphate (dCTP) and is incorporated
into DNA during replication, thereby inhibiting DNA chain
elongation and cell death by apoptosis.
As shown in Fig. 3,
exposure of the cells to different PNAs significantly affected cell
viability. Increasing PNA concentrations resulted in greater
inhibition of cell viability.
Effects of PNAs on the cell cycle of
AsPC-1 cells
PNA binding to the mutated KRAS gene may cause
cell-cycle arrest and cell apoptosis (an increase in the number of
cells in the sub-G1 phase). To assess this possibility, AsPC-1
cells were treated with 2 µM of PNAs for 24 and 48 h and incubated
with propidium iodide (PI) to determine the DNA content, which
reflects the cell number. PI signals were then read using a flow
cytometer, and the data were analyzed using Kaluza Software.
Gemcitabine (20 µM) and siRNA specific to the mRNA sequence of the
mutant KRAS gene (20 µM) served as positive controls. The results,
which are presented in Fig. 4,
show that, compared to the two positive controls, the PNAs induced
apoptosis (an increase in the number of cells in the sub-G1 phase),
and the number of cells in the G1 phase decreased.
The percentage of PNA-treated cells (after 48 h
treatment) in the G1 phase decreased by approximately 15% relative
to the control, and this was mirrored by an approximately 15%
increase in the percentage of cells in the sub-G1 phase (see
Fig. 4). To verify whether this
cell-cycle arrest was due to the induction of apoptosis, the
percentage of cells in the sub-G1 phase was quantified, and an
apoptosis test was performed (Fig.
5), as discussed below. We also looked at the effect of the
PNAs on the percentage of cells in the G2/M phase. At 24 h, the
percentage of PNA-treated cells in the G2/M phase decreased by
2–10% relative to the control, and this was mirrored by a 1–10%
increase in the percentage of cells in the sub-G1 phase. After the
same treatment for 48 h, there was almost a cessation in the G2/M
stage of the cell cycle, while the percentage of cells in the
sub-G1 phase increased by 9–16% (Figs.
4 and 5). These results
strongly indicate that the PNAs induced a halt in cell division and
an increase in sub-G1, which triggers cell apoptosis.
Effect of PNAs on apoptosis in AsPC-1
cells
As most cancer cells can escape apoptosis, we set
out to determine whether PNAs triggered AsPC-1 apoptosis in
vitro. The exposure of cells to 2 µM of PNA1, PNA3, or PNA14
for 48 h resulted in 18, 20, and 20% apoptosis, respectively,
whereas in the positive controls treated with 20 µM siRNA or 20 µM
gemcitabine, approximately 18 and 22% of the cells were found in
the apoptotic state, respectively (Fig. 5). In the untreated control samples,
7% of the cells were apoptotic.
Effect of PNAs on the expression of
the KRAS mutant gene in AsPC-1 cells
The PNAs were designed to specifically target the
mRNA sequence of the KRAS mutant gene and thus were expected to
affect its transcription and translation. To confirm this effect,
AsPC-1 cells were transfected with PNAs for 48 h, and the
expression of KRAS mRNA and its protein were analyzed. The results
are presented in the next two sections.
1. Effect of PNAs on KRAS mutant gene
transcriptional levels
As shown in Fig. 6,
PNA3 and PNA14 are dose-dependent and have a significant effect on
the translation of the mutant KRAS gene. The weaker effect of PNA1
at 4 µm compared to PNA1 at 1 µM can be explained by steric
hindrance. Collectively, the results clearly show that the PNAs
significantly affected the expression of the mutant KRAS gene.
2. Effect of PNAs on KRAS mutant gene
translational levels
Total protein lysates were extracted from the
PNA-treated cells, resolved with SDS/PAGE, and then transferred to
PVDF membranes that had been incubated with mouse anti-human RAS
antibodies and mouse anti-human GAPDH antibodies. Treatment with
PNA1 resulted in a decrease in total RAS protein levels (Fig. 7).
The results of the western blot analysis show an
evident effect of PNAs on KRAS protein levels. Overall, it is clear
that the three PNAs inhibit gene expression at the level of
translation in a dose-dependent manner, as shown in Fig. 7B.
Discussion
PNAs have been proposed as a potential anti-cancer
therapy (36). Mutations in the
genes of RAS proteins are very common in malignant tumors,
especially in gastrointestinal, colorectal, biliary tract, and
pancreatic tumors (43). The life
expectancy of patients suffering from these tumors is relatively
short, as these tumors are generally resistant to current chemical
and biological treatments. Even though KRAS is among the most
common oncogenes in human cancer, it is a difficult oncogene to
target (44,45). Antisense oligonucleotides for the
KRAS gene (18), and siRNA
products (17) may become
therapeutic options for RAS-mutated cancers; however, siRNA
technology is restricted by the need for systemic delivery
(46).
In this study, the PNAs in question were chemically
designed with aqueous solubility and an enhanced ability for cell
penetration in mind, owing to the unique distribution of charges
over the backbone and the nucleotide-like monomers (rings), and
they were attached to a positively charged peptide consisting of
three amino acids of lysine (KKK). These structural characteristics
not only enhance cell penetration, but also help PNAs to
selectively and specifically bind to the KRAS mRNA sequence. Each
of the three synthesized PNA has its own chemical unique
characteristics, but all three carry a net positive charge that
attracts them to the negatively charged cell membrane,
hypothetically enabling them to easily penetrate the membrane into
the cytoplasm. However, despite these characteristics, repeated
experiments have shown that the three PNAs are unable to penetrate
cells and thus cannot affect cell functionality. Nevertheless, we
were able to introduce them into the cells using lipofectamine, a
cationic lipid-based chemical. As demonstrated, lipofectamine
effectively and successfully transfected FITC-fluorescently labeled
PNA into the cytoplasm of cells. Lipofectamine is a cationic,
lipid-based chemical transfectant which forms liposomes that
complex with negatively charged nucleic acids, and, through the
endocytosis pathway, deliver DNA or RNA into the cell cytosol
(47). Despite their positive
charge, the PNAs were efficiently delivered into cells with the use
of cationic lipofectamine, as demonstrated by florescence
measurements and microscopy. It is plausible that lipid subunits of
the lipofectamine formed liposomes in the aqueous environment of
the growth medium, which transfected the PNAs into the cells. Since
lipofectamine cannot be used clinically, in future studies, we
ought to investigate the systemic delivery of PNA treatments by
other means, possibly using lipid or nanoparticle technology.
Not only did the PNAs examined penetrate cell
membranes, they also reached targets and affected important
parameters linked to cell viability. Among the measurable
parameters were cell membrane integrity and permeability, and the
activity of cellular enzymes. The effects of PNAs on cell viability
were evaluated using the XTT assay, and a significant,
concentration-dependent reduction in cell viability was found. This
can be attributed mainly to reduced activity of the mitochondrial
dehydrogenase enzyme, and it points to the cytotoxicity of PNAs and
their effects on cell membrane leakage. The effects of the three
PNAs on cell viability and their maximum inhibition was clearly
apparent after two days. Surprisingly, when the cells were exposed
to gemcitabine, there was total death with no dose response.
Gemcitabine, like other chemotherapies, has a narrow therapeutic
window, which is reflected in the results of this experiment.
The affinity of the antisense oligonucleotides for
the G12D-mutated KRAS mRNA, and their specificity, were evaluated
using melting-temperature (Tm) shift assays; the Tm of the gene
fragment increased by 4°C in the presence of PNA (data not shown).
In addition, antisense oligonucleotides specific to gene fragments
of KRAS inhibited their amplification by a PCR, as could be seen in
the agarose gel electrophoresis of the KRAS PCR products (data not
shown). To validate their specificity at the cellular level, it is
advisable to test the efficacy of PNA-based antisense molecules on
cells from a different pancreatic cancer cell line that have a
mutation other than the KRAS G12D mutation, such as on HPAF-II
cells or on KRAS wild-type pancreatic cancer cells.
KRAS is the predominant mutated RAS gene in cancers
and is involved in 84% of all RAS missense mutations; in
particular, KRAS mutation is the initiating genetic event for
pancreatic ductal adenocarcinoma (PDAC) (43). In general, the proliferation of
cells decreases under stressful or damaging conditions, and
cell-cycle arrest occurs through the activation of checkpoints in
an attempt to repair the damage and ensure proper cell division
(48,49). If the damage is too extensive to be
repaired, the cells undergo cell death in the form of apoptosis
(50–52). Mutated KRAS is persistently
GTP-bound and thus remains constitutively active, resulting in an
overstimulation of effectors, which causes cells to evade apoptotic
signals and to continue proliferating, regardless of extracellular
stimuli (48). In this experiment,
PNA-based antisense molecules were designed to target the mutation
effect by specifically and selectively binding to the G12D region
of the KRAS gene sequence. Our results suggest that upon
penetrating the cell membrane, PNA-based antisense leads to a
significant increase in the percentage of sub-G1 phase (apoptotic)
cells in AsPC-1 cells. Targeting the G12D KRAS mutation with
PNA-based antisense may cause cells to revert to controlled cell
division and make them more responsive to cell-cycle checkpoints,
thereby halting uncontrolled division and diverting them to
apoptosis in the absence of physiological growth stimuli. As seen
in the study, PNA-based antisense strikingly induced apoptosis,
compared to the control cells. Moreover, in addition to affecting
cell viability, cell-cycle progression, and apoptosis, PNA
treatment disrupted the integrity of the typical structure of the
AsPC-1 cells, which normally exhibit an epithelial cell-like
morphology (data not shown).
Our attempt to design PNA-based antisense that
specifically inhibits the expression of the G12D KRAS mutant gene
was successful, as evidenced by the inhibition of KRAS mutant gene
translation (Fig. 6). We observed
a decrease in total RAS protein, probably reflecting the decrease
in KRAS gene translation. Since AsPC-1 cells are homozygous for the
G12D KRAS mutation (involving two mutant alleles), the PNA-based
antisense targets both gene copies; thus, inhibiting the production
of the protein product of the KRAS mutant gene would clearly
decrease its activity. Like the findings of other studies (53,54),
these findings suggest that PNA-based antisense can serve as a
powerful tool for sequence-specific inhibition of the oncogenic
mutant allele, resulting in the suppression of proliferation
pathways and a decrease in cell growth and viability, and this
points to the potential usefulness of mutant KRAS as a target for
anti-cancer therapy.
PNA-based antisense therapeutics are still limited
by lack of an effective method of systemic delivery. More work is
needed to develop means of systemic delivery, and possibly
administration in conjunction with other drugs, such as
gemcitabine. The development of new PNA formulations, for example,
utilizing nanoparticle-based delivery methods (55), may offer ways to sustain the
delivery of PNA-based therapeutics. Furthermore, in vivo
studies will increase our understanding of the delivery and
activity of PNAs, as well as of the biological mechanisms involved
in the tissue distribution, cellular uptake, and intracellular
trafficking of the molecules. Future strategies that refine this
approach will be important to maximizing the potential of PNA-based
antisense therapeutics to treat a broad spectrum of human
diseases.
Furthermore, in order to establish the specificity
of PNAs for the G12D KRAS mutated gene sequence in cell-line, other
cancer cell lines that do not carry the G12D KRAS mutation must be
investigated with PNA-based antisense, since PNAs designed to
affect the mutated KRAS gene, which have only a single-point
mutation (GGT >GAT, a glycine-to-aspartate substitution in codon
12, G12D), might also affect normal variants of the gene in other
cells or other genes that have similar sequences. As well, it is
advisable to assess the toxicity of the PNAs by testing their
effects on normal pancreatic cells.
Our results suggest that antisense PNAs are capable
of inhibiting the expression and downstream function of mutant KRAS
in pancreatic cancer cells, exhibiting robust antitumor activity at
micromolar doses. PNAs are attractive candidates for the
therapeutic targeting of KRAS mutant genes. However, critical
issues, such as how to deliver PNAs to target tissues and how to
prevent their tendency to aggregate, need to be resolved before
they can be tested for pre-clinical and clinical application.
Acknowledgements
Not applicable.
Funding
This research was partially supported by the Israel Innovation
Authority (an incubator project that was granted to GeneArrest,
Ltd., between the years 2009 and 2015) and The Ministry for
Development of the Periphery, Negev and Galilee, 2016 (AS, RA and
MF).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AR, AS and MF conceived the study, raised the funds
and performed the analysis. EG, RM, HT, MS, MZ and MF carried out
the experiments. AR and MF carried out the statistical analysis of
the data, interpreted the data and wrote up the conclusions. AS,
MS, MZ, AR and MF wrote the first draft of the manuscript. AS, AR
and MF edited the manuscript. AR and MF confirm the authenticity of
all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
AR and MF are the founders of GeneArrest, Ltd., the
IP owner of the novel monomers. We declare that the funders had no
role in the study design, data collection and analysis, decision to
publish or preparation of the manuscript. The other authors declare
no conflicts of interest.
References
|
1
|
Prior IA, Lewis PD and Mattos C: A
comprehensive survey of Ras mutations in cancer. Cancer Res.
72:2457–2467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fernández-Medarde A and Santos E: Ras in
cancer and developmental diseases. Genes Cancer. 2:344–358. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cox AD, Fesik SW, Kimmelman AC, Luo J and
Der CJ: Drugging the undruggable RAS: Mission possible? Nat Rev
Drug Discov. 13:828–851. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tsiatis AC, Norris-Kirby A, Rich RG, Hafez
MJ, Gocke CD, Eshleman JR and Murphy KM: Comparison of Sanger
sequencing, pyrosequencing, and melting curve analysis for the
detection of KRAS mutations: Diagnostic and clinical implications.
J Mol Diagn. 12:425–432. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pylayeva-Gupta Y, Grabocka E and Bar-Sagi
D: RAS oncogenes: Weaving a tumorigenic web. Nat Rev Cancer.
11:761–774. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singh H, Longo DL and Chabner BA:
Improving prospects for targeting RAS. J Clin Oncol. 33:3650–3659.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu P, Wang Y and Li X: Targeting the
untargetable KRAS in cancer therapy. Acta Pharm Sin B. 9:871–879.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Buscail L, Bournet B and Cordelier P: Role
of oncogenic KRAS in the diagnosis, prognosis and treatment of
pancreatic cancer. Nat Rev Gastroenterol Hepatol. 17:153–168. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Simanshu DK, Nissley DV and McCormick F:
RAS proteins and their regulators in human disease. Cell.
170:17–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scheffzek K, Ahmadian MR, Kabsch W,
Wiesmüller L, Lautwein A, Schmitz F and Wittinghofer A: The
Ras-RasGAP complex: Structural basis for GTPase activation and its
loss in oncogenic Ras mutants. Science. 277:333–338. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bos JL, Rehmann H and Wittinghofer A: GEFs
and GAPs: Critical elements in the control of small G proteins.
Cell. 129:865–877. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hong DS, Fakih MG, Strickler JH, Desai J,
Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS,
et al: KRASG12C inhibition with sotorasib in advanced
solid tumors. N Engl J Med. 383:1207–1217. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu F, Yang X, Geng M and Huang M:
Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer
therapy. Acta Pharm Sin B. 8:552–562. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vallejo A, Perurena N, Guruceaga E, Mazur
PK, Martinez-Canarias S, Zandueta C, Valencia K, Arricibita A,
Gwinn D, Sayles LC, et al: An integrative approach unveils FOSL1 as
an oncogene vulnerability in KRAS-driven lung and pancreatic
cancer. Nat Commun. 8:142942017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ryan MB and Corcoran RB: Therapeutic
strategies to target RAS-mutant cancers. Nat Rev Clin Oncol.
15:709–720. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takashima A and Faller DV: Targeting the
RAS oncogene. Expert Opin Ther Targets. 17:507–531. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zorde Khvalevsky E, Gabai R, Rachmut IH,
Horwitz E, Brunschwig Z, Orbach A, Shemi A, Golan T, Domb AJ, Yavin
E, et al: Mutant KRAS is a druggable target for pancreatic cancer.
Proc Natl Acad Sci USA. 110:20723–20728. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ross SJ, Revenko AS, Hanson LL, Ellston R,
Staniszewska A, Whalley N, Pandey SK, Revill M, Rooney C, Buckett
LK, et al: Targeting KRAS-dependent tumors with AZD4785, a
high-affinity therapeutic antisense oligonucleotide inhibitor of
KRAS. Sci Transl Med. 9:eaal52532017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marin VL, Roy S and Armitage BA: Recent
advances in the development of peptide nucleic acid as a
gene-targeted drug. Expert Opin Biol Ther. 4:337–348. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gambari R: Peptide-nucleic acids (PNAs): A
tool for the development of gene expression modifiers. Curr Pharm
Des. 7:1839–1862. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nielsen PE, Egholm M, Berg RH and Buchardt
O: Sequence-selective recognition of DNA by strand displacement
with a thymine-substituted polyamide. Science. 254:1497–1500. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rajasekaran P, Alexander JC, Seleem MN,
Jain N, Sriranganathan N, Wattam AR, Setubal JC and Boyle SM:
Peptide nucleic acids inhibit growth of Brucella suis in pure
culture and in infected murine macrophages. Int J Antimicrob
Agents. 41:358–362. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Abdel-Aziz M, Yamasaki T and Otsuka M:
Synthesis and hybridization property of novel 2′,5′-isoDNA mimic
chiral peptide nucleic acids. Bioorg Med Chem Lett. 13:1041–1043.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koppelhus U and Nielsen PE: Cellular
delivery of peptide nucleic acid (PNA). Adv Drug Deliv Rev.
55:267–280. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dean DA: Peptide nucleic acids: Versatile
tools for gene therapy strategies. Adv Drug Deliv Rev. 44:81–95.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
De Cola C, Manicardi A, Corradini R, Izzo
I and De Riccardis F: Carboxyalkyl peptoid PNAs: Synthesis and
hybridization properties. Tetrahedron. 68:499–506. 2012. View Article : Google Scholar
|
|
27
|
Narenji H, Gholizadeh P, Aghazadeh M,
Rezaee MA, Asgharzadeh M and Kafil HS: Peptide nucleic acids
(PNAs): Currently potential bactericidal agents. Biomed
Pharmacother. 93:580–588. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang G and Xu XS: Peptide nucleic acid
(PNA) binding-mediated gene regulation. Cell Res. 14:111–116. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Quijano E, Bahal R, Ricciardi A, Saltzman
WM and Glazer PM: Therapeutic peptide nucleic acids: Principles,
limitations, and opportunities. Yale J Biol Med. 90:583–598.
2017.PubMed/NCBI
|
|
30
|
Zhao XL, Chen BC, Han JC, Wei L and Pan
XB: Delivery of cell-penetrating peptide-peptide nucleic acid
conjugates by assembly on an oligonucleotide scaffold. Sci Rep.
5:176402015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lucarelli F, Marrazza G, Turner AP and
Mascini M: Carbon and gold electrodes as electrochemical
transducers for DNA hybridisation sensors. Biosens Bioelectron.
19:515–530. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Barluenga S and Winssinger N: PNA as a
biosupramolecular tag for programmable assemblies and reactions.
Acc Chem Res. 48:1319–1331. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brandén LJ, Mohamed AJ and Smith CI: A
peptide nucleic acid-nuclear localization signal fusion that
mediates nuclear transport of DNA. Nat Biotechnol. 17:784–787.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liang KW, Hoffman EP and Huang L: Targeted
delivery of plasmid DNA to myogenic cells via
transferrin-conjugated peptide nucleic acid. Mol Ther. 1:236–243.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chiarantini L, Cerasi A, Fraternale A,
Millo E, Benatti U, Sparnacci K, Laus M, Ballestri M and Tondelli
L: Comparison of novel delivery systems for antisense peptide
nucleic acids. J Control Release. 109:24–36. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Montazersaheb S, Hejazi MS and Nozad
Charoudeh H: Potential of peptide nucleic acids in future
therapeutic applications. Adv Pharm Bull. 8:551–563. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Elbashir SM, Harborth J, Lendeckel W,
Yalcin A, Weber K and Tuschl T: Duplexes of 21-nucleotide RNAs
mediate RNA interference in cultured mammalian cells. Nature.
411:494–498. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fire A, Xu S, Montgomery MK, Kostas SA,
Driver SE and Mello CC: Potent and specific genetic interference by
double-stranded RNA in Caenorhabditis elegans. Nature. 391:806–811.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kennerdell JR and Carthew RW: Use of
dsRNA-mediated genetic interference to demonstrate that frizzled
and frizzled 2 act in the wingless pathway. Cell. 95:1017–1026.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rayan A and Falah M: Sequence specific
double-stranded DNA binding compounds. Patent Application Number
WO2009093188. Date Filed. January 22–2009.
|
|
41
|
Rayan A and Falah M: Sequence specific
double-stranded DNA/RNA binding compounds and uses thereof. Patent
Application Number WO2012011114. Date Filed. July 21–2011.
|
|
42
|
Komiyama M, Aiba Y, Ishizuka T and Sumaoka
J: Solid-phase synthesis of pseudo-complementary peptide nucleic
acids. Nat Protoc. 3:646–654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Waters AM and Der CJ: KRAS: The critical
driver and therapeutic target for pancreatic cancer. Cold Spring
Harb Perspect Med. 8:a0314352018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ostrem JM, Peters U, Sos ML, Wells JA and
Shokat KM: K-Ras(G12C) inhibitors allosterically control GTP
affinity and effector interactions. Nature. 503:548–551. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lim SM, Westover KD, Ficarro SB, Harrison
RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, et al:
Therapeutic targeting of oncogenic K-Ras by a covalent catalytic
site inhibitor. Angew Chem Int Ed Engl. 53:199–204. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xu CF and Wang J: Delivery systems for
siRNA drug development in cancer therapy. Asian J Pharm Sci.
10:1–12. 2015. View Article : Google Scholar
|
|
47
|
Llovera L, Berthold P, Nielsen PE and
Shiraishi T: Cell number and transfection volume dependent peptide
nucleic acid antisense activity by cationic delivery methods. Artif
DNA PNA XNA. 3:22–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hartwell LH and Weinert TA: Checkpoints:
Controls that ensure the order of cell cycle events. Science.
246:629–634. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chinnasamy S, Zameer F and Muthuchelian K:
Molecular and biological mechanisms of apoptosis and its detection
techniques. J Oncol Sci. 6:49–64. 2020. View Article : Google Scholar
|
|
51
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wong RS: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Rachagani S, Senapati S, Chakraborty S,
Ponnusamy MP, Kumar S, Smith LM, Jain M and Batra SK: Activated
KrasG¹2D is associated with invasion and metastasis of
pancreatic cancer cells through inhibition of E-cadherin. Br J
Cancer. 104:1038–1048. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Smakman N, Veenendaal LM, van Diest P, Bos
R, Offringa R, Borel Rinkes IH and Kranenburg O: Dual effect of
Kras(D12) knockdown on tumorigenesis: Increased immune-mediated
tumor clearance and abrogation of tumor malignancy. Oncogene.
24:8338–8342. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gupta A, Bahal R, Gupta M, Glazer PM and
Saltzman WM: Nanotechnology for delivery of peptide nucleic acids
(PNAs). J Control Release. 240:302–311. 2016. View Article : Google Scholar : PubMed/NCBI
|