Introduction
Thyroid carcinoma (TC) is the most common endocrine
malignancy (1), with increasing
incidence worldwide (2–4). Epidemiological data from China show
that the age standardized incidence rate of thyroid cancer
increased from 3.21/105 in 2005 to 9.61/105
in 2015, with an annual increasing incidence rate of 12.4%
(5). There are four primary types
of TC according to the cellular derivation and state of
differentiation (6): Papillary
thyroid cancer (PTC), follicular thyroid cancer (FTC) and
anaplastic thyroid cancer (ATC) arise from follicular cells of the
thyroid, while medullary thyroid cancer (MTC) arises from
parafollicular C cells of the thyroid. Although PTC and FTC, the
most common forms of TC, usually have good prognoses (7,8),
~10% of patients suffer disease recurrence and metastasis following
treatment (9). Furthermore, MTC
and ATC have worse prognoses due to their aggressive behavior
(10), with a 1-year survival rate
of 5–30% for ATC (11). A major
challenge in treatment of TC is failure to respond to radiotherapy
and chemotherapy (8,12). Therefore, it is urgent to identify
new biomarkers and therapeutic targets.
Cadherins (CDHs) are members of a family of
homozygous and Ca2+-dependent cell adhesion
glycoproteins, which perform essential roles in embryonic
development, normal cell function and tissue integrity preservation
by mediating cell-cell adhesion (13). Abnormal expression of CDHs has been
reported in carcinogenesis and is associated with tumor initiation
and progression (14). Notably,
CDH16 has been implicated in differentiation of the kidney
(15,16) and thyroid (17–19).
Furthermore, decreased CDH16 has been observed in renal cell
carcinoma (16) and studies
suggest that CDH16 may be downregulated in TC (20,21).
In one study (21), downregulation
of CDH16 was shown to be correlated with unfavorable
clinicopathological features of PTC and was inversely associated
with expression of cancer-associated genes; however, the analysis
did not distinguish between TC subtypes and did not consider the
potential contribution of gene copy number variation (CNV).
Furthermore, bioinformatic analysis was limited to a single cohort
in The Cancer Genome Atlas (TCGA) database and direct expression
analysis was limited to a single 16 patient cohort. Though the
results of the previous studies (20,21)
are consistent with a tumor suppressor role of CDH16, the
biological activity of CDH16 in TC has not been directly
demonstrated and its role in TC remains hypothetical.
In the present study, the expression and CNV of
CDH16 in TC was investigated. Functional assays were also performed
to probe its role and molecular mechanism in TC. These studies
provide a molecular basis for the role of CDH16 in TC, as well as
confirming the potential of CDH16 as a target for TC prognosis and
treatment.
Materials and methods
Database analysis
mRNA expression and DNA CN of CDH16 in TC were
investigated using the Oncomine 4.5 database (oncomine.org), an
integrated data mining platform for collecting, analyzing and
delivering cancer transcriptome data (22). The present study analysis focused
on four TC studies, including He (accession no. GSE3467), Vasko
(accession no. GSE6004), Giordano (accession no. GSE27155) and TCGA
thyroid (23–25). The CDH16 mRNA expression and DNA CN
were assessed between TC and normal thyroid tissue. P<0.05 was
considered to indicate a statistically significant difference.
LinkedOmics database (linkedomics.org/login.php), a
publicly available portal analyzing multi-omics data from 32 types
of cancer in TCGA (26), was used
to visualize genes that were co-expressed with CDH16 and perform
KEGG pathway analysis of these genes by Gene Set Enrichment
Analysis (GSEA).
Tissue specimens and cell culture
A total of 35 paired PTC surgical and corresponding
adjacent normal thyroid specimens were obtained from Tangshan
Gongren Hospital and Tangshan Renmin Hospital between January 2017
and November 2021. The inclusion criteria were: i) histologically
confirmed PTC; ii) voluntary participation in the research; iii) no
preoperative radiotherapy, chemotherapy or other treatment for TC.
The exclusion criteria were: i) presence of tumor other than TC;
ii) heart, lung, liver, kidney or hematopoietic system disease;
iii) pregnant or lactating. Patient characteristics are presented
in Table I. The samples were
stored in liquid nitrogen for reverse transcription-quantitative
(RT-q)PCR or fixed in 4% paraformaldehyde solution for 48 h at room
temperature and embedded in paraffin for immunohistochemistry
(IHC). The present study was approved by the Ethical Committee of
Tangshan Gongren Hospital (Hebei, China; approval no.
GRYY-LL-2020-104) and all patients provided written informed
consent.
 | Table I.Association of CDH16 protein
expression with clinicopathological features of 35 patients with
papillary thyroid cancer. |
Table I.
Association of CDH16 protein
expression with clinicopathological features of 35 patients with
papillary thyroid cancer.
|
|
| CDH16
expression |
|
|---|
|
|
|
|
|
|---|
| Characteristic | n | + | - | % | P-value |
|---|
| Sex |
|
|
|
|
|
|
Male | 10 | 4 | 6 | 60.0 | 0.471a |
|
Female | 25 | 14 | 11 | 44.0 |
|
| Age, years |
|
|
|
|
|
|
<55 | 20 | 11 | 9 | 45.0 | 0.625 |
|
≥55 | 15 | 7 | 8 | 53.3 |
|
| Tumor size, cm |
|
|
|
|
|
| ≥2 | 11 | 1 | 10 | 90.9 | 0.001 |
|
<2 | 24 | 17 | 7 | 29.2 |
|
| Lymph node
metastasis |
|
|
|
|
|
|
Present | 18 | 5 | 13 | 72.2 | 0.004 |
|
Absent | 17 | 13 | 4 | 23.5 |
|
| Disease stage |
|
|
|
|
|
| I,
II | 28 | 17 | 11 | 39.3 | 0.041a |
| III,
IV | 7 | 1 | 6 | 85.7 |
|
Human TC BCPAP and TPC1 cells were obtained from
American Type Culture Collection and the human thyroid follicular
cell line Nthy-ori3-1 was purchased from Shanghai Institute of Cell
Biology of the Chinese Academy of Sciences (Shanghai, China).
BCPAP, TPC1 and Nthy-ori3-1 cells were cultured in RPMI-1640
(Gibco; Thermo Fisher Scientific, Inc.) containing 10% fetal calf
serum (Gibco; Thermo Fisher Scientific, Inc.). All cell lines were
cultured in a 5% CO2 humidified incubator at 37°C.
IHC staining
Tissue fixation, paraffin embedding, sectioning and
dewaxing were performed as previously described (27). Tissue samples were incubated with
mouse anti-CDH16 (1:100; cat. no. ab215769; Abcam) overnight at
4°C. Subsequently, sections were incubated with horseradish
peroxidase (HRP)-conjugated goat anti-mouse IgG secondary antibody
(1:2,000; cat. no. ab205719; Abcam) for 30 min at 37°C. The slides
were examined by two blinded senior pathologists from Tangshan
Gongren Hospital. Cells within five randomly selected fields of
view were counted under a light microscope (magnification, ×20) and
those with brown or yellow staining on the cell membrane were
considered CDH16-positive. The percentage of positive tumor cells
was calculated. A threshold of >25% positively stained tumor
cells was used to define CDH16 positivity.
RT-qPCR
RNA was isolated from thyroid tissue samples or
cells using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.). RNA was reverse transcribed into cDNA using cDNA
Synthesis kit (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The prepared cDNA was
subjected to RT-qPCR using a SYBR Green PCR Supermix kit
(Invitrogen; Thermo Fisher Scientific, Inc.) with the Rotor
Gene-3000 instrument (Corbett Research Ltd.). Reactions were
conducted in a 20 µl volume with 1 µl cDNA according to the
manufacturer's protocol of the SYBR Green PCR Supermix kit. Primer
sequences for CDH16 were forward, 5′-CCCTGAGTTCATCACTTCCC-3′ and
reverse, 5′-AGAGTCTGGCTCCCAATCC-3′. Primer sequences for GAPDH were
forward, 5′- GAAAGCCTGCCGGTGACTAA-3′ and reverse,
5′-AGGAAAAGCATCACCCGGAG-3′. The PCR protocol was as follows:
Initial denaturation at 95°C for 2 min, followed by 45 cycles of
95°C for 15 sec and 60°C for 30 sec. Relative expression was
calculated using the 2−ΔΔCq method (28) with GAPDH as a reference gene for
normalization.
Western blot analysis
Protein was isolated from cells using RIPA buffer
(Beyotime Institute of Biotechnology) with protease inhibitor
phenylmethylsulfonyl fluoride (1:200; Beyotime Institute of
Biotechnology). Total protein concentration was determined using a
BCA kit (Beyotime Institute of Biotechnology), according to the
manufacturer's protocol. Aliquots of 25 µg protein were
electrophoresed on 10% polyacrylamide gels and transferred onto
polyvinylidene fluoride membranes. Membranes were blocked in 5%
non-fat milk in TBS containing 0.05% Tween-20 (TBST) for 2 h at
room temperature. The membranes were incubated overnight at 4°C
with primary antibodies against CDH16 (1:1,000, cat. no.
15107-1-AP, ProteinTech Group, Inc.), DNA polymerase (POL)D1
(1:1,000, cat. no. 15646-1-AP, ProteinTech Group, Inc),
minichromosome maintenance (MCM)6 (1:2,000, cat. no. 13347-2-AP;
ProteinTech Group, Inc), claudin (CLDN)1 (1:1,500, cat. no.
13050-1-AP, ProteinTech Group, Inc), intercellular adhesion
molecule (ICAM)1 (1:2,000, cat. no. 60299-1-Ig, ProteinTech Group,
Inc), syndecan (SDC)4 (1:1,000, cat. no. 11820-1-AP, ProteinTech
Group, Inc) or β-actin (1:1,000, cat. no. TA-09; OriGene
Technologies, Inc.). The membranes were washed with PBS three times
and subsequently probed with anti-rabbit (cat. no. ZB-2301) or
anti-mouse horseradish peroxidase-linked secondary antibody (both
1:2500; cat. no. ZB-2305) for 60 min at room temperature and
visualized with ECL reagent (cat. no. sc-2048; all OriGene
Technologies, Inc.). The protein bands were scanned and quantified
by Tanon Gis software v4.2 (Tanon Science and Technology Co.,
Ltd.). Expression of β-actin served as the loading control.
Gene transfection
A full length CDH16 cDNA clone was chemically
synthesized by Sino Biological, Inc. and ligated into pCMV3 vector
(Sino Biological, Inc.). Empty pCMV3 vector was used as the
negative control. Untransfected cells were used as the blank
control group. To introduce the vector, BCPAP cells were seeded in
6-well plates at a density of 5×105 cells per well.
After incubation at 37°C overnight, the degree of cell fusion was
50–70%. The cells were transiently transfected with plasmid
carrying CDH16 or negative control plasmid (both 4 µg/well) using
TransIntro EL (TransGen Biotech Co., Ltd.) according to the
manufacturer's instructions. The effect of pCMV3-CDH16 on CDH16
mRNA and protein expression was analyzed by RT-qPCR and western
blotting, as aforementioned, at 48 h post-transfection.
MTT analysis
Cells (1×104 per well) were seeded in
96-well plates. At 0, 24, 48, 72, and 96 h post-transfection, cell
viability was determined by adding 10 µl 5 mg/ml MTT solution
(Sigma-Aldrich; Merck KGaA) to each well and incubating the samples
for 4 h at 37°C. The cell culture medium was removed and 150 µl
DMSO (Sigma-Aldrich; Merck KGaA) was added to dissolve the formazan
crystals. The absorbance at 490 nm was measured using a microplate
reader (Bio-Rad Laboratories, Inc.). The experiment was repeated
three times.
Flow cytometry analysis
Effect of CDH16 on BCPAP cell apoptosis was
determined by flow cytometry. Cells (5×105 per well)
were seeded in 6-well plates. At 48 h post-transfection, the cells
were trypsinized using 0.25% trypsin (Beijing Solarbio Science
& Technology Co., Ltd.) at room temperature for 1 min and
collected by centrifugation at 800 × g for 5 min at room
temperature. The cells were incubated with 0.5 ml binding buffer
and 1.0 µl Annexin V-FITC (Merck KGaA) at room temperature for 15
min and resuspended in fresh 0.5 ml binding buffer containing 5 µl
PI. Early apoptosis was measured using a Beckman Coulter FC500 flow
cytometer (Beckman Coulter, Inc.) and MXP 2.2 software (Beckman
Coulter, Inc.).
Scratch test
Effect of CDH16 on BCPAP cell migration was
determined by scratch test. The cells were inoculated on 6-well
plates at a density of 5×105 cells/well and incubated in
RPMI-1640 (Gibco) containing 10% FBS (Gibco) at 37°C for 24 h. When
confluency reached 80%, the medium was replaced with serum-free
RPMI-1640 and the cells were starved for 24 h. A scratch was then
drawn with a 200-µl plastic pipette tip and the plates were rinsed
with PBS to remove any cells suspended in culture medium after
scratching. The gap distance of each scratch wound was photographed
at 0 and 48 h after scratching using an light microscope (Olympus
Corporation IX71; ×40 magnification) and scratch area was measured
with ImageJ v1.50 (National Institutes of Health). Wound healing
was calculated as a percentage.
Transwell invasion assay
Pre-coated Matrigel chambers (BD Biosciences) were
used for the invasion assays according to the manufacturer's
instructions. After transfection of pCMV3-CDH16 plasmid and empty
pCMV3 plasmid at 37°C for 48 h, the cells were collected. A total
of 1×104 BCPAP cells were seeded into the upper chamber
of the Transwell apparatus in serum-free RPMI-1640, while RPMI-1640
supplemented with 10% FBS was added to the lower chamber. After
incubation at 37°C for 24 h, cells adhering to the lower surface
were fixed with 100% methanol for 20 min at room temperature and
stained with 5% Giemsa solution for 20 min at room temperature. The
number of infiltrated cells was calculated under a light microscope
(Olympus Corporation IX71) on five randomly selected fields (×100
magnification).
Statistical analysis
All experiments were performed ≥3 times. Statistical
analysis was performed using SPSS 17.0 software (SPSS, Inc.). The
χ2 or Fisher's exact test was used to analyze
differences in CDH16 levels between PTC and normal thyroid tissue
in IHC staining. Data are presented as the mean ± standard
deviation. The paired t-test was used to compare CDH16 mRNA
expression between 35 paired thyroid tissue samples. Pearson
correlation test was used in correlation analysis. Single factor
analysis of variance (one-way ANOVA) followed by post hoc Fisher's
least significant difference test was used to compare >2
samples. P<0.05 was considered to indicate a statistically
significant difference.
Results
CDH16 is downregulated in human TC
tissue and cell lines independent of CNV
To verify that CDH16 is downregulated in TC, CDH16
transcript levels were evaluated in three independent TC studies
from GEO. Data from all three studies in the Oncomine database
showed that mRNA expression of CDH16 was significantly lower in PTC
than in normal thyroid tissue (P<0.05; Fig. 1A-C). CDH16 expression was also
significantly lower in FTC (P<0.05; Fig. 1D) and ATC (P<0.05; Fig. 1E) than in normal thyroid tissue.
Notably, CDH16 ranked in the top 5% of all genes downregulated in
each of these studies and the fold difference for PTC was >2. To
determine whether decreased expression of CDH16 in PTC may be
accounted for by DNA CNV, a dataset from TCGA that has previously
been shown to have lower CDH16 expression in PTC (21) compared with matched normal tissue
was used. The DNA CN of CDH16 showed no significant difference in
blood, normal thyroid or TC tissue (P>0.05; Fig. 1F). Collectively, these findings
indicated that CDH16 expression was decreased in TC in three PTC
datasets as well as FTC and ATC, and also demonstrated that lower
expression of CDH16 in TC was not accounted the result of CNV.
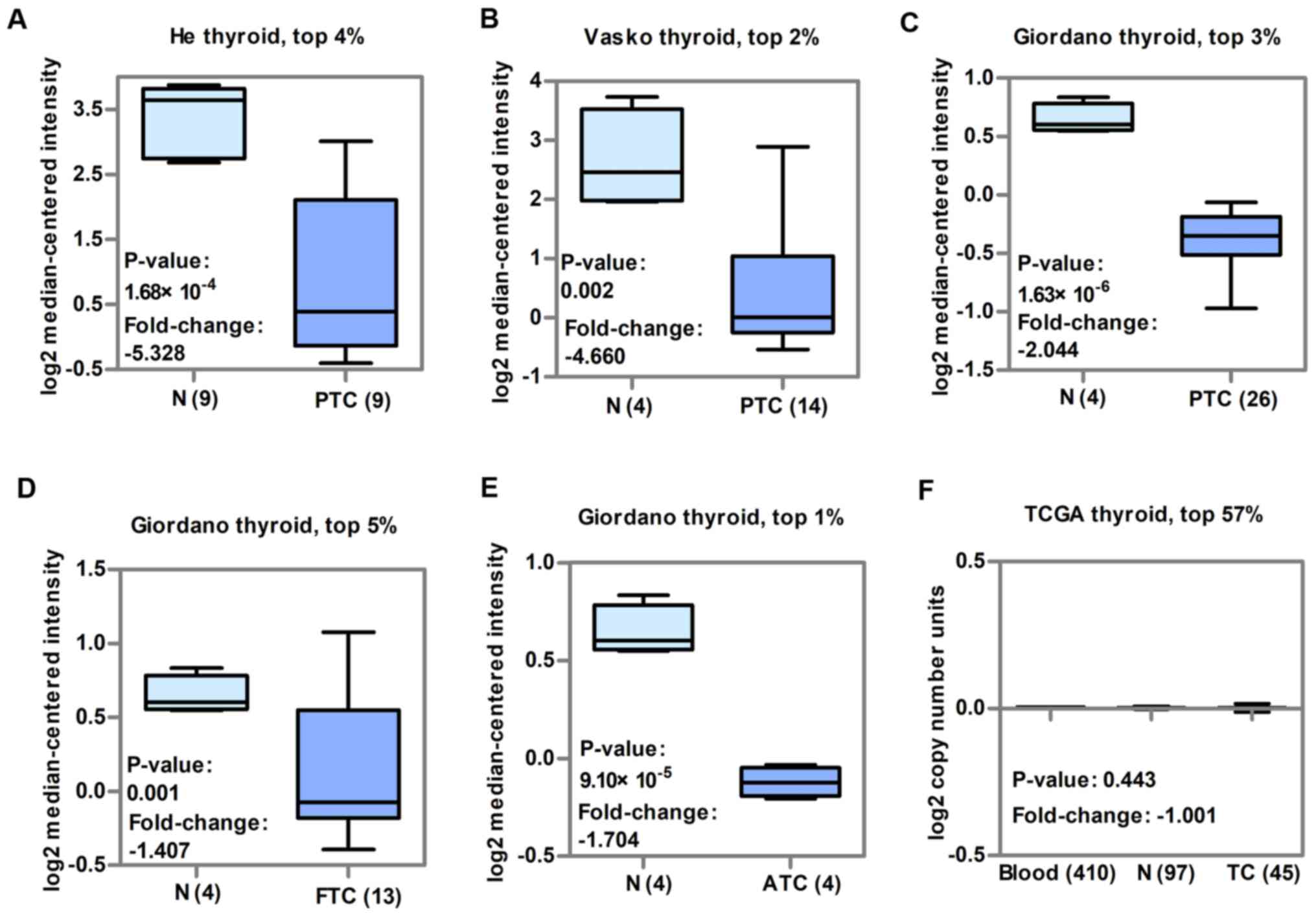 | Figure 1.CDH16 is downregulated in TC from
three GEO datasets and there is no significant variation in copy
number in TCGA dataset. The fold-change, P-value and
underexpression gene ranks are based on Oncomine 4.5 analysis. Box
plots showing CDH16 mRNA levels in patients with PTC in (A) He, (B)
Vasko and (C) Giordano thyroid datasets from GEO. Box plot showing
CDH16 mRNA levels in patients with (D) FTC and (E) ATC in the
Giordano thyroid dataset. (F) Box plot showing CDH16 copy number in
TCGA thyroid dataset. N, normal; CDH, cadherin; TC, thyroid
carcinoma; GEO, Gene Expression Omnibus; TCGA, The Cancer Genome
Atlas; PTC, papillary thyroid cancer; FTC, follicular thyroid
cancer; ATC, anaplastic thyroid cancer. |
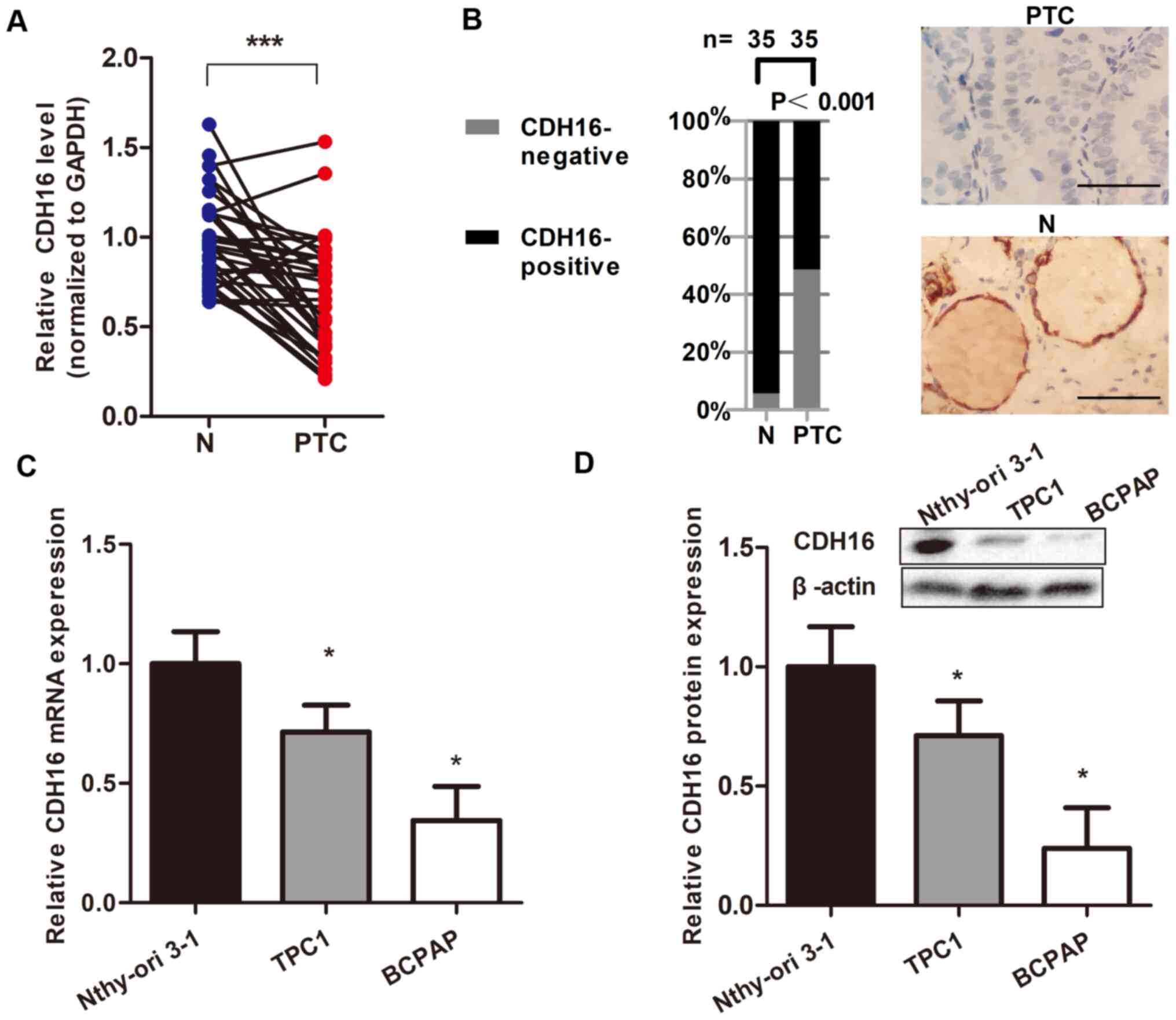
To support these findings, RT-qPCR and IHC were
performed for 35 paired thyroid tissue samples collected from
patients with PTC. CDH16 mRNA expression was significantly
decreased in PTC compared with normal tissue (P<0.05; Fig. 2A). Furthermore, IHC showed that
CDH16 was localized to the cell membrane of PTC cells. Of 35 PTC
tissue samples, 17 (48.6%) stained negative for CDH16, while only
two (5.7%) of the normal thyroid tissue samples stained negative
for CDH16 (P<0.05; Fig. 2B).
Therefore, decreased expression of CDH16 in PTC was observed at
both the mRNA and protein level.
Next, to determine whether downregulation of CDH16
was correlated with the outcome of PTC, the association between
CDH16 expression and clinicopathological characteristics of 35
patients with PTC was assessed. CDH16 protein expression was
associated with tumor size, lymph node metastasis and pathological
stage of PTC but there was no significant association with sex or
age (Table I).
To confirm the downregulation of CDH16 in TC, CDH16
levels in TC-derived cell lines (BCPAP and TPC1) and the human
thyroid follicular cell line Nthy-ori3-1 were measured. CDH16
expression was lower in TC cell lines, particularly BCPAP cells,
both at the mRNA and protein level (P<0.05; Fig. 2C and D). These results were
consistent with the aforementioned downregulation of CDH16 in
patients with advanced PTC and support the use of BCPAP cells as a
model for evaluating CDH16 biological activity.
CDH16 expression is negatively
associated with expression of proliferation and invasion
pathway-associated genes
To determine the role of CDH16 in TC, genes
co-expressed with CDH16 in TC samples from TCGA were investigated
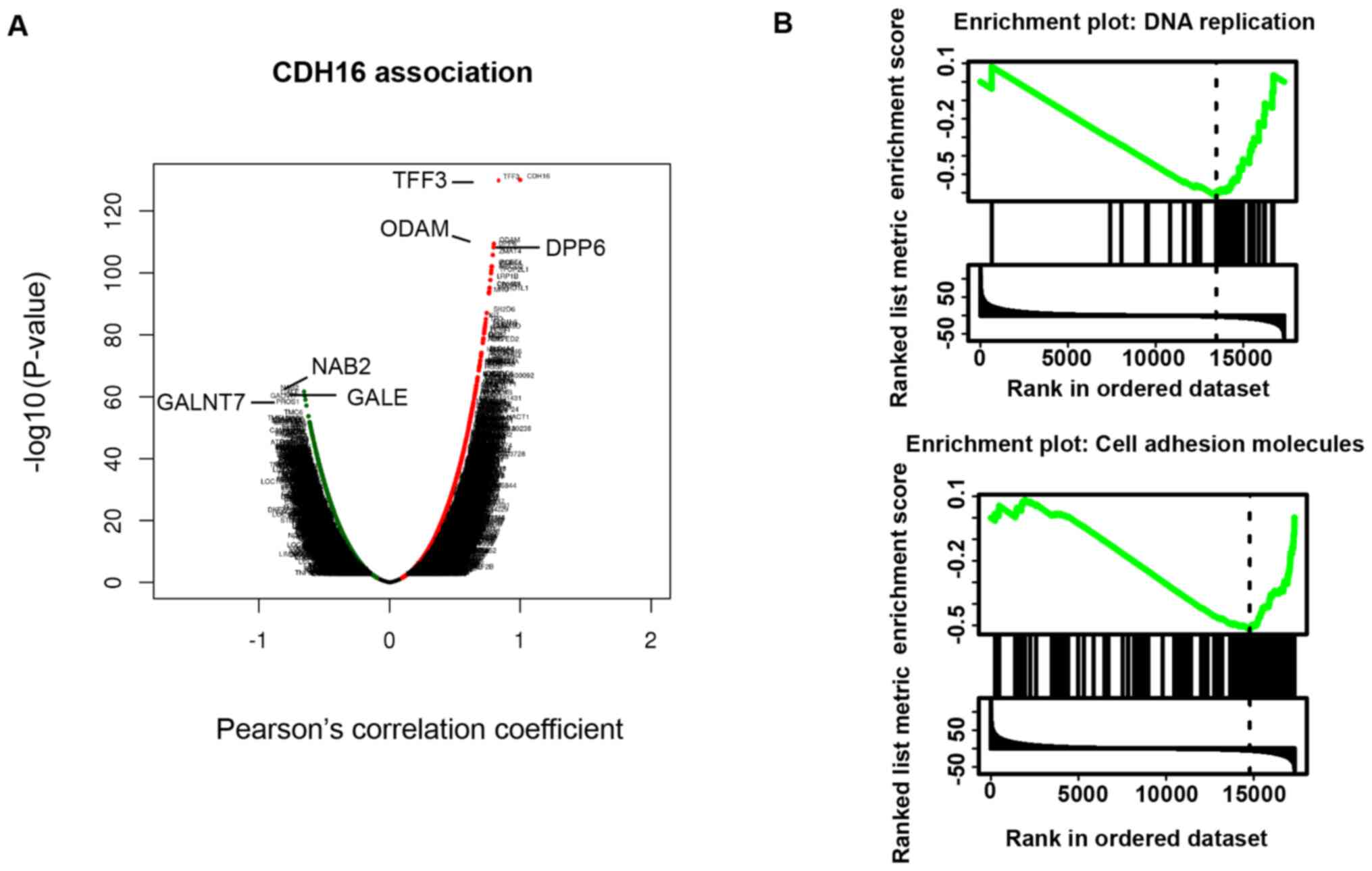
using the LinkedOmics database. Volcano plot suggested that the
most highly co-expressed genes were polypeptide
N-acetylgalactosaminyltransferase 7 (GALNT7), NGFI-A binding
protein 2 (NAB2) and UDP-galactose-4-epimerase (GALE), and the most
highly inversely co-expressed genes are trefoil factor 3 (TFF3),
odontogenic, ameloblast associated (ODAM) and dipeptidyl peptidase
like 6 (DPP6) (P<0.05; Fig.
3A). KEGG pathway analysis by GSEA showed that negatively
CDH16-associated genes were enriched in DNA replication and cell
adhesion pathways (Fig. 3B;
Table II). Correlation between
expression levels of POLD1, MCM6, CLDN1, ICAM1 and SDC4 and those
of CDH16 in 503 patients with PTC in TCGA was analyzed. The
expression of each of these genes was significantly negatively
correlated with expression of CDH16 in PTC (r=−0.290, −0.395,
−0.594, −0.552, −0.583, respectively; P<0.001; Fig. 4).
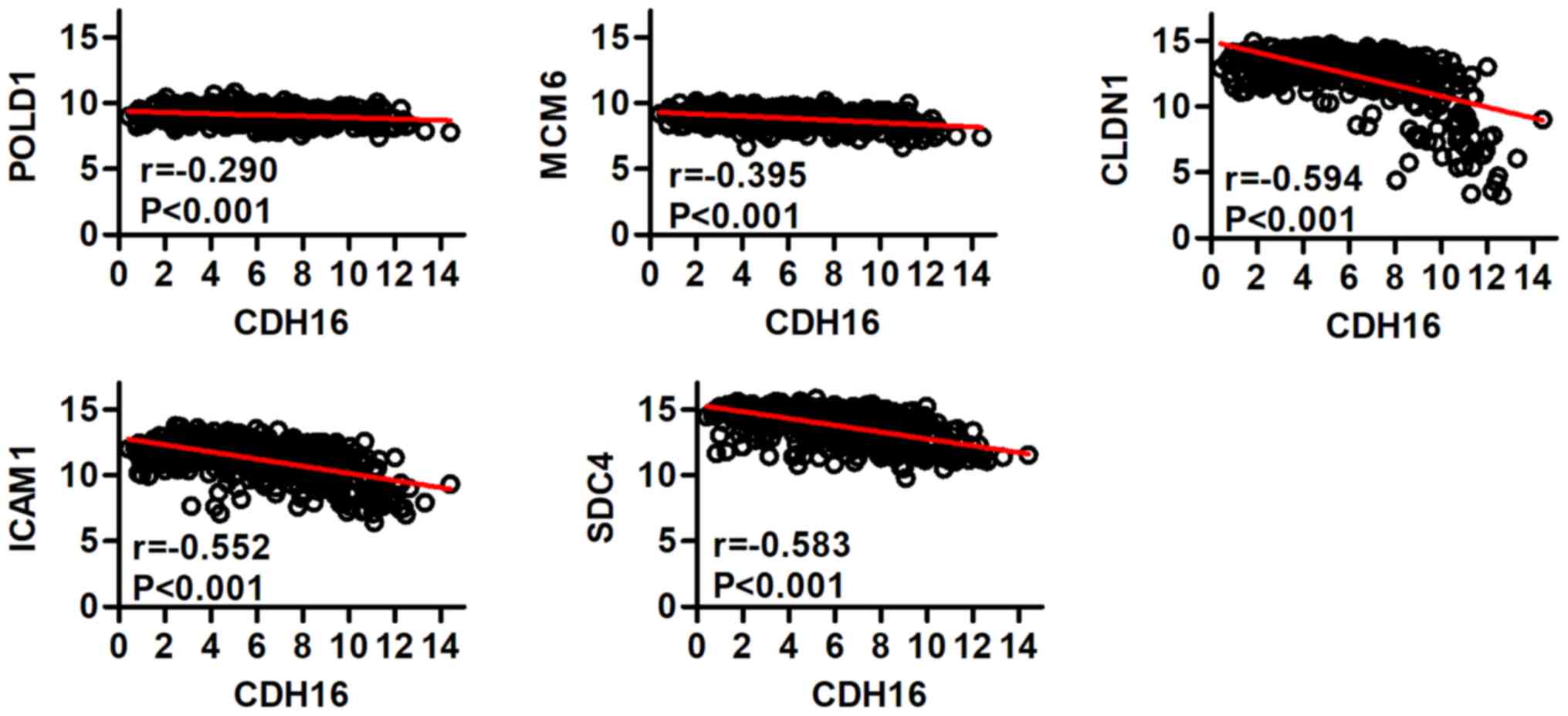 | Figure 4.Correlation between POLD1, MCM6,
CLDN1, ICAM1, SDC4 and CDH16 expression was analyzed using the
LinkedOmics database. POL, polymerase; MCM, minichromosome
maintenance; CLDN, claudin; ICAM, intercellular adhesion molecule;
SDC, syndecan; CDH, cadherin. |
| Table II.Significant enrichment of Kyoto
Encyclopedia of Genes and Genomes pathways of cadherin 16 in
thyroid carcinoma (LinkedOmics). |
Table II.
Significant enrichment of Kyoto
Encyclopedia of Genes and Genomes pathways of cadherin 16 in
thyroid carcinoma (LinkedOmics).
| Description | Leading edge genes,
n | Leading edge
gene |
|---|
| DNA
replication | 24 | MCM6; MCM2; MCM7;
MCM4; MCM5; POLD1; RFC2; MCM3; LIG1; POLE3; POLE; RFC3; POLE2;
PCNA; RNASEHIA; POLD3; POLA2; PRIM1; RFC4; POLA1; FEN1; RNASEH1;
RPA2; PRIM2 |
| Cell adhesion
molecules | 54 | CLDN1; SDC4; ICAM1;
CDH3; NRCAM; CD276; CLDN16; SDC3; CLDN10; CLDN7; CD58; CLDN4;
ITGB8; CDH4; PTPRF; NLGN2; ALCAM; CLDN9; VTCN1; CNTNAP1; F11R; MAG;
ITGAM; HLA-8; CDH15; CLDN2; ITGB7; HLA-DQB1; ITGA9; HLA-DRA;
HLA-DQA1; HLA-G; CD22; HLA-DOB; HLA-A; HLA-C; HLA-DRB1; ITGB1;
HLA-DOA; SELL; SELPLG; CD86; ITGB2; CD274; HLA-DPA1; NCAM2;
HLA-DRB5; CD40; PTPRM;ICOSLG; HLA-DQA2; HLA-DPB1; SPN; CLDN3 |
Overexpression of CDH16 inhibits
proliferation, migration and invasion of BCPAP cells in vitro
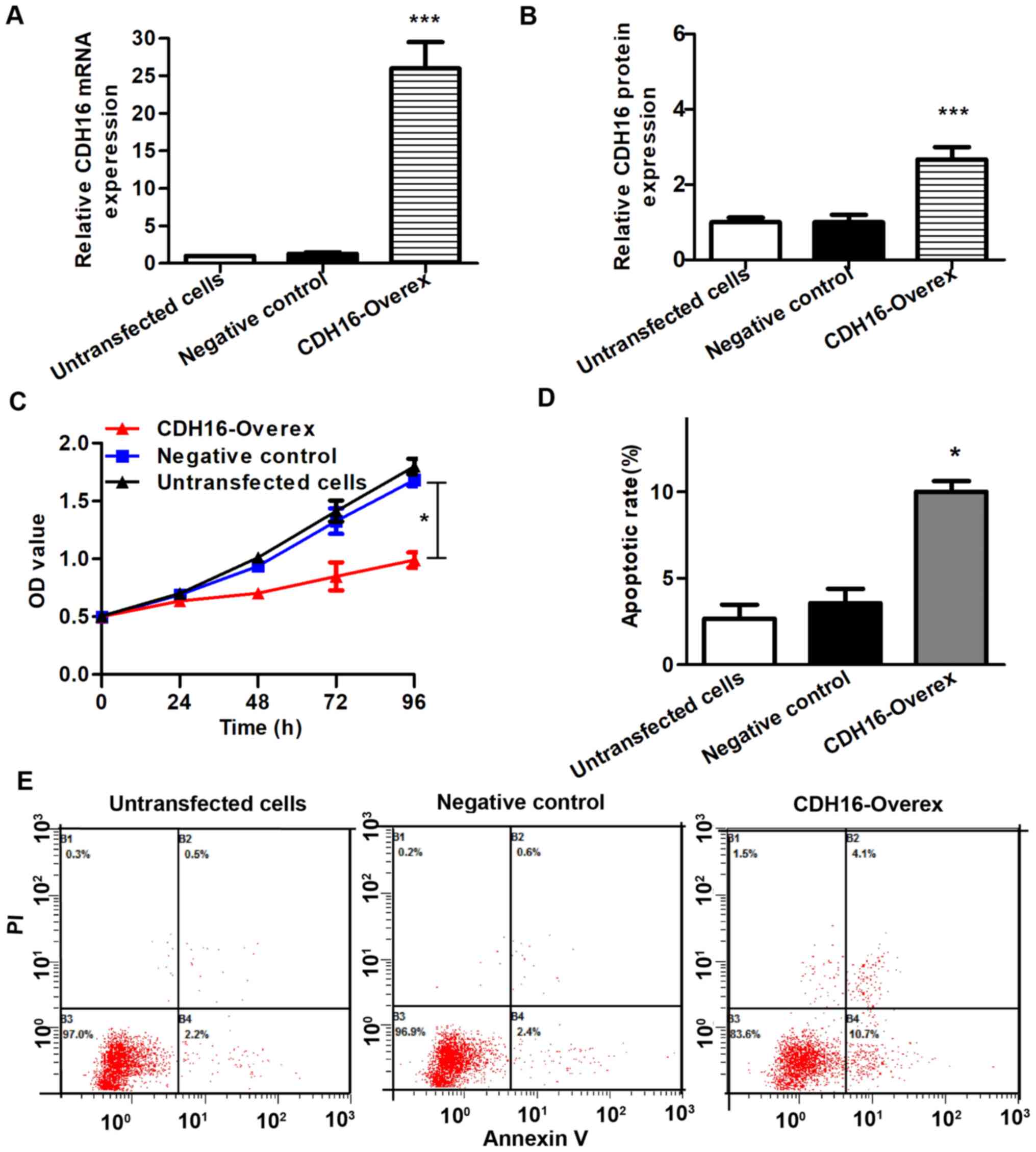
To verify the function of CDH16 in TC cells, BCPAP
cells were transfected with CDH16 expression vector. RT-qPCR and
western blot assay verified higher mRNA and protein CDH16 levels in
CDH16 overexpression compared with negative control BCPAP cells
(P<0.05; Fig. 5A and B). To
evaluate the effect of CDH16 on proliferation, MTT assay was
performed. The proliferation of BCPAP cells was significantly
decreased in overexpression compared with negative control cells
(P<0.05; Fig. 5C).
To determine whether CDH16 expression affects
tumor-associated functions, we the effect of CDH16 overexpression
on cell apoptosis, migration and invasion was assessed. Compared
with negative control, the CDH16 overexpression group exhibited a
significantly higher apoptosis rate, as evaluated by flow cytometry
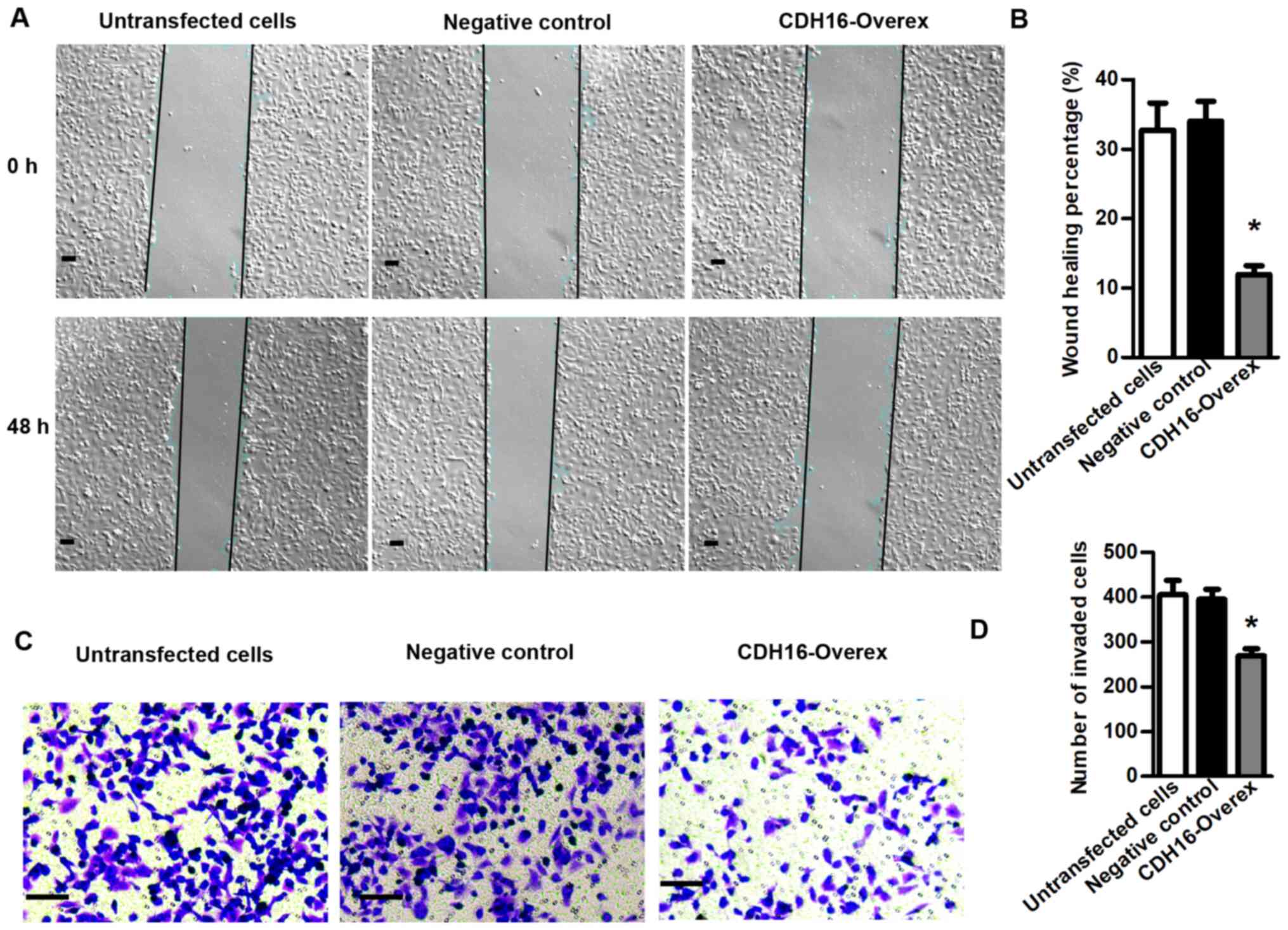
with Annexin V and PI staining (P<0.05; Fig. 5D and E). Cell scratch test
indicated that overexpression of CDH16 significantly inhibited
migration of BCPAP cells (P<0.05; Fig. 6A and B). Furthermore, Transwell
invasion assay indicated that overexpression of CDH16 significantly
inhibited invasion capacity of BCPAP cells (P<0.05; Fig. 6C and D). Overall, these results
demonstrated that CDH16 promoted apoptosis and inhibited TC cell
proliferation, migration and invasion in vitro, which is
consistent with the pattern of expression in patients with PTC.
Overexpression of CDH16 downregulates
POLD1, MCM6, CLDN1, ICAM1 and SDC4 expression in BCPAP cells in
vitro
To verify that CDH16 inhibited cell proliferation,
migration and invasion via DNA replication and cell adhesion
molecular pathways, western blot assay was used to detect protein
expression levels. Following overexpression of CDH16 in TC cells,
expression of POLD1, MCM6, CLDN1, ICAM1 and SDC4 was significantly
downregulated (53.42±6.38, 37.67±14.27, 42.17±3.36, 37.04±9.34 and
47.83±9.23% decrease, respectively; P<0.05; Fig. 7) compared with negative control.
These data indicated that CDH16 may regulate proliferation,
migration and invasion of TC cells via DNA replication and cell
adhesion pathways.
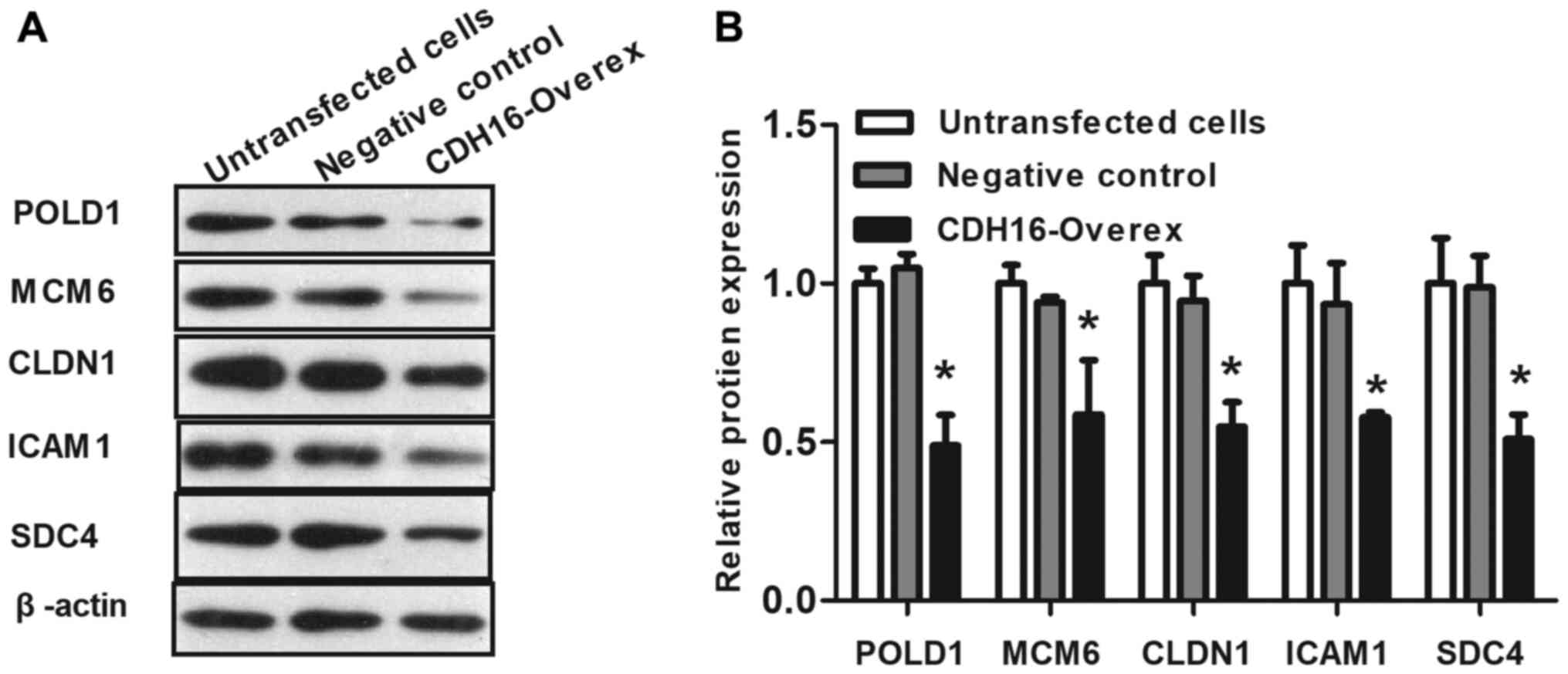 | Figure 7.Overexpression of CDH16 downregulates
DNA replication and cell adhesion pathway genes. (A) Representative
western blots showing downregulation of POLD1, MCM6, CLDN1, ICAM1
and SDC4 following overexpression of CDH16. (B) Relative protein
levels were quantified by densitometry of western blots (three
replicates). Data are presented as the mean ± SD of three
independent experiments. n=3. *P<0.05 vs. negative control.
CDH16, cadherin; Overex, overexpression, POL, polymerase; MCM,
minichromosome maintenance; CLDN, claudin; ICAM, intercellular
adhesion molecule; SDC, syndecan. |
Discussion
The human genome encodes 115 members of the CDH
superfamily and the expression of these family members is
tissue-specific (29). CDH1/E-CDH,
one of the most classical CDHs, is primarily expressed in
epithelial tissue and has been characterized as a tumor suppressor
involved in epithelial-mesenchymal transition (30–33).
Expression of CDH16 was first observed in the rabbit kidney
(34) and has since been shown to
be expressed in mouse and human thyroid (17,20).
CDH16 co-localizes with CDH1 in thyroid follicular cells and serves
a key role in thyroid cell polarity acquisition and follicle
formation (18,20). Calì et al (20) demonstrated that CDH16 promotes
intercellular adhesion to a similar extent as CDH1 and loss of
CDH16 precedes loss of CDH1 in TC. CDH1 expression decreases in FTC
and has been suggested as a marker for prognosis of TC (35,36).
To the best of our knowledge, however, the mechanism of CDH16 in TC
has not previously been determined.
The present study characterized CDH16 expression in
different forms of TC. The results demonstrated that CDH16 mRNA
levels were significantly downregulated in PTC, FTC and ATC
compared with normal thyroid tissue. Furthermore, CDH16 ranked
within the top 5% of downregulated genes, with the highest rank
(<2%) for PTC. These data demonstrated that CDH16 downregulation
of expression was a common feature of TC. CDH16 protein expression
in TC was correlated with tumor size, disease stage and nodal
metastasis in an independent cohort of 35 patients. Consistent with
the present results, Li et al (21) demonstrated an association between
CDH16 downregulation and lymph node metastasis using TCGA cohorts.
These results confirm previous findings suggesting that CDH16 may
serve as a biomarker for PTC diagnosis and prognosis (20,21).
CNV affects gene expression and is a key pathogenic factor in types
of cancer, such as ovarian cancer (37). However, in the present study, no
significant CN loss of CDH16 was detected in TC. These results
suggested that CDH16 may be a potential diagnostic and prognostic
marker for TC and that downregulation of CDH16 was independent of
CNV.
CDH16 serves as a tumor suppressor in TC (20,21)
but the specific pathogenesis remains largely unclear. Here, CDH16
was co-expressed with genes involved in DNA replication and cell
adhesion molecule pathways. To verify the role of CDH16 in TC,
CDH16 was overexpressed in BCPAP cells and MTT, flow cytometry,
cell scratch and Transwell invasion assays were performed to
determine whether overexpression of CDH16 affected proliferation,
apoptosis, migration and invasion. The results demonstrated that
overexpression of CDH16 resulted in inhibition of proliferation,
migration and invasion of TC cells and increased apoptosis.
KEGG analysis demonstrated that CDH16 expression was
negatively associated with expression of DNA replication genes. DNA
replication is a key event for cell proliferation that is separated
into three stages: Initiation, extension and termination (38). At the beginning of replication, the
double strands of the DNA helix unwind under the action of helicase
(38). Then, using each parent
chain as a template and four deoxynucleotides in the surrounding
environment as raw materials, a chain complementary to the parent
chain is synthesized under the action of DNA polymerase according
to complementary base pairing (39). The accuracy of DNA replication in
eukaryotes requires proteins such as helicase and DNA POL (40,41).
MCM protein complex, which consists of six highly conserved
proteins (MCM2-7), initiates DNA replication and unwinding via its
replicative helicase activity (42). MCM2-7 proteins are present in
proliferating cells (43) and
overexpression of MCM2, MCM4 and MCM6 is associated with
tumorigenesis (44–46). DNA POL α, δ, and ε are key
mediators of DNA replication in eukaryotes (41); their mutation or abnormal
expression affects the occurrence, development and invasion of
human colorectal cancer, stomach adenocarcinoma and pancreatic
adenocarcinoma (47–49). POLD1 encodes DNA POL δ (50); an increase in its protein
expression or activity has been demonstrated to be associated with
tumorigenesis in colorectal carcinoma and endometrial carcinoma
(51–53). In addition, it is associated with
the invasive ability of cancer cells. For example, Sanefuji et
al (54) reported that POLD1
is a key indicator of the activity and invasion of hepatoma cells
and its expression is associated with the degree of vascular
invasion of cancer cells. Sigurdson et al (55) found that increased POLD1 expression
increases the risk of breast cancer. The present study showed that
expression of MCM6 and POLD1 in TC was significantly negatively
correlated with CDH16 by analyzing TCGA thyroid cancer data and
that expression of MCM6 and POLD1 was significantly downregulated
and cell proliferation inhibited after overexpression of CDH16 in
BCPAP cells. This suggested that CDH16 inhibited proliferation of
TC cells via downregulation of proteins associated with DNA
replication.
KEGG analysis showed that CDH16 was associated with
the cell adhesion molecular pathway. Adhesion molecules are divided
into five groups: Integrins, selectins, CDHs, hyaladherin and
immunoglobulin superfamily members (56). Cell adhesion molecules regulate
tumor invasion and metastasis (57). By binding to surface adhesion
molecules and extracellular matrix or cell ligands, they activate
intracellular signaling pathways and endow tumor cells with
metastatic ability (57). CLDNs
are cytoskeletal proteins of tight junction between cells that not
only regulate paracellular transepithelial/transendothelial
transport but are also key for cell proliferation and
differentiation (58). The
abnormal expression of CLDNs can lead to structural and functional
damage of epithelial and endothelial cells, which leads to tumor
invasion and metastasis (59).
CLDN1 is the most studied CLDN in cancer and its overexpression or
loss of expression is observed in different types of cancer
(60). CLDN1 expression is low in
invasive breast, esophageal and prostate cancer (59–61).
However, CLDN1 is highly expressed in ovarian, colon and thyroid
cancer (60,62–64).
Furthermore, the mechanism of CLDN1 in promoting tumor invasion may
involve activation of matrix metalloproteinase 2 (65). Considering the role of CLDN1 in TC,
targeting CLDN1 expression may be a promising treatment for TC
(66).
Other cell adhesion molecules identified in the
present study included ICAM1 and SDC4. ICAM1 is an immune protein
superfamily adhesion molecule that is widely expressed on the
surface of monocytes, lymphocytes and vascular endothelial and
cancer cells (67). Its primary
function is to regulate adhesion between cells and between cells
and the extracellular matrix. In addition, it is involved in cell
signal transduction, proliferation and migration, immune
inflammatory response, vascular growth, tumor invasion and
metastasis, as well as other physiological and pathological
processes (68). ICAM1 is
overexpressed in a number of tumor types, including TC (69,70).
The binding of ICAM1 and its ligand lymphocyte-associated antigen 1
(LFA1) inhibits natural killer and cytotoxic T cells in the
occurrence and development of malignant tumor, helps tumor cells
escape immune surveillance and attack and promotes invasion and
metastasis of tumor cells (71).
SDC, a family of four transmembrane proteoglycans, has been
reported to serve key roles in cell proliferation, migration and
differentiation (72). SDC4 is
highly expressed in TC compared with normal tissue (73). Chen et al (73) revealed that SDC4 expression levels
are upregulated in PTC and SDC4 gene silencing suppresses PTC cell
migration and invasion by suppressing activation of the
Wnt/β-catenin signaling pathway.
The present study confirmed that expression of
CLDN1, ICAM1 and SDC4 was negatively correlated with CDH16
expression in TCGA databases. Furthermore, expression of CLDN1,
ICAM1 and SDC4 was verified by western blotting following
overexpression of CDH16. The present results demonstrated that
CDH16-overexpressing TC cells exhibited significantly downregulated
levels of CLDN1, ICAM1 and SDC4, which lead to inhibition of cell
migration and invasion. Thus, CDH16 and cell adhesion pathways may
serve a role in TC via migration and invasion, which is consistent
with results of KEGG analysis. These results are in accordance with
bioinformatics analysis results and support the findings of the
present study, confirming that CDH16 may be a functional tumor
suppressor in TC.
In conclusion, the present study used an extended
dataset to demonstrate that CDH16 is downregulated in PTC, FTC and
ATC and demonstrated that its expression was significantly
correlated with tumor size, lymph node metastasis and disease stage
in a cohort of 35 patients with PTC. Furthermore, bioinformatics
analysis showed that downregulation of CDH16 may promote TC via DNA
replication and cell adhesion pathways. The present study also
demonstrated that overexpression of CDH16 inhibited proliferation,
migration and invasion and induced apoptosis of BCPAP cells. These
findings were demonstrated using human tumor samples and cell
lines; however, studies in mice would provide in vivo
verification. To the best of our knowledge, the present study is
the first to provide direct evidence for the tumor suppressive
effect of CDH16 in TC. While the molecular mechanism of CDH16
disruption in TC remains unknown, elucidating the signaling
mechanism underlying CDH16 downregulation in TC may inform the
design of novel therapeutic strategies to treat TC.
Acknowledgements
The authors would like to thank Professor Jun Li
from Tangshan Renmin Hospital (Hebei, China) for collecting tissue
specimens.
Funding
The present study was supported by The Science and Technology
Program of Hebei (grant no. H2018105066).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XY, YuL and GL designed the study. XY, WZ and YiL
performed the experiments. GL and XY performed data analysis. XY
and YuL confirm the authenticity of all the raw data. XY drafted
the manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethical
Committee of Tangshan Gongren Hospital (approval no.
GRYY-LL-2020-104). All population-related studies were carried out
according to the World Medical Association Declaration of Helsinki.
All patients provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pellegriti G, Frasca F, Regalbuto C,
Squatrito S and Vigneri R: Worldwide increasing incidence of
thyroid cancer: Update on epidemiology and risk factors. J Cancer
Epidemiol. 2013:9652122013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim J, Gosnell JE and Roman SA: Geographic
influences in the global rise of thyroid cancer. Nat Rev
Endocrinol. 16:17–29. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig
AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and
deaths to 2030: The unexpected burden of thyroid, liver, and
pancreas cancers in the United States. Cancer Res. 74:2913–2921.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang J, Yu F, Shang Y, Ping Z and Liu L:
Thyroid cancer: Incidence and mortality trends in China, 2005–2015.
Endocrine. 68:163–173. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sherman SI: Thyroid carcinoma. Lancet.
361:501–511. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Burns WR and Zeiger MA: Differentiated
thyroid cancer. Semin Oncol. 37:557–566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rahmani N, Abbas Hashemi S, Fazli M and
Raisian M: Clinical management and outcomes of papillary,
follicular and medullary thyroid cancer surgery. Med Glas.
10:164–167. 2013.PubMed/NCBI
|
|
9
|
Grant CS: Papillary thyroid cancer:
Strategies for optimal individualized surgical management. Clin
Ther. 36:1117–1126. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Manoj G, Deepika K, Ryoko O, Saket J,
Vikas M and Wenwen C: Laminin-5γ-2 (LAMC2) is highly expressed in
anaplastic thyroid carcinoma and is associated with tumor
progression, migration, and invasion by modulating signaling of
EGFR. J Clin Endocrinol Metab. 99:E62–E72. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu H, Sun Y, Ye H, Yang S, Lee SL and de
las Morenas A: Anaplastic thyroid cancer: Outcome and the
mutation/expression profiles of potential targets. Pathol Oncol
Res. 21:695–701. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ernani V, Kumar M, Chen AY and Owonikoko
TK: Systemic treatment and management approaches for medullary
thyroid cancer. Cancer Treat Rev. 50:89–98. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hulpiau P and van Roy F: Molecular
evolution of the cadherin superfamily. Int J Biochem Cell Biol.
41:349–369. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Casal JI and Bartolomé RA: Beyond
N-Cadherin, Relevance of Cadherins 5, 6 and 17 in Cancer
Progression and Metastasis. Int J Mol Sci. 20:33732019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thomson RB, Ward DC, Quaggin SE, Igarashi
P, Muckler ZE and Aronson PS: cDNA cloning and chromosomal
localization of the human and mouse isoforms of Ksp-cadherin.
Genomics. 51:445–451. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thedieck C, Kuczyk M, Klingel K, Steiert
I, Müller CA and Klein G: Expression of Ksp-cadherin during kidney
development and in renal cell carcinoma. Br J Cancer. 92:2010–2017.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Calì G, Zannini M, Rubini P, Tacchetti C,
D'Andrea B, Affuso A, Wintermantel T, Boussadia O, Terracciano D,
Silberschmidt D, et al: Conditional inactivation of the E-cadherin
gene in thyroid follicular cells affects gland development but does
not impair junction formation. Endocrinology. 148:2737–2746. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Koumarianou P, Goméz-López G and
Santisteban P: Pax8 controls thyroid follicular polarity through
cadherin-16. J Cell Sci. 130:219–231. 2017.PubMed/NCBI
|
|
19
|
de Cristofaro T, Di Palma T, Fichera I,
Lucci V, Parrillo L, De Felice M and Zannini M: An essential role
for Pax8 in the transcriptional regulation of cadherin-16 in
thyroid cells. Mol Endocrinol. 26:67–78. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Calì G, Gentile F, Mogavero S, Pallante P,
Nitsch R, Ciancia G, Ferraro A, Fusco A and Nitsch L:
CDH16/Ksp-cadherin is expressed in the developing thyroid gland and
is strongly down-regulated in thyroid carcinomas. Endocrinology.
153:522–534. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li P, Wu Q, Sun Y, Pan X, Han Y, Ye B,
Zhang Y, Dong J and Zheng Z: Downregulation of cdh16 in papillary
thyroid cancer and its potential molecular mechanism analysed by
qRT-PCR, TCGA and in silico analysis. Cancer Manag Res.
11:10719–10729. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rhodes DR, Kalyana-Sundaram S, Mahavisno
V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ,
Kincead-Beal C, Kulkarni P, et al: Oncomine 3.0: Genes, pathways,
and networks in a collection of 18,000 cancer gene expression
profiles. Neoplasia. 9:166–180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He H, Jazdzewski K, Li W, Liyanarachchi S,
Nagy R, Volinia S, Calin GA, Liu CG, Franssila K, Suster S, et al:
The role of microRNA genes in papillary thyroid carcinoma. Proc
Natl Acad Sci USA. 102:19075–19080. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vasko V, Espinosa AV, Scouten W, He H,
Auer H, Liyanarachchi S, Larin A, Savchenko V, Francis GL, de la
Chapelle A, et al: Gene expression and functional evidence of
epithelial-to-mesenchymal transition in papillary thyroid carcinoma
invasion. Proc Natl Acad Sci USA. 104:2803–2808. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Giordano TJ, Au AYM, Kuick R, Thomas DG,
Rhodes DR, Wilhelm KG Jr, Vinco M, Misek DE, Sanders D, Zhu Z, et
al: Delineation, functional validation, and bioinformatic
evaluation of gene expression in thyroid follicular carcinomas with
the PAX8-PPARG translocation. Clin Cancer Res. 12:1983–1993. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vasaikar SV, Straub P, Wang J and Zhang B:
LinkedOmics: Analyzing multi-omics data within and across 32 cancer
types. Nucleic Acids Res. 46D:D956–D963. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang X, Liu G, Li W, Zang L, Li D, Wang Q,
Yu F and Xiang X: Silencing of zinc finger protein 703 inhibits
medullary thyroid carcinoma cell proliferation in vitro and
in vivo. Oncol Lett. 19:943–951. 2020.PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Suzuki ST: Structural and functional
diversity of cadherin superfamily: Are new members of cadherin
superfamily involved in signal transduction pathway? J Cell
Biochem. 61:531–542. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vleminckx K, Vakaet L Jr, Mareel M, Fiers
W and van Roy F: Genetic manipulation of E-cadherin expression by
epithelial tumor cells reveals an invasion suppressor role. Cell.
66:107–119. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Perl AK, Wilgenbus P, Dahl U, Semb H and
Christofori G: A causal role for E-cadherin in the transition from
adenoma to carcinoma. Nature. 392:190–193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shiozaki H, Oka H, Inoue M, Tamura S and
Monden M: E-cadherin mediated adhesion system in cancer cells.
Cancer. 77 (Suppl):1605–1613. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pal M, Bhattacharya S, Kalyan G and Hazra
S: Cadherin profiling for therapeutic interventions in Epithelial
Mesenchymal Transition (EMT) and tumorigenesis. Exp Cell Res.
368:137–146. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Thomson RB, Igarashi P, Biemesderfer D,
Kim R, Abu-Alfa A, Soleimani M and Aronson PS: Isolation and cDNA
cloning of Ksp-cadherin, a novel kidney-specific member of the
cadherin multigene family. J Biol Chem. 270:17594–17601. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brabant G, Hoang-Vu C, Cetin Y, Dralle H,
Scheumann G, Mölne J, Hansson G, Jansson S, Ericson LE and Nilsson
M: E-cadherin: A differentiation marker in thyroid malignancies.
Cancer Res. 53:4987–4993. 1993.PubMed/NCBI
|
|
36
|
Mitselou A, Ioachim E, Peschos D,
Charalabopoulos K, Michael M, Agnantis NJ and Vougiouklakis T:
E-cadherin adhesion molecule and syndecan-1 expression in various
thyroid pathologies. Exp Oncol. 29:54–60. 2007.PubMed/NCBI
|
|
37
|
Reid BM, Permuth JB, Chen YA, Fridley BL,
Iversen ES, Chen Z, Jim H, Vierkant RA, Cunningham JM,
Barnholtz-Sloan JS, et al: Genome-wide analysis of common copy
number variation and epithelial ovarian cancer risk. Cancer
Epidemiol Biomarkers Prev. 28:1117–1126. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fragkos M, Ganier O, Coulombe P and
Méchali M: DNA replication origin activation in space and time. Nat
Rev Mol Cell Biol. 16:360–374. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ganai RA and Johansson E: DNA
replication-A matter of fidelity. Mol Cell. 62:745–755. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Burgers PMJ and Kunkel TA: Eukaryotic DNA
replication fork. Annu Rev Biochem. 86:417–438. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Syväoja J, Suomensaari S, Nishida C,
Goldsmith JS, Chui GS, Jain S and Linn S: DNA polymerases alpha,
delta, and epsilon: Three distinct enzymes from HeLa cells. Proc
Natl Acad Sci USA. 87:6664–6668. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Forsburg SL: Eukaryotic MCM proteins:
Beyond replication initiation. Microbiol Mol Biol Rev. 68:109–131.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stoeber K, Tlsty TD, Happerfield L, Thomas
GA, Romanov S, Bobrow L, Williams ED and Williams GH: DNA
replication licensing and human cell proliferation. J Cell Sci.
114:2027–2041. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Das M, Prasad SB, Yadav SS, Govardhan HB,
Pandey LK, Singh S, Pradhan S and Narayan G: Over expression of
minichromosome maintenance genes is clinically correlated to
cervical carcinogenesis. PLoS One. 8:e696072013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kwok HF, Zhang SD, McCrudden CM, Yuen HF,
Ting KP, Wen Q, Khoo US and Chan KY: Prognostic significance of
minichromosome maintenance proteins in breast cancer. Am J Cancer
Res. 5:52–71. 2014.PubMed/NCBI
|
|
46
|
Issac MSM, Yousef E, Tahir MR and Gaboury
LA: MCM2, MCM4, and MCM6 in Breast Cancer: Clinical Utility in
Diagnosis and Prognosis. Neoplasia. 21:1015–1035. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shinbrot E, Henninger EE, Weinhold N,
Covington KR, Göksenin AY, Schultz N, Chao H, Doddapaneni H, Muzny
DM, Gibbs RA, et al: Exonuclease mutations in DNA polymerase
epsilon reveal replication strand specific mutation patterns and
human origins of replication. Genome Res. 24:1740–1750. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rayner E, van Gool IC, Palles C, Kearsey
SE, Bosse T, Tomlinson I and Church DN: A panoply of errors:
Polymerase proofreading domain mutations in cancer. Nat Rev Cancer.
16:71–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang Y, Chen Y, Wang C, Yang M, Wang Y,
Bao L, Wang JE, Kim B, Chan KY, Xu W, et al: MIF is a 3′ flap
nuclease that facilitates DNA replication and promotes tumor
growth. Nat Commun. 12:29542021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Preston BD, Albertson TM and Herr AJ: DNA
replication fidelity and cancer. Semin Cancer Biol. 20:281–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Palles C, Cazier JB, Howarth KM, Domingo
E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I,
et al CORGI Consortium; WGS500 Consortium, : Germline mutations
affecting the proofreading domains of POLE and POLD1 predispose to
colorectal adenomas and carcinomas. Nat Genet. 45:136–144. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Siraj AK, Parvathareddy SK, Bu R, Iqbal K,
Siraj S, Masoodi T, Concepcion RM, Ghazwani LO, AlBadawi I,
Al-Dayel F, et al: Germline POLE and POLD1 proofreading domain
mutations in endometrial carcinoma from Middle Eastern region.
Cancer Cell Int. 19:3342019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nicolas E, Golemis EA and Arora S: POLD1:
Central mediator of DNA replication and repair, and implication in
cancer and other pathologies. Gene. 590:128–141. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sanefuji K, Taketomi A, Iguchi T,
Sugimachi K, Ikegami T, Yamashita Y, Gion T, Soejima Y, Shirabe K
and Maehara Y: Significance of DNA polymerase delta catalytic
subunit p125 induced by mutant p53 in the invasive potential of
human hepatocellular carcinoma. Oncology. 79:229–237. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sigurdson AJ, Hauptmann M, Chatterjee N,
Alexander BH, Doody MM, Rutter JL and Struewing JP: Kin-cohort
estimates for familial breast cancer risk in relation to variants
in DNA base excision repair, BRCA1 interacting and growth factor
genes. BMC Cancer. 4:92004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Samanta D and Almo SC: Nectin family of
cell-adhesion molecules: Structural and molecular aspects of
function and specificity. Cell Mol Life Sci. 72:645–658. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Harjunpää H, Llort Asens M, Guenther C and
Fagerholm SC: Cell Adhesion Molecules and Their Roles and
Regulation in the Immune and Tumor Microenvironment. Front Immunol.
10:10782019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Krause G, Winkler L, Mueller SL, Haseloff
RF, Piontek J and Blasig IE: Structure and function of claudins.
Biochim Biophys Acta. 1778:631–645. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Singh AB, Sharma A and Dhawan P: Claudin
family of proteins and cancer: An overview. J Oncol.
2010:5419572010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bhat AA, Syed N, Therachiyil L, Nisar S,
Hashem S, Macha MA, Yadav SK, Krishnankutty R, Muralitharan S,
Al-Naemi H, et al: Claudin-1, A Double-Edged Sword in Cancer. Int J
Mol Sci. 21:5692020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhou B, Moodie A, Blanchard AA, Leygue E
and Myal Y: Claudin 1 in breast cancer: New insights. J Clin Med.
4:1960–1976. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Németh J, Németh Z, Tátrai P, Péter I,
Somorácz A, Szász AM, Kiss A and Schaff Z: High expression of
claudin-1 protein in papillary thyroid tumor and its regional lymph
node metastasis. Pathol Oncol Res. 16:19–27. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hucz J, Kowalska M, Jarzab M and Wiench M:
Gene expression of metalloproteinase 11, claudin 1 and selected
adhesion related genes in papillary thyroid cancer. Endokrynol Pol.
57 (Suppl A):18–25. 2006.(In Polish). PubMed/NCBI
|
|
64
|
Fluge Ø, Bruland O, Akslen LA, Lillehaug
JR and Varhaug JE: Gene expression in poorly differentiated
papillary thyroid carcinomas. Thyroid. 16:161–175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Li J, Chigurupati S, Agarwal R, Mughal MR,
Mattson MP, Becker KG, Wood WH III, Zhang Y and Morin PJ: Possible
angiogenic roles for claudin-4 in ovarian cancer. Cancer Biol Ther.
8:1806–1814. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Piontek A, Eichner M, Zwanziger D, Beier
LS, Protze J, Walther W, Theurer S, Schmid KW, Führer-Sakel D,
Piontek J, et al: Targeting claudin-overexpressing thyroid and lung
cancer by modified Clostridium perfringens enterotoxin. Mol Oncol.
14:261–276. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Pober JS, Gimbrone MA Jr, Lapierre LA,
Mendrick DL, Fiers W, Rothlein R and Springer TA: Overlapping
patterns of activation of human endothelial cells by interleukin 1,
tumor necrosis factor, and immune interferon. J Immunol.
137:1893–1896. 1986.PubMed/NCBI
|
|
68
|
Kotteas EA, Boulas P, Gkiozos I, Tsagkouli
S, Tsoukalas G and Syrigos KN: The intercellular cell adhesion
molecule-1 (ICAM-1) in lung cancer: Implications for disease
progression and prognosis. Anticancer Res. 34:4665–4672.
2014.PubMed/NCBI
|
|
69
|
Buitrago D, Keutgen XM, Crowley M,
Filicori F, Aldailami H, Hoda R, Liu YF, Hoda RS, Scognamiglio T,
Jin M, et al: Intercellular adhesion molecule-1 (ICAM-1) is
upregulated in aggressive papillary thyroid carcinoma. Ann Surg
Oncol. 19:973–980. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Min IM, Shevlin E, Vedvyas Y, Zaman M,
Wyrwas B, Scognamiglio T, Moore MD, Wang W, Park S, Park S, et al:
CAR T Therapy Targeting ICAM-1 Eliminates Advanced Human Thyroid
Tumors. Clin Cancer Res. 23:7569–7583. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Gray KD, McCloskey JE, Vedvyas Y, Kalloo
OR, Eshaky SE, Yang Y, Shevlin E, Zaman M, Ullmann TM, Liang H, et
al: PD1 blockade enhances ICAM1-directed CAR T therapeutic efficacy
in advanced thyroid cancer. Clin Cancer Res. 26:6003–6016. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ohkawara B, Glinka A and Niehrs C: Rspo3
binds syndecan 4 and induces Wnt/PCP signaling via
clathrin-mediated endocytosis to promote morphogenesis. Dev Cell.
20:303–314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Chen LL, Gao GX, Shen FX, Chen X, Gong XH
and Wu WJ: SDC4 gene silencing favors human papillary thyroid
carcinoma cell apoptosis and inhibits epithelial mesenchymal
transition via Wnt/beta-catenin pathway. Mol Cells. 41:853–867.
2018.PubMed/NCBI
|