Introduction
Renal cell carcinoma (RCC) is one of the most common
malignant tumors in urinary system, third only to prostate cancer
and bladder cancer (1). According
to the latest survey, there were ~76,000 new cases in the United
States, including 48,780 men and 27,300 women, which accounts for
3–5% of all malignant tumors (1,2).
Prognosis for RCC is relatively favorable when identified at an
early stage with five-year survival rates for localized and locally
advanced RCC ranging from 70–90% (1). However, once distant metastasis takes
place, the five-year survival rate decreases dramatically and can
be as little as 10% (1). It is
therefore necessary to investigate all possible methods to stratify
the RCC patients upon disease initiation and make more
individualized follow-up strategies. Clear cell renal cell
carcinoma (ccRCC) is the most common pathological type and accounts
for between 60–80% of all types (3). With radiographic imaging becoming
common place in routine diagnostics, a dramatic rise in the number
of small and early-stage ccRCC cases has also been observed. Tumor,
Node, Metastasis (TNM) staging system remains the most commonly
used method for determining prognosis of ccRCC. However, for
early-stage RCC, the TNM staging system alone is insufficient to
determine postoperative treatment and survival prognostics due to
its lack of sensitivity and specificity (4). According to the NCCN clinical
practice guidelines, for stage I and II RCC, post-operative
surveillance is recommended (5).
However, if postoperative surveillance or treatment could be more
precise for ccRCC patients, the clinical outcomes could be more
favorable. That's the reason why certain other staging systems,
such as the UISS, Kattan, Yaycioglu and Cindolo model, are being
explored and investigated in clinical research (4). It remains of the utmost importance to
construct and validate a prognostic model for early-stage RCC.
Recently, accumulating evidence has suggested that
as the most common type of RNA modification, N6-methyladenosine
(m6A) affects tumorigenesis in various types of cancers and could
even hold a function in regulating the tumor immune
microenvironment (6–10). In addition, they could also serve
as diagnostic and prognostic biomarkers (11). Thus, the gene mutation data and
transcriptional sequencing data of The Cancer Genome Atlas (TCGA)
database as well as immunohistochemistry staining were combined
with our proteomic data detected by high-performance liquid
chromatography-mass spectrometry (HPLC-MS) to construct a
prognostic model consisting of m6A methylation regulators for
early-stage RCC. The relationship between the model and tumor
infiltrating immune cells was evaluated, so as to preciously
stratify the ccRCC patients and provide new ideas for the inner
mechanism related to RCC.
Materials and methods
Data collection and analyzation of 19
m6A methylation regulators from the TCGA database
The gene mutation files and RNA sequencing data
(count value) with the corresponding clinical information from
ccRCC patients were downloaded from TCGA database (https://portal.gdc.cancer.gov/repository). Based on
the clinical data, the 192 somatic mutation data and 261 RNA
transcriptomes with early-stage ccRCC (pT1) were selected for
inclusion. Data with VarScan2 analysis of somatic mutation and
tumor mutation burden were downloaded from TCGA database. The
result was displayed with the R packages ‘maftools’ (12). RNA sequencing counts in tumor
tissues and adjacent relatively normal tissues were standardized
and analyzed using the DESeq2 package (13).
Proteomic examination of 27 paired
early-stage ccRCC tissues
To assess the reliability of the m6A methylation
regulator expression across protein levels, 27 paired early-stage
ccRCC tissues were then collected and analyzed by means of HPLC-MS.
The present study was approved (approval no. KS2021034) by the
Institutional Review Board of Peking Union Medical College
Hospital, Chinese Academy of Medical Science and Peking Union
Medical College (Beijing, China). Formal written consent was
requested and signed by all patients to signify approval to
participate. All patients received the partial or radical
nephrectomy during the period of June 2019 to September 2019 and
the ccRCC diagnosis was determined by at least two independent
specialists for pathological analysis in our center. The HPLC-MS
process was divided into the following steps:
i) Sample collection and preparation: A total
of 27 paired ccRCC tissues, including 27 paired tumor and
non-cancerous normal tissues (NATs), were collected for further
analysis. The ccRCC tissues were resected from the center of the
tumor without necrotic regions. NATs were defined as tissues at
least 5 mm away from the cancer capsule and no tumor invasion was
identified under microscopy. All tissue samples were collected from
the operation room and transferred directly into the −80°C
refrigerator for storage until final analysis took place.
A total of ~25–120 mg of each cryo-pulverized renal
tumor tissue or NATs were homogenized separately in an appropriate
volume of lysis buffer [i.e. 2% SDS, 20 mM Tris, Cocktail (1:100),
DNAse (1:100), RNAse (1:1,000)] by repeated vortexing. Protein
concentration was determined using Pierce™ BCA assay protein assay
kit (Thermo Fisher Scientific, Inc.). A total of 100 mg of protein
was reduced with 20 mM dithiothreitol (DTT) for 5 min at 95°C and
subsequently alkylated with 50 mM iodoacetamide for 45 min at room
temperature in the dark. Protein digestion was carried out using a
filter-aided sample preparation technique method. Proteins were
loaded onto 30-kDa filter devices (Pall Life Sciences). Trypsin
(Trypsin Gold, mass spec grade, Promega Corporation) was added at
an enzyme to protein ratio of 1:50, and samples were incubated at
37°C overnight.
ii) ESI-LC-MS/MS for proteome library
generation: The pooled peptide samples (30 µl) of each group
were separated by high-pH RPLC columns (4.6×250 mm, C18, 3 µm;
Waters Corporation). Each pooled sample was loaded onto a column in
buffer A1 (‘H2O’, pH 10). The elution gradient was
between 5–30% buffer B1 (90% ACN, pH 10; flow rate, 1 ml/min) for
30 min. Eluted peptides were collected at one fraction per min.
After lyophilization, 30 fractions were resuspended in 0.1% formic
acid, before being concatenated into 10 fractions by combining
fractions 1, 11, 21 and so forth.
In order to generate a spectral library, fractions
from RPLC were analyzed in the DDA mode. Parameters were set as
follows: the MS was recorded at 350-1,500 m/z at a resolution of
60,000 m/z; the maximum injection time was 50 ms, the auto gain
control (AGC) was 1e6, and the cycle time was 3 sec. MS/MS scans
were performed at a resolution of 15,000 with an isolation window
of 1.6 Da and a collision energy at 32% (HCD); the AGC target was
50,000, and the maximum injection time was 30 ms.
iii) ESI-LC-MS/MS for proteome data-independent
acquisition analysis: Digested peptides were dissolved in 0.1%
formic acid and separated on an RP C18 self-packing capillary LC
column (75 µm × 150 mm, 3 µm). The eluted gradient was 5–30% buffer
B2 (0.1% formic acid, 99.9% ACN; flow rate, 0.3 µl/min) for 60 min.
For MS acquisition, the variable isolation window DIA method with
38 windows was developed. The specific window lists were
constructed based on the DDA experiment of the pooled sample. The
full scan was set at a resolution of 120,000 over the m/z range of
400 to 900, followed by DIA scans with a resolution of 30,000; the
HCD collision energy was 32%, the AGC target was 1E6 and the
maximal injection time was 50 ms.
iv) Spectral library generation: To generate
a comprehensive spectral library, a pooled sample from each group
was processed. DDA data were processed using Proteome Discoverer
software (Thermo Fisher Scientific, Inc.) and searched against the
human UniProt database (https://sparql.uniprot.org/) appended with the iRT
fusion protein sequence (Biognosys AG).
A maximum of two missed cleavages for trypsin were
used, cysteine carbamidomethylation was set as a fixed
modification, and methionine oxidation deamination and +43 on Kn
(Carbamyl) were used as variable modifications. Parent and fragment
ion mass tolerances were set to 10 ppm and 0.02 Da, respectively.
The applied false discovery rate (FDR) cutoff was 0.01 at the
protein level. The results were then imported to Spectronaut Pulsar
software (Biognosys AG) to generate the spectral library.
Additionally, DIA data were imported into
Spectronaut Pulsar software and searched against the human UniProt
database to generate DIA library. The final library was generated
by combining DDA and DIA libraries of ccRCC and controls.
v) Data analysis: DIA-MS data were analyzed
using Spectronaut Pulsar (Biognosys AG) with default settings. All
results were filtered with a Q-value cutoff of 0.01 which
corresponded to an FDR of 1%. Proteins identified in more than 50%
of the samples in each group were retained for further analysis.
Missing values were imputed based on the k-nearest neighbor method.
Raw proteomics data were transformed using log2 and then
centralized. Wilcoxon signed rank test was implemented with the
software R version 4.1.1 (R-project.org).
Selection of m6A RNA methylation regulators:
After integrating RNA sequencing and proteomic data, 19 m6A RNA
methylation regulators were included for further analysis and
included: ALKBH5, CBLL1, ELAVL1, FMR1, FTO, FXR2, HNRNPA2B1,
HNRNPC, METTL 14, METTL 3, RBM15, RBM15B, RBMX, RBMXL1, WTAP,
YTHDC1, YTHDC2, YTHDF2 and YTHDF3.
Uni-variate survival analysis and
establishment of prognostic model based on the 19 m6A methylation
regulators
To assess the survival impact of every single m6A
methylation regulators on early stage ccRCC, univariate survival
analysis was applied. Firstly, pROC package (14) was applied to determine the optimal
threshold of every single gene expression and then all the ccRCC
patients were divided into two subgroups, namely high and low
subgroups. Then, Kaplan-Meier survival analysis with log rank test
was calculated and displayed with package ‘survminer’ (15).
Aiming to construct a comprehensive prognostic
model, all 19 m6A methylation regulators were included into
multivariate Cox's regression analysis with risk scores for each
sample being calculated. Prognostic risk scores for ccRCC patients
were calculated using the following formula: Risk score=∑ðβi × Exp
iÞ, with i representing the number of prognostic m6A expression
levels.
According to the median of risk score, all samples
were divided into two subgroups: high risk and low risk, and
survival curves and time-dependent ROC curves were generated. The
Human Protein Atlas (16) was
searched to assess the protein expression level of m6A methylation
regulators within the prognostic model.
Multivariate Cox analysis and nomogram
development for survival predictions
In order to assess the independence of the
diagnostic model, certain critical clinical parameters of the
patients were included into the multivariate analysis, including
sex, age, tumor side, pathological T stage, N stage and tissue
grade. Furthermore, these factors were applied using the ‘rms’
package (17) to develop a
survival time nomogram.
Correlations between the prognostic
model and tumor infiltrating immune cells
To assess the relationship between prognostic model
and tumor purity, R package ‘estimate’ (18) was used to calculate tumor purity,
the scores of stromal cells and the infiltration level of immune
cells. To further explore the immune cell composition within the
tumor microenvironment, Tumor Immune Estimation Resource (TIMER,
http://timer.cistrome.org/) was used to
compute the proportion of six kinds of immune cells. Spearman's
correlation analysis was used to investigate correlations between
risk score and immune infiltrating cells. In addition, associations
between clinical characteristics, tumor purity and risk scores were
analyzed using the standard Kruskal-Wallis test for multi-group
(Dunn's test was used for the post hoc analysis) and Wilcoxon
signed rank test for two-group comparisons, respectively.
Statistical analysis
All the data analysis and the figures were completed
by R (version 4.1.1) as well as corresponding R packages
illustrated in the Methods. The non-parametric Spearman's rank
correlation analysis was used to calculate the correlation
coefficient. All the tests were two-sided, and P<0.05 was
considered to indicate a statistically significant difference.
Results
Baseline information
Clinical characteristics for 261 TCGA participants
and 27 paired proteomic samples have been provided in Table I. At the end of the follow-up
period, 44 of 261 patients had succumbed while none of the 27
patients had experienced tumor recurrence or succumbed. The number
of men in this cohort was slightly greater than women with a ratio
of 158:103 for TCGA and 20:7 in the proteomic sample. In addition,
the average age within the two datasets was similar at 59 years for
the TCGA group and 57 years for the proteomic sample.
 | Table I.Baseline information of enrolled
patients from TCGA database and our cohort. |
Table I.
Baseline information of enrolled
patients from TCGA database and our cohort.
| Clinicopathological
characteristics | TCGA (n=261) | Our paired sample
(n=27) |
|---|
| Survival
status |
|
|
|
Alive | 217 | 27 |
|
Dead | 44 | 0 |
| Sex |
|
|
|
Male | 158 | 20 |
|
Female | 103 | 7 |
| Age | 59 (26–90) | 57 (24–71) |
| Laterality |
|
|
|
Left | 116 | 10 |
|
Right | 145 | 7 |
| Pathological
stage |
|
|
| G1 | 13 | 4 |
| G2 | 155 | 19 |
| G3 | 86 | 4 |
| G4 | 7 | 0 |
| T stage |
|
|
|
T1a | 135 | 13 |
|
T1b | 105 | 14 |
| T1 | 21 | 0 |
| N stage |
|
|
| NX | 159 | 0 |
| N0 | 102 | 27 |
| Neoadjuvant
therapy |
|
|
|
Yes | 7 | 0 |
| No | 254 | 27 |
Expression levels of 19 m6A RNA
methylation regulators in genome, transcriptome and proteome
Firstly, the gene mutation pattern of the 19 m6A
methylation regulator genes was examined (Fig. 1A). It was identified that the
mutation frequency was relatively low with a rate of 4.69%. Among
the 9 mutated samples, YTHDC2 demonstrated the highest mutation
frequency at 2% and was followed by WTAP with 1%. Furthermore,
missense mutation was the most common mutation type with a rate of
3.6%.
Expression levels for RNA sequencing and proteomic
data were presented in Fig. 1B and
C, respectively. It was revealed that there existed 11
differentially expressed genes, including 4 upregulated and 7
downregulated m6A regulators (Fig.
1B). According to the level of proteins, 15 aberrantly
expressed m6A regulators were detected, except for FMR1, FTO,
METTLE14 and METTLE3 (Fig.
1C).
Identification of survival-related m6A
methylation regulators
In order to identify survival-related m6A
methylation regulators, single-variate Cox analysis was performed
and the results were provided in a forest plot (Fig. 2A). It could be clearly observed
that the expression of FMR 1, METTL14, RBMXL1 and YTHDF 2 were
significantly associated with early-stage ccRCC survival. Notably,
all 4 regulators were negatively associated with ccRCC overall
survival having hazard ratios (HR) of less than 1. After
calculating the best cut-off value of each gene with ROC curves,
the survival curves for m6A regulators were plotted. The results
suggested that 8 gene expression levels, namely RBMXL1, YTHDF3,
FMR1, METTLE14, YTHDF2, ELAVL1, FXR2 and RBM 15 were associated
with ccRCC patient survival and the top three are revealed in
Fig. 2B-D.
Construction and validation of m6A
methylation regulators—based prognostic signature
In order to comprehensively describe the
predictability of m6A methylation regulators, all 19 m6A regulators
were included to construct a prognostic model. After applying
multivariate Cox regression analysis, a risk score algorithm was
established: Risk score=(−0.54) × expr (YTHDF2) + 0.67 × expr
(YTHDC2) + (−1.05) × expr (FMR1) + 1.43 × expr (ELAVL1). Then, all
261 patients were classified into two subgroups according to the
median risk score, namely high risk and low risk. Through survival
analysis plots, it could be clearly observed that a higher risk
score was significantly associated with poorer ccRCC survival
(Fig. 3A). To assess the YTHDF2,
YTHDC2, FMR1 and ELAVL1 expression, it was found that the
immunohistochemistry results from the Human Protein Atlas were in
accordance with the risk score. Compared with normal kidney tissue,
the expression levels of YTHDF2 and FMR1 within tumor cells were
low. In contrast to this situation, the YTHDC2 and ELAVL1
expression levels were upregulated, suggesting the unfavorable role
in predicting ccRCC survival (Fig.
S1).
To further assess the accuracy of the prognostic
model, a ROC curve was generated with an AUC of 65.46% (Fig. 3B). To examine the independency of
the model, multivariate Cox analysis with several confirmed
critical clinical characteristics was performed and the results
showed that the model remained an independent indicator of the
survival risk (Fig. 3C).
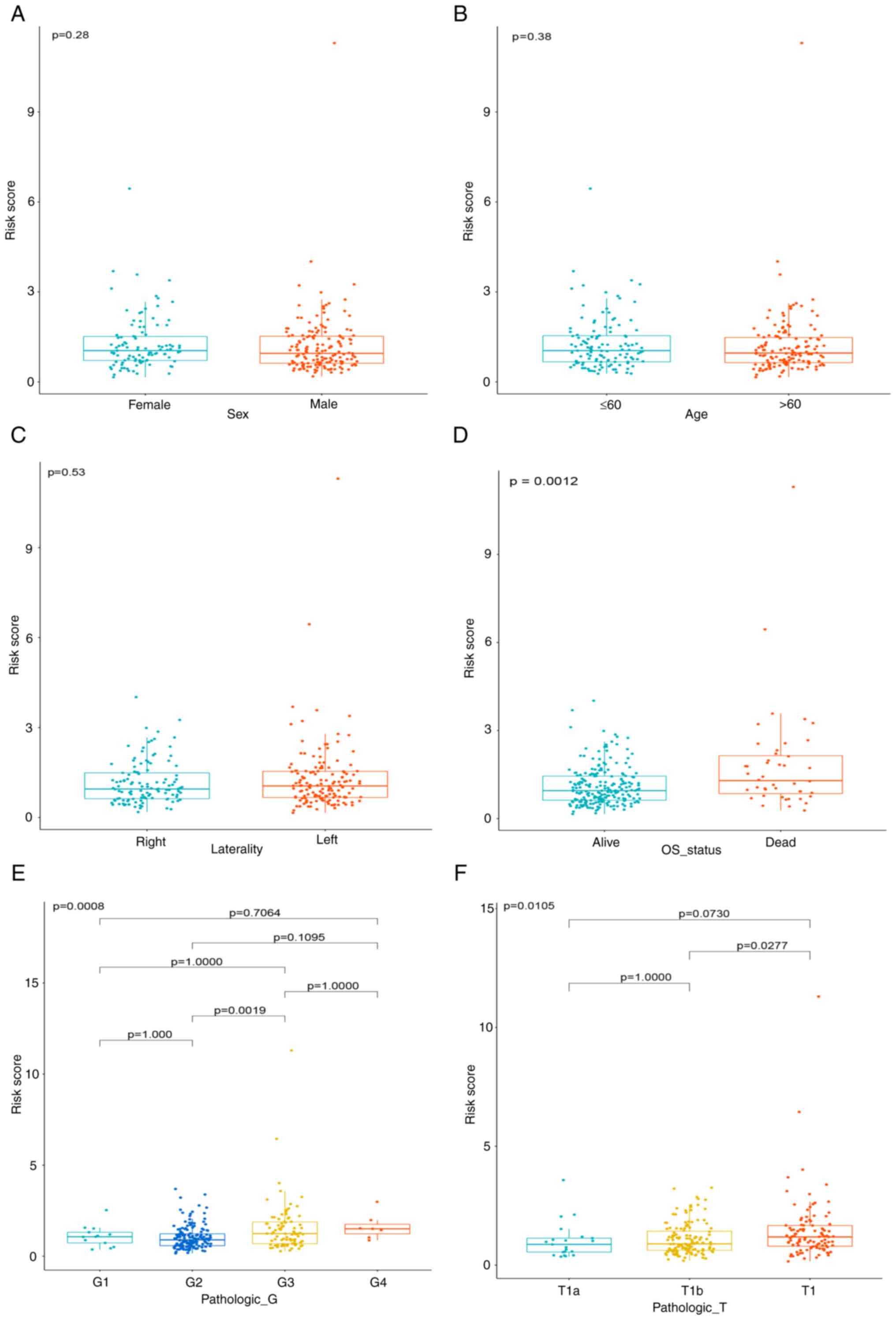
Furthermore, the association between risk scores and
clinical characteristics were also assessed (Fig. 4A-F). The results suggested that
risk scores are significantly associated with survival status. A
cluster heat map was generated to show RNA expression level for all
regulators included in the model (Fig.
5).
Establishing a prognostic nomogram for
early-stage ccRCC
To predict survival status more comprehensively and
easily, a nomogram was constructed after integrating prognostic
risk score and clinical information such as sex, age, laterality,
pathological grade, T stage and N stage (Fig. 6). It was revealed that the nomogram
is suitable for predicting 1-year, 2-year, 3-year and 5-year
survival for ccRCC patients.
Correlations between risk scores and
immune cells in tumor microenvironment
m6A methylation regulators participate in the
regulation of tumor microenvironment immune status during
tumorigenesis (6). After comparing
the tumor purity within different risk subgroups, it was found that
the higher risk group was significantly associated with higher
immune score (P<0.001) and lower tumor purity (P=0.0066).
However, the stromal score within high and low risk groups did not
show significant difference (P=0.68) (Fig. 7A-C).
To further confirm which kind of immune cells maybe
associated with the prognostic model, Spearman's correlation
analysis was implemented to assess the association between risk
scores and immune cell composition (Fig. 7D-I). The results revealed that risk
scores were statistically associated with various immune cells,
including dendritic cells, CD4+ T cells, CD8+
T cells and macrophages as opposed to B cells and neutrophils.
Discussion
As the most common type of renal cancer, the
prognosis of ccRCC remains poor with both a high incidence and
mortality. Epigenetic modifications, particularly RNA modifications
have proven to play various functions in the bioprocess of
tumorigenesis and therefore produce a frame of tumor biomarkers
(19). This should not be
overlooked as a key element in the quest to identify elements which
will enable clinicians to identify cases early and to determine
those who are more likely to require swift interventions. As the
most prevalent type of RNA post-translational modification, m6A has
drawn an increasing attention since the identification of the first
RNA demethylase in 2011, the fat mass and obesity—associated
protein (FTO), revealing that the RNA methylation is a reversible
process (20). Numerous studies
(21,22) have revealed that FTO is closely
related to BMI, which is closely related to the tumorigenesis of
ccRCC. But after assessing the relationship between BMI and the
proteomic expression of FTO with our cohort, no significant
correlation was observed (Fig.
S2). Due to the incomplete clinical data of TCGA database, the
correlation within the 261 patients was not analyzed, which is a
disadvantage of the aforementioned analysis.
In the present study, multi-omics were applied to
reveal the prognostic role of m6A methylation regulators in
early-stage ccRCC. To the best of our knowledge, the present study
is the first one to suggest that m6A could predict the prognosis
for pT1 stage ccRCC patients. From the 19 m6A methylation
regulators, the expression of four genes were significantly
associated with overall survival for ccRCC patients, namely: FMR1,
METTL14, RBMXL1 and YTHDF2. To further predict survival, a mode
consisting of YTHDF2, YTHDC2, FMR1 and ELAVL1 was constructed based
on Cox's regression analysis, which was verified from
immunohistochemistry. Additionally, multivariate Cox analysis
revealed that the signature was an independent predictor of ccRCC.
In order to render our model more applicable, a nomogram that could
be more easily adopted into clinical practice was established,
although this is also only an initial study. Previous studies found
that ccRCC is an immune-rich tumor since multiple kinds of
immune—infiltrating cells exist in the tumor microenvironment
(10,23,24).
Therefore, the relationship between the generated prognostic model
and immune cells were also assessed with the help of online TIMER
software and ‘ESTIMATE’ package. The results suggested that the m6A
methylation prognostic model was significantly associated with
dendritic cells, CD4+ T cells, CD8+ T cells
and macrophages. This may provide new evidence for the role of m6A
methylation in regulating tumor immune-infiltrating
microenvironment; however, of course further interdisciplinary
research is required.
In our analysis and relative study (25), the molecule YTHDF2 appears to play
a critical role since this was not only associated with survival
status but also made up a proportion of the prognostic signature.
Additionally, protein expression for YTHDF2 between cancer and
non-cancerous normal tissues remains differential in the samples
examined in the present study. As a member of m6A-binding proteins
(i.e. readers), YTHDF2 possesses a YTH binding capability which may
interact with m6A modifications for specific RNAs. Therefore, this
ability may play a critical role in improving target mRNA
translation efficiency (26).
Previous studies have reported that YTHDF2 may
participate in the progression of various malignant tumors. For
example, Zhang et al (27)
found that by maintaining m6A methylation of the OCT4 mRNA, YTHDF2
enhances its protein expression and therefore promotes liver cancer
stem cell phenotype. This appears to suggest that YTHDF2 acts as an
oncogene in the progression of hepatic cell carcinoma (HCC).
However conversely, another study has demonstrated that YTHDF2 is a
novel tumor suppressor for HCC since it mechanistically suppresses
STAT3 phosphorylation and tumor growth by degrading IL-6 family
cytokine-encoded mRNAs (28).
Similarly, in glioma studies, YTHDF2 has been found to downregulate
the expression of LXRα and HIVEP2 through m6A-dependent mRNA decay
and therefore may impact on the survival of glioma patients
(29). Xie et al (30) revealed that METTL3/YTHDF2 m6A axis
may directly degrade mRNAs of tumor suppressors such as SETD7 and
KLF4, which can lead to carcinogenesis and progression of bladder
cancer.
Another important molecule in the prognostic model
is FMR1, which has been found to be involved in the negative
regulation of mRNA translation. Recently, the FMR1 was identified
as a putative m6A reader which means that it binds to m6A
containing transcripts. For example, Edens et al (31) showed that FMRP, the protein encoded
by FMR1, preferentially binds m6A-modified mRNAs and thereby
promotes their nuclear export through CMR1, thus regulating neutral
differentiation (32). Notably,
FMRP could also bind to m6A sites in targets and interact with
YTHDF2, which is yet another ‘m6A reader’, in an RNA-independent
manner. The results suggested that the function of FMR1 was
maintaining the stability of its mRNA targets while YTHDF2 focused
on the degradation of these transcripts (33).
As an m6A writer, METTL14 mainly performed its
function through combining with METTL3 and forming an m6A-METTL
complex (MAC). In contrast to the evidenced catalytic function of
METTL3, METTL14 appears to be mainly responsible for maintaining
MAC integrity and mediating RNA bind (34). Although recently, METTL14 has also
been reported to participate in crosstalk between histone
modification and RNA methylation. METTL14 thereby could recognize
H3K36me3 and facilitate binding between m6A MTC and the adjacent
RNA polymerase II, thereby writing m6A into actively transcribed
nascent RNAs (35). Zhang et
al (36) reported that
METTL14-mediated m6A modification negatively regulates mRNA
stability of bromodomain PHD finger transcription factor, therefore
facilitating the tumor metastasis and reprogramming glycolysis of
RCC. This suggested that the functions of METTL14-mediated m6A are
likely to be multifaceted and demand further research.
Another reader, ELAVL1 also participated in the
composition of the prognostic model. Embryonic lethal abnormal
visual protein 1 (ELAVL1), known as RNA binding proteins, also
called human antigen R (HuR) has been reported to participate in
various processes, including nervous system development, cellular
proliferation and migration (37).
Through binding to specific RNAs, ELAVL1 could increase mRNA
stability of target genes. Ronkainen et al (38) reported that ELAVL1 was associated
with reduced RCC-specific survival and inner mechanism study
revealed that the HuR protein could regulate the expression of
COX-2 in RCC cells, which maybe a putative mechanism of action for
the progression of RCC. In addition, Danilin et al (39,40)
identified that knockdown of HuR in RCC cell lines could inhibit
proliferation and induce apoptosis, the process of which was
mediated by inhibiting the PI3K/Akt and MAPK oncogenic signaling
pathways. All the evidences suggested that the molecular ‘HuR’ may
be potential drug target for ccRCC.
As a retrospective study, it is necessary to discuss
the limitations involved. First, the sample size for the present
study was relatively small which inhibits our ability to generalize
findings to a broader community. A total of 261 cases with
early-stage ccRCC were initially enrolled from the TCGA database
and all the subsequent analysis was based on this data. Even though
the data is reliable, the sample size remains small for
high-throughput omics research. Second, the favorable prognosis,
which may be due to a short follow-up period and pathologically pT1
ccRCC, is another shortcoming of the present study. Regarding the
survival of enrolled patients from our center, the final event was
not found until now since they received the surgery within the
period of June 2019 to September 2019. In addition, all the
enrolled patients were pT1 ccRCC with pT2 excluded due to the fact
that tumorigenesis is a gradual process and pT1 ccRCC may reflect
the original status of ccRCC, highlighting the significance of
molecular changes of pT1 ccRCC. In the future, our follow-up time
will be extended, more patients will be enrolled and the clinical
outcomes will be reported to the journal if necessary. To sum up, a
larger cohort with much longer follow-up period is required. Third,
the lack of external validation of the established prognostic model
is also another limitation. GEO dataset, one of the most widely
used dataset, has been searched. Nevertheless, it was found that no
studies have complete prognostic information as well as RNA
sequencing data regarding pT1 ccRCC. Finally, all our analysis was
based on the multi-omics data. In order to explore the concrete
effect of critical m6A RNA methylation regulators on ccRCC
tumorigenesis, more in vitro and in vivo experiments
are required to validate the result in the future.
In conclusion, a prognostic model of early-stage
ccRCC with m6A methylation regulators was established. The
evidence-based model could predict survival for early stage ccRCC
patients and is closely related to various tumor infiltrating
immune cells.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article. To protect the privacy of
patients, other information could be only acquired with the
permission of the corresponding author.
Authors' contributions
ZW and YuZ designed the study, wrote and revised the
manuscript. ZW and MZ performed experiments, data analysis and
collection and wrote the manuscript. GZ, WW, YaZ, SS and XW
collected data and revised the paper. SS conducted statistical
analysis and language editing. All authors have read and approved
the final manuscript. ZW and YuZ confirm the authenticity of all
the raw data.
Ethics approval and consent to
participate
The present study was conducted in accordance with
the Declaration of Helsinki and was approved (approval no.
KS2021034) by the Institutional Review Board of Peking Union
Medical College Hospital, Chinese Academy of Medical Science and
Peking Union Medical College (Beijing, China). Formal written
consent was provided by all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Capitanio U, Bensalah K, Bex A, Boorjian
SA, Bray F, Coleman J, Gore JL, Sun M, Wood C and Russo P:
Epidemiology of renal cell carcinoma. Eur Urol. 75:74–84. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Obeng RC, Arnold RS, Ogan K, Master VA,
Pattaras JG, Petros JA and Osunkoya AO: Molecular characteristics
and markers of advanced clear cell renal cell carcinoma: Pitfalls
due to intratumoral heterogeneity and identification of genetic
alterations associated with metastasis. Int J Urol. 27:790–797.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Downs TM, Schultzel M, Shi H, Sanders C,
Tahir Z and Sadler GR: Renal cell carcinoma: Risk assessment and
prognostic factors for newly diagnosed patients. Crit Rev Oncol
Hematol. 70:59–70. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Motzer RJ, Jonasch E, Agarwal N, Alva A,
Baine M, Beckermann K, Carlo MI, Choueiri TK, Costello BA, Derweesh
IA, et al: Kidney cancer, version 3.2022, nccn clinical practice
guidelines in oncology. J Natl Compr Canc Netw. 20:71–90. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li N, Kang Y, Wang L, Huff S, Tang R, Hui
H, Agrawal K, Gonzalez GM, Wang Y, Patel SP and Rana TM: ALKBH5
regulates anti-PD-1 therapy response by modulating lactate and
suppressive immune cell accumulation in tumor microenvironment.
Proc Natl Acad Sci USA. 117:20159–20170. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu H, Xu Z, Wang Z, Ren Z, Li L and Ruan
Y: Exosomes from dendritic cells with Mettl3 gene knockdown prevent
immune rejection in a mouse cardiac allograft model.
Immunogenetics. 72:423–430. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang L, Hui H, Agrawal K, Kang Y, Li N,
Tang R, Yuan J and Rana TM: m6A RNA methyltransferases
METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J.
39:e1045142020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu X, Wang P, Teng X, Zhang Z and Song S:
Comprehensive analysis of expression regulation for RNA m6A
regulators with clinical significance in human cancers. Front
Oncol. 11:6243952021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fang J, Hu M, Sun Y, Zhou S and Li H:
Expression profile analysis of m6A RNA methylation regulators
indicates they are immune signature associated and can predict
survival in kidney renal cell carcinoma. DNA Cell Biol.
39:2194–2211. 2020. View Article : Google Scholar
|
|
11
|
Wang J, Zhang C, He W and Gou X: Effect of
m6A RNA methylation regulators on malignant progression
and prognosis in renal clear cell carcinoma. Front Oncol. 10:32020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mayakonda A, Lin DC, Assenov Y, Plass C
and Koeffler HP: Maftools: Efficient and comprehensive analysis of
somatic variants in cancer. Genome Res. 28:1747–1756. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Robin X, Turck N, Hainard A, Tiberti N,
Lisacek F, Sanchez JC and Müller M: pROC: An open-source package
for R and S+ to analyze and compare ROC curves. BMC Bioinformatics.
12:772011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kassambara A, Kosinski M and Biecek P:
Survminer: Drawing survival curves using ‘ggplot2’. R package
version 0.4.9. 2021.https://CRAN.R-project.org/package=survminer
|
|
16
|
Uhlén M, Fagerberg L, Hallström BM,
Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C,
Sjöstedt E, Asplund A, et al: Tissue-based map of the human
proteome. Science. 347:12604192015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Harrell FE Jr: Rms: Regression modeling
strategies. R package version 6.2-0. 2021.https://cran.r-project.org/web/packages/rms/index.html
|
|
18
|
Yoshihara K, Kim H and Verhaak RG:
Estimate: Estimate of stromal and immune cells in malignant tumor
tissues from expression data. R package version 1.0.13/r21.
2016.https://r-forge.r-project.org/projects/estimate/
|
|
19
|
Shen C, Xuan B, Yan T, Ma Y, Xu P, Tian X,
Zhang X, Cao Y, Ma D, Zhu X, et al: m6A-dependent
glycolysis enhances colorectal cancer progression. Mol Cancer.
19:722020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang T, Kong S, Tao M and Ju S: The
potential role of RNA N6-methyladenosine in cancer progression. Mol
Cancer. 19:882020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wojciechowski P, Lipowska A, Rys P, Ewens
KG, Franks S, Tan S, Lerchbaum E, Vcelak J, Attaoua R, Straczkowski
M, et al: Impact of FTO genotypes on BMI and weight in polycystic
ovary syndrome: A systematic review and meta-analysis.
Diabetologia. 55:2636–2645. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zarza-Rebollo JA, Molina E and Rivera M:
The role of the FTO gene in the relationship between depression and
obesity. A systematic review. Neurosci Biobehav Rev. 127:630–637.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qiu Y, Wang X, Fan Z, Zhan S, Jiang X and
Huang J: Integrated analysis on the N6-methyladenosine-related long
noncoding RNAs prognostic signature, immune checkpoints, and immune
cell infiltration in clear cell renal cell carcinoma. Immun Inflamm
Dis. 9:1596–1612. 2021. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu T, Gao S, Ruan H, Liu J, Liu Y, Liu D,
Tong J, Shi J, Yang H, Chen K and Zhang X: METTL14 Acts as a
potential regulator of tumor immune and progression in clear cell
renal cell carcinoma. Front Genet. 12:6091742021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mu Z, Dong D, Sun M, Li L, Wei N and Hu B:
Prognostic value of YTHDF2 in clear cell renal cell carcinoma.
Front Oncol. 10:15662020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lan Q, Liu PY, Haase J, Bell JL,
Hüttelmaier S and Liu T: The critical role of RNA m6A
methylation in cancer. Cancer Res. 79:1285–1292. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang C, Huang S, Zhuang H, Ruan S, Zhou
Z, Huang K, Ji F, Ma Z, Hou B and He X: YTHDF2 promotes the liver
cancer stem cell phenotype and cancer metastasis by regulating OCT4
expression via m6A RNA methylation. Oncogene. 39:4507–4518. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hou J, Zhang H, Liu J, Zhao Z, Wang J, Lu
Z, Hu B, Zhou J, Zhao Z, Feng M, et al: YTHDF2 reduction fuels
inflammation and vascular abnormalization in hepatocellular
carcinoma. Mol Cancer. 18:1632019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fang R, Chen X, Zhang S, Shi H, Ye Y, Shi
H, Zou Z, Li P, Guo Q, Ma L, et al: EGFR/SRC/ERK-stabilized YTHDF2
promotes cholesterol dysregulation and invasive growth of
glioblastoma. Nat Commun. 12:1772021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xie H, Li J, Ying Y, Yan H, Jin K, Ma X,
He L, Xu X, Liu B, Wang X, et al: METTL3/YTHDF2 m(6)A axis promotes
tumorigenesis by degrading SETD7 and KLF4 mRNAs in bladder cancer.
J Cell Mol Med. 24:4092–4104. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Edens BM, Vissers C, Su J, Arumugam S, Xu
Z, Shi H, Miller N, Ringeling FR, Ming GL, He C and Song H: FMRP
modulates neural differentiation through m(6)a-dependent mRNA
nuclear export. Cell Rep. 28:845–854.e845. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hsu PJ, Shi H, Zhu AC, Lu Z, Miller N,
Edens BM, Ma YC and He C: The RNA-binding protein FMRP facilitates
the nuclear export of N6-methyladenosine-containing
mRNAs. J Biol Chem. 294:19889–19895. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang
W, Song H, Wu H, Shu Q and Jin P: Fragile X mental retardation
protein modulates the stability of its m6A-marked messenger RNA
targets. Hum Mol Genet. 27:3936–3950. 2018.PubMed/NCBI
|
|
34
|
Liu L, Wang Y, Wu J, Liu J, Qin Z and Fan
H: N 6-Methyladenosine: A potential breakthrough for
human cancer. Mol Ther Nucleic Acids. 19:804–813. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang H, Weng H, Zhou K, Wu T, Zhao BS,
Sun M, Chen Z, Deng X, Xiao G, Auer F, et al: Histone H3
trimethylation at lysine 36 guides m6A RNA modification
co-transcriptionally. Nature. 567:414–419. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang C, Chen L, Liu Y, Huang J, Liu A, Xu
Y, Shen Y, He H and Xu D: Downregulated METTL14 accumulates BPTF
that reinforces super-enhancers and distal lung metastasis via
glycolytic reprogramming in renal cell carcinoma. Theranostics.
11:3676–3693. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang F, Hu A, Li D, Wang J, Guo Y, Liu Y,
Li H, Chen Y, Wang X, Huang K, et al: Circ-HuR suppresses HuR
expression and gastric cancer progression by inhibiting CNBP
transactivation. Mol Cancer. 18:1582019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ronkainen H, Vaarala MH, Hirvikoski P and
Ristimäki A: HuR expression is a marker of poor prognosis in renal
cell carcinoma. Tumour Biol. 32:481–487. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Danilin S, Sourbier C, Thomas L, Rothhut
S, Lindner V, Helwig JJ, Jacqmin D, Lang H and Massfelder T: Von
hippel-lindau tumor suppressor gene-dependent mRNA stabilization of
the survival factor parathyroid hormone-related protein in human
renal cell carcinoma by the RNA-binding protein HuR.
Carcinogenesis. 30:387–396. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Danilin S, Sourbier C, Thomas L, Lindner
V, Rothhut S, Dormoy V, Helwig JJ, Jacqmin D, Lang H and Massfelder
T: Role of the RNA-binding protein HuR in human renal cell
carcinoma. Carcinogenesis. 31:1018–1026. 2010. View Article : Google Scholar : PubMed/NCBI
|