Introduction
Liver cancer is the sixth most commonly diagnosed
cancer in the world, with ~906,000 new cases and 830,000 cases of
mortality in 2020 (1). Moreover,
liver cancer is characterized by late onset, rapid progression and
high rates of metastasis (2). As
the majority of patients newly diagnosed with liver cancer are
already at advanced stages of disease, this disease is associated
with a poor prognosis and high fatality rate (3). Comorbidities such as cirrhosis,
hydroperitoneum and severe liver dysfunction render advanced liver
cancer almost untreatable, even using surgical resection or
ablation (4).
Epithelial-mesenchymal transition (EMT) is defined as the
transformation of epithelial cells into a phenotype with
mesenchymal cells, increasing the metastatic capacity and
therapeutic resistance of the tumor (5). Furthermore, the application of
chemotherapy is limited by the occurrence of adverse side effects
and the development of resistance (6). Therefore, novel treatment options are
in high demand. Over the past decade, naturally occurring compounds
have received widespread attention as potential anti-cancer drugs
or sensitization agents (7).
Cisplatin (DDP) is a chemotherapeutic agent that is
widely applied in clinical practice and induces cytotoxicity via
binding to DNA to induce cell apoptosis (8). The Fas receptor and mitochondrial
signaling pathways serve key roles in the apoptotic process of
tumor cells following DNA damage induced by DDP (9). DDP is highly effective in the
treatment of various tumors, such as testicular, ovarian, bladder,
cervical, esophageal, small-cell lung cancers and liver cancer
(8). However, it has significant
disadvantages, with the most important being cancer cell resistance
and numerous harmful side effects (10–12).
High DDP doses are one of the main causes of drug resistance and
side effects, which frequently result in treatment failure
(13). Therefore, enhancing the
anti-tumor activity of DDP, whilst decreasing the potency of its
adverse effects, has become a key focus of cancer research.
Artemisinin and its derivatives have been widely
used for the treatment of malaria since it was discovered and
extracted by Tu Yoyo from Artemisia annua L., Asteraceae, a
herb that has been recorded and used in ancient China for >2,000
years (14). Dihydroartemisinin
(DHA) is produced from artemisinin via the reduction of the C-10
carbonyl group, which improves bioavailability and solubility
(15,16). In addition to its anti-malarial
properties, previous studies have reported the anti-cancer
potential of DHA, including against lung cancer, breast cancer and
digestive system tumors (17,18).
The induction of apoptosis in tumor cells is an important mechanism
underlying the anti-tumor effects of DHA. Previous study
demonstrated that blocking the induction of cell apoptosis after
the formation of the DDP-DNA adducts is responsible for DDP
resistance (9). Therefore, it can
be hypothesized that DDP combined with DHA may increase the
apoptotic rate of tumor cells. Moreover, the ability of tumor cells
to proliferate and migrate should be explored following treatment
with DDP combined with DHA.
In the present study, HepG2 cells were used as an
in vitro model of liver cancer to investigate the effects of
DHA combined with DDP. It was hypothesized that the efficacy of a
lower dose of DDP combined with DHA was equal to or even greater
than that of a higher dose of DDP alone. The aim of the present
study was to investigate the role of DHA in combination with DDP
for the treatment of liver cancer and to provide experimental data
for the clinical use of DHA.
Materials and methods
Cell culture and reagents
The liver cancer HepG2 cell line was purchased from
The Cell Bank of Type Culture Collection of The Chinese Academy of
Sciences. The cells were cultured in RPMI-1640 medium (HyClone;
Cytiva), supplemented with 10% FBS (Hangzhou Sijiqing Biological
Engineering Materials Co., Ltd.), 100 U/ml penicillin and 100 U/ml
streptomycin at 37°C in a humidified incubator supplied with 5%
CO2. The culture medium was replaced every 1–2 days. DHA
(Fig. 1A) was purchased from
Dalian Meilun Biology Technology Co., Ltd. DDP was purchased from
Qilu Pharmaceutical Co., Ltd.
Cell grouping
As previously described (19,20),
HepG2 cells were treated at 37°C for 24 h with six treatment groups
as follows. i) Control (0 µM DHA and 0 µg/ml DDP); ii) DHA (100 µM
DHA); iii) medium-dose (MD) DDP (15 µg/ml DDP); iv) high-dose (HD)
DDP (20 µg/ml DDP); v) DHA + low-dose (LD) DDP (100 µM DHA combined
with 10 µg/ml DDP); and vi) DHA + MD DDP (100 µM DHA combined with
15 µg/ml DDP).
Cell counting kit-8 (CCK-8) assay
Cells were treated with different concentrations of
DHA (37.5, 50, 75, 100, 150, 300 and 400 µM) and DDP (5, 7.5, 10,
15, 20, 30 and 40 µg/ml) for 24 h or aforementioned concentrations
of DHA and/or DDP 24–48 h at 37°C and their effects on cell
viability was assayed by CCK-8 assay. Specifically, cell suspension
(200 µl) containing a total of 4×103 cells was seeded
into each well of a 96-well plate. After 12 h of incubation at 37°C
in a humidified atmosphere containing 5% CO2, the
culture medium in each well was replaced with indicated
concentration of DHA or DDP. In total, six duplicate wells were
used for each group. The cells were treated for 24 h at 37°C,
following which the culture medium was replaced with 200 µl PBS
containing 10% CCK-8 (Dalian Meilun Biology Technology Co., Ltd.).
Following 2 h of incubation, the viability of HepG2 cells was
assessed at an absorbance of 450 nm using a microplate reader. Cell
viability was quantified using the following formula: Inhibitory
rate (%)=[(1-optical density at 450 nm for the treatment)/optical
density at 450 nm for the control] ×100.
Colony formation assay
HepG2 cells were seeded into a six-well plate at a
density of 500 cells/well for 2 days at 37°C, the aforementioned
concentrations of DHA and/or DDP were added to the wells followed
by incubation for 12 h at 37°C, before the medium was replaced with
fresh RPMI-1640 medium supplemented with 10% FBS, 100 U/ml
penicillin and 100 U/ml streptomycin. After 2 weeks of incubation
at 37°C with 5% CO2, the colonies were fixed using
anhydrous ethanol (≥99.7%) for 15 min at room temperature and
stained with 10% Giemsa (Beyotime Institute of Biotechnology) for
15 min at room temperature, before the number of colonies (>50
cells) in each well was quantified using ImageJ 1.8.0. (National
Institutes of Health).
Flow cytometry
The FITC-Annexin V/PI Apoptosis Kit was purchased
from Tianjin Sungene Biotech Co., Ltd. 1×106 per well
HepG2 cells were treated with the aforementioned concentrations of
DHA and/or DDP for 24 h at 37°C. Following treatment, cells were
collected using trypsin without EDTA, Cells were resuspended in 400
µl binding buffer followed by incubation with 5 µl FITC-Annexin V
for 30 min and 10 µl PI for 30 min at room temperature, which were
added in turn, in the dark. The fluorescent cells were then
detected using a BD FACSCalibur™ flow cytometer (BD
Biosciences) and the results were analyzed using FlowJo X 10.0.7
(FlowJo LLC).
Acridine orange (AO)/ethidium bromide
(EB) dual staining assay
AO/EB fluorescence staining was used to detect any
morphological changes associated with cell apoptosis. HepG2 cells
were seeded onto a round coverslip placed in six-well plates at a
density of 5×105 cells per well. After 12 h of
incubation at 37°C, the medium in each group was replaced with the
aforementioned concentrations of DHA and/or DDP before further
incubation for 24 h at 37°C. Cells that attached to the coverslips
were stained using AO/EB (AO, cat. no. 158550-10G; EB, cat. no.
E8751-5G; Sigma-Aldrich; Merck KGaA) for 5 min at room temperature.
Fluorescence microscopy (model 80i; Nikon Corporation) was used to
observe the morphology of any apoptotic cells in five randomly
selected fields of view. The apoptotic rate was assessed as a
percentage from the number of apoptotic cells among 1,000 cells
counted per well.
Wound healing assay
A total of 1×106 per well HepG2 cells
were seeded in six-well plate. Following incubation for 12 h at
37°C, confluent cell monolayers were scratched using 20-µl pipette
tips, before any cells detached were washed away with PBS. Images
were then captured of each group at 0 h using an inverted light
microscope (model CK-40; Olympus Corporation) before the cells were
incubated with serum-free medium at 37°C containing the
aforementioned concentrations of DHA and/or DDP for 24 h. Wound
healing images were obtained at the same location in the well,
which was marked. The migration rate was determined via
quantification of the area change of the wound using ImageJ 1.8.0.
(National Institutes of Health).
Transwell migration assay
The migratory ability of HepG2 cells was assessed
using a Transwell chamber (8 µm; Corning, Inc.). Cells were seeded
into the upper chamber at a density of 2×105 cells/well
and were suspended in serum-free medium after being treated with
the aforementioned concentrations of DHA and/or DDP in a six-well
plate for 24 h at 37°C. The bottom chamber contained 600 µl medium
containing 10% FBS. After being incubated at 37°C in 5%
CO2 for 24 h, cells that had failed to migrate and
remained on the upper membrane surface were wiped away using cotton
swabs. Migrated cells were fixed in anhydrous ethanol (≥99.7%) at
room temperature for 10 min and stained with 10% Giemsa (Beyotime
Institute of Biotechnology) at room temperature for 15 min.
Migratory cells were then manual counted in five random fields
using an inverted light microscope (model CK-40; Olympus
Corporation) at ×200 magnification.
Gelatin zymography for the detection
of MMP-9
HepG2 cells were seeded into a six-well plate at
density of 1×106 per well and treated with the
aforementioned concentrations of DHA and/or DDP for 24 h at 37°C.
Subsequently, 30 µl supernatant from each group was collected and
resuspended in non-reducing Laemmli sample buffer and separated
using SDS-PAGE on an 8% gel containing 1 mg/ml gelatin (Shanghai
Yuanye Bio-technology, Co., Ltd.). Following electrophoresis, gels
were washed using 2.5% Triton X-100 to remove any SDS before being
incubated in substrate buffer (50 mM Tris buffer containing 5 mM
CaCl2; pH 8) for 18 h at 37°C. Gels were then stained
with 0.5% Coomassie Brilliant Blue R-250 (Wuhan Servicebio
Technology Co., Ltd.) for 3 h at room temperature, followed by
de-staining for 2 h at room temperature. Gelatinolytic activity was
visualized via imaging the negative staining bands using a digital
camera. The acquired image results are analyzed by ImageJ 1.8.0.
(National Institutes of Health).
Western blotting
HepG2 cells were treated with the aforementioned
concentrations of DHA and/or DDP for 24 h at 37°C before being
lysed on ice using RIPA lysis buffer (Beyotime Institute of
Biotechnology) containing proteinase inhibitors. Protein samples in
the lysate were quantified using a BCA kit (Beyotime Institute of
Biotechnology) before being mixed with 5X loading buffer and 20 µg
of protein in each lane was denatured for separation using SDS-PAGE
on a 10% gel. The PVDF membrane onto which the protein was
transferred was blocked with 5% skimmed milk at room temperature
for 1 h. The membranes were incubated with primary antibodies
against the following proteins at 4°C for 12 h: Fas (1:1,000; cat.
no. K008079P; Beijing Solarbio Science & Technology Co., Ltd.),
Fas-associated death domain (FADD; 1:1,000; cat. no. K008372P;
Beijing Solarbio Science & Technology Co., Ltd.), pro-caspase-3
(1:1,000; cat. no. sc-7272; Santa Cruz Biotechnology, Inc.),
cleaved caspase-3 (1:1,000; cat. no. Asp175; Cell Signaling
Technology, Inc.), pro-caspase-8 (1:1,000; cat. no. bsm33190M;
Thermo Fisher Scientific, Inc.), cleaved caspase-8 (1:1,000; cat.
no. Asp384; Cell Signaling Technology, Inc.), Bax (1:1,000; cat.
no. K008076P; Beijing Solarbio Science & Technology Co., Ltd.),
Bcl-2 (1:1,000; cat. no. K003505P; Beijing Solarbio Science &
Technology Co., Ltd.), N-cadherin (1:1,000; cat. no. bs-1172R;
BIOSS) and E-cadherin (1:1,000; cat. no. bs-1519R; BIOSS). The
membranes were then washed by TBST (0.1% Tween-20) and incubated
with goat anti-rabbit IgG secondary antibody (1:1,000; cat. no.
SE134; Beijing Solarbio Science & Technology Co., Ltd.) and
goat anti-mouse IgG secondary antibody (1:1,000; cat. no.
bs-40296G-HRP; BIOSS) conjugated with HRP at room temperature for 2
h. β-actin was detected using anti-β-actin antibody (1:1,000; cat.
no. K200058M; Beijing Solarbio Science & Technology Co., Ltd.)
and was used as the loading control. Following incubation with the
secondary antibody and ECL reagent (Wuhan Servicebio Biotechnology
Co., Ltd.) the blots were imaged using a gel imaging system
(ChemiDoc XRS+; Bio-Rad Laboratories, Inc.). The blots were
semi-quantified using Image Lab™ software (version 5.0;
MCM DESIGN).
Statistical analysis
SPSS 26.0 (IBM Corp.) software was used for data
analysis. Data are presented as the mean ± SD. One-way ANOVA
followed by Tukey's post-hoc test was used for statistical
comparisons among more than two groups. An unpaired Student's
t-test were used for comparisons between two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
DHA enhances the effect of DDP on
inhibiting HepG2 cell viability and proliferation
CCK-8 assay was used to detected the cell viability
and the result showed that Both DHA and DDP exerted significant
inhibitory effects on HepG2 cells compared with the untreated
control group (P<0.05; Fig. 1B and
C). When the combination group was compared with the
monotherapy group, there was no significant difference between the
MD DDP group and the DHA + LD DDP group after treatment for 24 h
(P>0.05). The inhibition rate was significantly enhanced in the
DHA + MD DDP group compared with the HD DDP group following
treatment for 24 h (P<0.05). After treatment for 48 h, the
inhibition rate of both combination groups was significantly
enhanced compared with the DDP-only groups (P<0.05; DHA + LD DDP
vs. MD DDP and DHA + MD DDP vs. HD DDP) (Fig. 1D). Furthermore, the number of
colonies formed was significantly decreased in the combination
groups compared with that of the DDP-only groups (Fig. 1E and F; P<0.05; DHA + LD DDP vs.
MD DDP and DHA + MD DDP vs. HD DDP). These data suggested that DHA
may have enhanced the inhibitory effects of DDP on HepG2 cell
proliferation.
DHA combined with DDP induces
apoptosis and morphological changes in HepG2 cells
AO/EB dual-staining was used to detect the apoptosis
of HepG2 cells. Typically, green/yellow fluorescence is emitted in
healthy cells stained with AO/EB, whereas apoptotic cells emit
orange/red fluorescence. The results demonstrated that fluorescent
green cells were the majority in the control group, with the number
of cells fluorescing red or orange being markedly increased after
treatment with DHA or DDP and increasing particularly in the
combination groups treated with DHA and DDP (Fig. 2A). Furthermore, combined treatment
significantly increased the apoptosis of HepG2 cells compared with
the DDP-only groups (P<0.05; DHA + LD DDP vs. MD DDP and DHA +
MD DDP vs. HD DDP; Fig. 2B). As
the percentage of apoptotic cells increased, the density of cells
in the DHA and/or DDP treatment groups markedly decreased and the
cell morphology became rounded and shrunken (data not shown).
 | Figure 2.Apoptosis of HepG2 cells treated with
DHA (100 µM) and/or DDP. (A) Representative image of HepG2 cells
treated with DHA and/or DDP for 24 h and dual-stained with AO/EB.
Scale bar, 50 µm, and arrows point to apoptotic cells.
(magnification, ×100). (B) Apoptotic rate of HepG2 cells was
assessed using the AO/EB dual-staining assay. (C) Effect of DHA
and/or DDP on the apoptosis of HepG2 cells was quantified via
Annexin V-FITC/PI-staining flow cytometry. (D) Apoptotic rate of
HepG2 cells assessed using the Annexin V-FITC/PI-staining flow
cytometry assay. Data are presented as the mean ± SD of more than
three independent experiments. *P<0.05 vs. control; #P<0.05.
DHA, dihydroartemisinin; DDP, cisplatin; AO, acridine orange; EB,
ethidium bromide; LD, low dose (10 µg/ml); MD, medium dose, (15
µg/ml); HD, high dose, (20 µg/ml). |
Cell apoptosis was also detected using flow
cytometry. Significantly increased apoptosis was detected in all
treatment groups compared with the control group (P<0.05).
Compared with the DDP-only groups, the apoptotic rate was
significantly increased in the combined groups (P<0.05; DHA + LD
DDP vs. MD DDP and DHA + MD DDP vs. HD DDP) (Fig. 2C and D). These results were
consistent with the results of the dual AO/EB staining. In summary,
DHA + LD DDP treatment resulted in apoptotic effects significantly
greater than those observed with MD DDP, whereas DHA + MD DDP
exerted a significantly greater apoptotic effect compared with HD
DDP. These observations suggested that the extent of apoptosis in
HepG2 cells induced by DDP could be enhanced by DHA.
DHA combined with DDP suppresses the
migration of HepG2 cells
The migratory ability of HepG2 cells was detected
using the wound healing and Transwell assays. Cells migrated to the
center of the wound in the control group but this was significantly
inhibited in the other treatment groups compared with the control
(P<0.05; Fig. 3A). Moreover,
the migration rate of HepG2 cells in the combined treatment groups
were significantly more inhibited compared with the DDP-only
treatment groups (P<0.05; DHA + LD DDP vs. MD DDP and DHA + MD
DDP vs. HD DDP; Fig. 3B).
Furthermore, the Transwell assay results demonstrated that a large
number of cells in the control group migrated to the lower
compartment, which demonstrated a potent migratory ability of HepG2
cells. However, the number of migratory cells decreased
significantly when treated with DHA and/or DDP compared with the
control. Combination treatment groups exhibited significantly
greater reductions in migratory ability compared with the DDP-only
treatment groups (P<0.05; DHA + LD DDP vs. MD DDP and DHA + MD
DDP vs. HD DDP) (Fig. 3C and D).
Taken together, these data suggested that DHA combined with DDP may
have inhibited the migration of HepG2 cells.
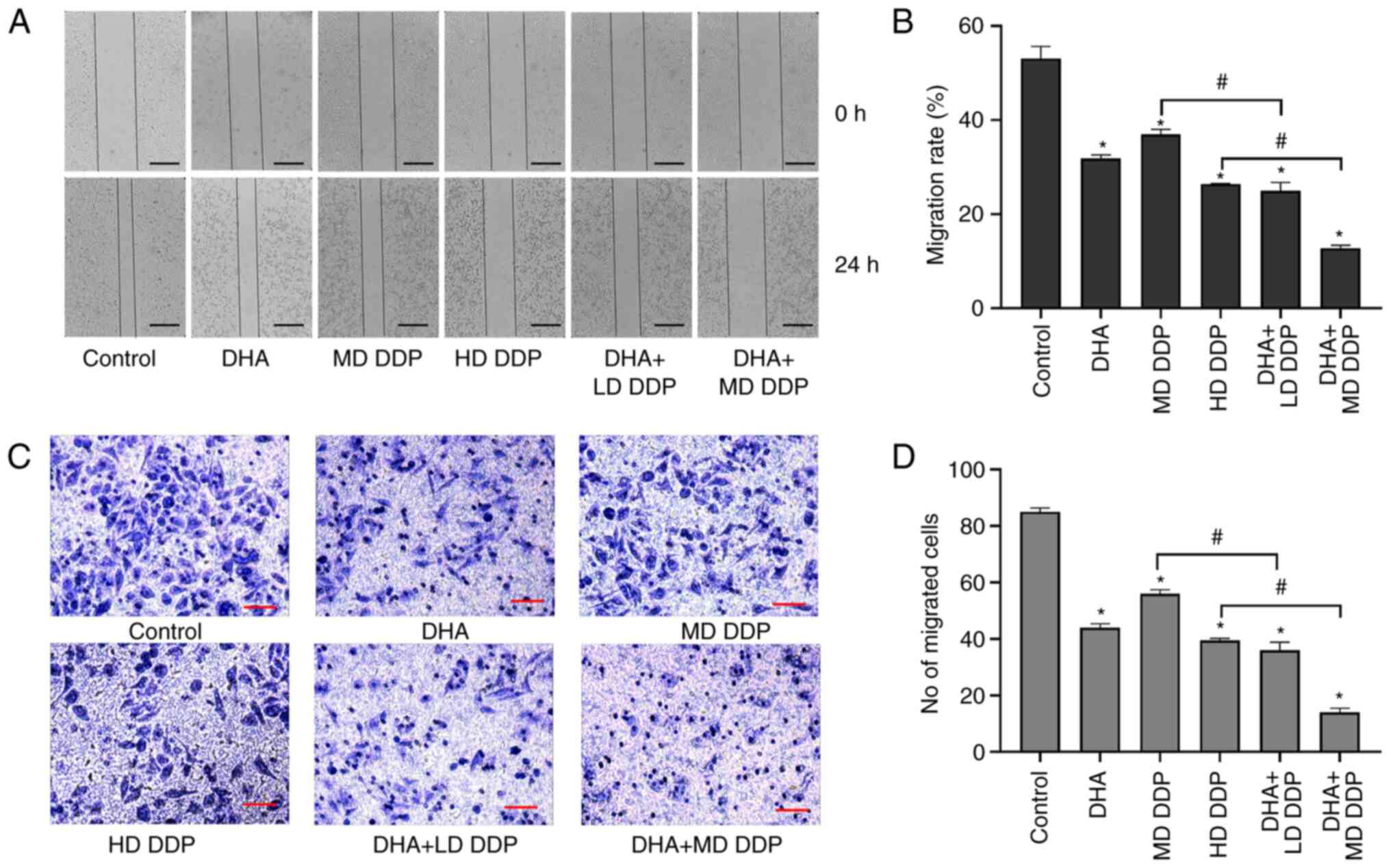 | Figure 3.Migration abilities of HepG2 cells
treated with DHA (100 µM) and/or DDP were detected using the wound
healing assay and Transwell assay. (A) Representative images of the
wound healing assay at 0 and 24 h of HepG2 cells being treated with
DHA and/or DDP. Scale bar, 2 mm. (B) Migration rate of HepG2 cells
assessed using the wound healing assay. (C) Images of HepG2 cells
that have migrated to the lower Transwell chamber following
treatment with DHA and/or DDP. Scale bar is 25 µm (magnification,
×200). (D) Mean number of migrated cells in five fields of view per
group. Data are presented as the mean ± SD of more than three
independent experiments. *P<0.05, vs. control; #P<0.05. DHA,
dihydroartemisinin; DDP, cisplatin; LD, low dose (10 µg/ml); MD,
medium dose, (15 µg/ml); HD, high dose, (20 µg/ml). |
DHA in combination with DDP modulates
the expression of apoptosis-related proteins
To determine if the expression levels of proteins
associated with cell apoptosis were affected by DHA combined with
DDP treatment in HepG2 cells, protein samples from HepG2 cells
treated with DHA and/or DDP were semi-quantified using western
blotting (Fig. 4A-C). The relative
protein expression levels were semi-quantified via gray value
analysis (Fig. 4D). Significant
increases in Fas FADD and Bax expression levels in DHA and/or DDP
treatment groups were detected compared with that of the control
group (P<0.05). Furthermore, this effect was significantly
greater in the combination groups compared with the DDP-only groups
(P<0.05; DHA + LD DDP vs. MD DDP and DHA + MD DDP vs. HD DDP).
The ratio of cleaved caspase-3 to pro-caspase-3 and the ratio of
cleaved caspase-8 to pro-caspase-8 were significantly increased in
the DHA and DDP combination groups compared with the DDP-only
groups (P<0.05). The relative protein expression levels of Bcl-2
were significantly decreased in the combination groups compared
with the DDP-only groups. Overall, these data suggested that DHA
potentially worked synergistically with DDP to enhance cell
apoptosis via the modulation of Fas death receptor signaling
pathway activity and the mitochondrial apoptosis signaling pathway
in HepG2 cells.
 | Figure 4.Apoptosis-associated proteins in
HepG2 cells treated with DHA (100 µM) and/or DDP were detected
using western blotting. Effect of DHA and/or DDP on the protein
expression levels of (A) Fas and FADD, (B) pro-caspase-3, cleaved
caspase-3, pro-caspase-8 and cleaved caspase-8 and (C) Bax and
Bcl-2 in HepG2 cells. β-actin was used as the loading control. (D)
Relative protein expression levels of Fas, FADD, pro-caspase-3,
cleaved caspase-3, pro-caspase-8, cleaved caspase-8, Bax and Bcl-2.
Data are presented as the mean ± SD of more than three independent
experiments. *P<0.05 vs. control; #P<0.05. DHA,
dihydroartemisinin; DDP, cisplatin; FADD, Fas-associated death
domain; LD, low dose (10 µg/ml); MD, medium dose, (15 µg/ml); HD,
high dose, (20 µg/ml). |
DHA in combination with DDP regulates
cadherin and MMP-9 expression
To further explore the mechanism underlying the
effects of DHA combined with DDP on the migration of HepG2 cells,
E-cadherin and N-cadherin protein expression levels were assessed
in HepG2 cells using western blotting. Furthermore, the expression
of MMP-9 in HepG2 cell culture supernatant was analyzed using
gelatin zymography. The combined use of DHA and DDP significantly
increased the protein expression levels of E-cadherin but
significantly decreased the protein expression levels of N-cadherin
compared with the untreated control and DDP-only groups (P<0.05;
DHA + LD DDP vs. MD DDP and DHA + MD DDP vs. HD DDP; Fig. 5A). The expression of MMP-9 was
significantly decreased when DHA was combined with DDP compared
with the DDP-only groups (P<0.05; DHA + LD DDP vs. MD DDP and
DHA + MD DDP vs. HD DDP) (Fig. 5E and
F). In conclusion, DHA combined with DDP regulated expression
of EMT-related protein and decreased the expression of MMP-9 in
HepG2 cells.
Discussion
DDP-based chemotherapy serves an important role in
the treatment of malignancies (21). However, drug resistance and adverse
side effects have largely limited its application and efficacy
(22). Therefore, the exploration
of a novel DDP sensitizer agent would be of great therapeutic
significance (23,24). Over the past decade, several
naturally-occurring compounds have been reported to exert
anti-tumor properties and can increase the efficacy of existing
chemotherapeutic agents, while conferring minimal side effects and
complications (25). A previous
study reported that berberine, a compound extracted from Huang Lian
and other Chinese medicinal herbs, displayed synergistic effects
with DDP in inducing necroptosis and apoptosis of ovarian cancer
cells. This was caused via the promotion of the expression of
caspase-3, caspase-8, receptor interacting serine/threonine kinase
3 and mixed lineage kinase domain-like pseudokinase (24). Moreover, Abe et al (26) reported that caffeine citrate
significantly improves the anti-cancer effects of DDP on
osteosarcoma and fibrosarcoma cells via the inhibition of DNA
repair induced by DDP. In the present study, DHA was demonstrated
to enhance the anti-cancer effects of DDP, which provided
additional evidence of the viability of DHA as a sensitizer of DDP
for liver cancer therapy.
DHA is a naturally-occurring compound, that is
primarily used for the treatment of malaria, and which exhibits
superior bioavailability compared with artemisinin (27). Previous clinical and laboratory
studies have demonstrated that DHA confers minimal adverse side
effects (28,29). Furthermore, a previous study
reported that DHA exerts potent cytotoxic effects on liver cancer
cells, including on the HepG2, Huh-7, BEL-7404 and Hep3B cell
lines, but not on normal non-cancerous human liver cells (30). DHA has been previously explored as
a potential sensitizer via synergy with other clinical anti-cancer
agents, which enhanced their anti-cancer activity (31–33).
Here, CCK-8 results revealed that both DHA and DDP alone inhibited
HepG2 cell viability in a dose-dependent manner. However, the
combination with DHA showed to achieve higher dose effects than
even the lower dose of DDP, suggesting the possibility of reducing
the dosage of chemotherapeutic agents by combination. Subsequent
colony formation assays further validated the role of this
combination in inhibiting HepG2 cell proliferation. Further study
demonstrated that combining DHA and DDP significantly enhanced
apoptosis and prevented cell migration in the HepG2 cell line,
which supported the action of DHA as a viable DDP sensitizer in the
aforementioned studies. These results provided primary experimental
evidence for the therapeutic application of DHA in the treatment of
liver cancer.
DNA damage and the activation of poly (ADP-ribose)
polymerase to induce tumor cell apoptosis are important mechanisms
underlying the anti-tumor effects of DDP (8). It has been previously reported that
mitochondria and the Fas signaling pathway serve a key role in this
type of apoptosis (34,35). However, suppression of apoptosis
increases tumor cell drug resistance to DDP (9,36,37).
According to previous studies, the Fas receptor signaling pathway
and the mitochondrial apoptosis signaling pathway are both closely
associated with DHA-induced tumor cell apoptosis (38,39).
In the present study, apoptosis of HepG2 cells treated with DHA
and/or DDP and the protein expression levels of Fas, FADD,
pro-caspase-3, cleaved caspase-3, pro-caspase-8, cleaved caspase-8,
Bax and Bcl-2 were all assessed. This suggested that the Fas
receptor signaling pathway and the mitochondrial apoptosis
signaling pathway potentially serve a vital role in DHA/DDP-induced
apoptosis in liver cancer cells.
The epithelial-mesenchymal transition (EMT) is
mainly associated with the migration and invasion of tumor cells
and is one of the main factors of tumor progression (40). Numerous studies have previously
demonstrated that the EMT serves a key contributing role in
chemoresistance (41,42). Furthermore, E-cadherin and
N-cadherin are known markers of the EMT, whereas MMP-9 can degrade
the extracellular matrix following the EMT (43). In the present study, the migratory
ability of liver cancer cells was significantly inhibited by DHA
combined with DDP. In studying the potential molecular mechanisms
of cell metastasis, enzyme profiling and protein blotting showed
that DDP combined with DHA treatment downregulated the expression
levels of N-calmodulin and MMP-9, as well as up-regulated
expression of E-calmodulin in HepG2 cells. Based on this, we
hypothesized that DDP can inhibit EMT in HepG2 cells in combination
with DHA, and is the mechanism by which DHA improves DDP drug
resistance.
In conclusion, DHA significantly enhanced the
effects of DDP on the proliferation, apoptosis and migration of
HepG2 cells. These results suggested that DHA has the potential for
application as a novel anti-tumor sensitizer of DDP and provided an
experimental basis for the use of DHA as a clinical anti-tumor drug
in combination with DDP. Further studies are required to assess DNA
damage and the effect on the cell cycle in liver cancer cells of
this combined treatment. Furthermore, in vivo study of this
combination therapy will also be necessary in subsequent
studies.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Chinese Academy of
Medical Sciences Innovation Fund for Medical Sciences (grant no.
2018-I2M-AI-015) and the National Science Foundation of China
(grant no. 81702920).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QR performed the experiments, analyzed the data and
drafted the manuscript. RL performed the western blotting
experiments. HY, LX and BH performed the cell culture assay and
analyzed the data. GZ and FW designed the study, conceived the
experiments, laid out the experimental scheme, confirm the
authenticity of all the raw data, and revised the manuscript. All
the authors read and approved the final version of the manuscript
to be published.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang Y, Liu Y, Liu Y, Zhou W, Wang H, Wan
G, Sun D, Zhang N and Wang Y: A polymeric prodrug of cisplatin
based on pullulan for the targeted therapy against hepatocellular
carcinoma. Int J Pharm. 483:89–100. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
El Dika I, Makki I and Abou-Alfa GK:
Hepatocellular carcinoma, novel therapies on the horizon. Chin Clin
Oncol. 10:122021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Anwanwan D, Singh SK, Singh S, Saikam V
and Singh R: Challenges in liver cancer and possible treatment
approaches. Biochim Biophys Acta Rev Cancer. 1873:1883142020.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gurzu S, Kobori L, Fodor D and Jung I:
Epithelial mesenchymal and endothelial mesenchymal transitions in
hepatocellular carcinoma: A review. Biomed Res Int.
2019:29625802019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Demir T, Lee SS and Kaseb AO: Systemic
therapy of liver cancer. Adv Cancer Res. 149:257–294. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dutta S, Mahalanobish S, Saha S, Ghosh S
and Sil PC: Natural products: An upcoming therapeutic approach to
cancer. Food Chem Toxicol. 128:240–255. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ghosh S: Cisplatin: The first metal based
anticancer drug. Bioorg Chem. 88:1029252019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fuertes MA, Alonso C and Pérez JM:
Biochemical modulation of cisplatin mechanisms of action:
Enhancement of antitumor activity and circumvention of drug
resistance. Chem Rev. 103:645–662. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oun R and Rowan E: Cisplatin induced
arrhythmia; electrolyte imbalance or disturbance of the SA node?
Eur J Pharmacol. 811:125–128. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pabla N and Dong Z: Cisplatin
nephrotoxicity: Mechanisms and renoprotective strategies. Kidney
Int. 73:994–1007. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Santos NAGD, Ferreira RS and Santos ACD:
Overview of cisplatin-induced neurotoxicity and ototoxicity, and
the protective agents. Food Chem Toxicol. 136:1110792020.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Astolfi L, Ghiselli S, Guaran V, Chicca M,
Simoni E, Olivetto E, Lelli G and Martini A: Correlation of adverse
effects of cisplatin administration in patients affected by solid
tumours: A retrospective evaluation. Oncol Rep. 29:1285–1292. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miller LH and Su X: Artemisinin: Discovery
from the Chinese herbal garden. Cell. 146:855–858. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma N, Zhang Z, Liao F, Jiang T and Tu Y:
The birth of artemisinin. Pharmacol Ther. 216:1076582020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Haynes RK: From artemisinin to new
artemisinin antimalarials: Biosynthesis, extraction, old and new
derivatives, stereochemistry and medicinal chemistry requirements.
Curr Top Med Chem. 6:509–537. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dai X, Zhang X, Chen W, Chen Y, Zhang Q,
Mo S and Lu J: Dihydroartemisinin: A potential natural anticancer
drug. Int J Biol Sci. 17:603–622. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen X, He LY, Lai S and He Y:
Dihydroartemisinin inhibits the migration of esophageal cancer
cells by inducing autophagy. Oncol Lett. 20:942020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu L, Pang Y, Qin G, Xi G, Wu S, Wang X
and Chen T: Farnesylthiosalicylic acid sensitizes hepatocarcinoma
cells to artemisinin derivatives. PLoS One. 12:e01718402017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang R, Niu Y and Zhou Y: Increase the
cisplatin cytotoxicity and cisplatin-induced DNA damage in HepG2
cells by XRCC1 abrogation related mechanisms. Toxicol Lett.
192:108–114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Raudenska M, Balvan J, Fojtu M, Gumulec J
and Masarik M: Unexpected therapeutic effects of cisplatin.
Metallomics. 11:1182–1199. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kiss RC, Xia F and Acklin S: Targeting DNA
damage response and repair to enhance therapeutic index in
cisplatin-based cancer treatment. Int J Mol Sci. 22:81992021.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun Y, Jiang W, Lu W, Song M, Liu K, Chen
P, Chang A, Ling J, Chiao PJ, Hu Y and Huang P: Identification of
cisplatin sensitizers through high-throughput combinatorial
screening. Int J Oncol. 53:1237–1246. 2018.PubMed/NCBI
|
|
24
|
Liu L, Fan J, Ai G, Liu J, Luo N, Li C and
Cheng Z: Berberine in combination with cisplatin induces
necroptosis and apoptosis in ovarian cancer cells. Biol Res.
52:372019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rejhová A, Opattová A, Čumová A, Slíva D
and Vodička P: Natural compounds and combination therapy in
colorectal cancer treatment. Eur J Med Chem. 144:582–594. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abe K, Yamamoto N, Hayashi K, Takeuchi A
and Tsuchiya H: Caffeine citrate enhanced cisplatin antitumor
effects in osteosarcoma and fibrosarcoma in vitro and in vivo. BMC
Cancer. 19:6892019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tu Y: The discovery of artemisinin
(qinghaosu) and gifts from Chinese medicine. Nat Med. 17:1217–1220.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Angus B: Novel anti-malarial combinations
and their toxicity. Expert Rev Clin Pharmacol. 7:299–316. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wan X, Zhong H, Pan W, Li Y, Chen Y, Li N
and Tang B: Programmed release of dihydroartemisinin for
synergistic cancer therapy using a CaCO3 mineralized
metal-organic framework. Angew Chem Int Ed Engl. 58:14134–14139.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Im E, Yeo C, Lee HJ and Lee EO:
Dihydroartemisinin induced caspase-dependent apoptosis through
inhibiting the specificity protein 1 pathway in hepatocellular
carcinoma SK-Hep-1 cells. Life Sci. 192:286–292. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li Q, Ma Q, Cheng J, Zhou X, Pu W, Zhong X
and Guo X: Dihydroartemisinin as a sensitizing agent in cancer
therapies. Onco Targets Ther. 14:2563–2573. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tai X, Cai XB, Zhang Z and Wei R: In
vitro and in vivo inhibition of tumor cell viability by
combined dihydroartemisinin and doxorubicin treatment, and the
underlying mechanism. Oncol Lett. 12:3701–3706. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jin H, Jiang AY, Wang H, Cao Y, Wu Y and
Jiang XF: Dihydroartemisinin and gefitinib synergistically inhibit
NSCLC cell growth and promote apoptosis via the Akt/mTOR/STAT3
pathway. Mol Med Rep. 16:3475–3481. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dasari S, Njiki S, Mbemi A, Yedjou CG and
Tchounwou PB: Pharmacological effects of cisplatin combination with
natural products in cancer chemotherapy. Int J Mol Sci.
23:15322022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qi L, Luo Q, Zhang Y, Jia F, Zhao Y and
Wang F: Advances in toxicological research of the anticancer drug
cisplatin. Chem Res Toxicol. 32:1469–1486. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Makovec T: Cisplatin and beyond: Molecular
mechanisms of action and drug resistance development in cancer
chemotherapy. Radiol Oncol. 53:148–158. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou J, Kang Y, Chen L, Wang H, Liu J,
Zeng S and Yu L: The drug-resistance mechanisms of five
platinum-based antitumor agents. Front Pharmacol. 11:3432020.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mao H, Gu H, Qu X, Sun J, Song B, Gao W,
Liu J and Shao Q: Involvement of the mitochondrial pathway and
Bim/Bcl-2 balance in dihydroartemisinin-induced apoptosis in human
breast cancer in vitro. Int J Mol Med. 31:213–218. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hu YJ, Zhang JY, Luo Q, Xu JR, Yan Y, Mu
LM, Bai J and Lu WL: Nanostructured dihydroartemisinin plus
epirubicin liposomes enhance treatment efficacy of breast cancer by
inducing autophagy and apoptosis. Nanomaterials (Basel). 8:8042018.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang Y, Liu Y, Xiang L, Han L, Yao X, Hu Y
and Wu F: Cyclin D1b induces changes in the macrophage phenotype
resulting in promotion of tumor metastasis. Exp Biol Med (Maywood).
246:2559–2569. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bao Y, Zhang Y, Lu Y, Guo H, Dong Z, Chen
Q, Zhang X, Shen W, Chen W and Wang X: Overexpression of microRNA-9
enhances cisplatin sensitivity in hepatocellular carcinoma by
regulating EIF5A2-mediated epithelial-mesenchymal transition. Int J
Biol Sci. 16:827–837. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Du B and Shim JS: Targeting
epithelial-mesenchymal transition (EMT) to overcome drug resistance
in cancer. Molecules. 21:9652016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pastushenko I and Blanpain C: EMT
transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
















