Introduction
Hemophagocytic syndrome (HPS), also known as
hemophagocytic lymphohistiocytosis (HLH), is an immune-mediated
syndrome with a typically rapid progression (1). HPS is a serious disease, the main
clinical manifestations of which are fever, hemocytopenia,
ferremia, hypertriglyceridemia, hepatosplenomegaly and
hemophagocytosis affecting the bone marrow, liver, spleen or lymph
nodes (2). HPS is classified into
two distinct forms: Primary or familial HPS and secondary HPS.
Secondary HPS may develop as a consequence of strong immune
activation due to severe infections, malignancies or autoimmune
diseases. Currently, the most common secondary HPS is
lymphoma-associated HPS (LAHS), which has been reported in 20–67%
of patients (3–5).
LAHS has been extensively described in occidental
countries (6,7). However, there maybe marked
differences between the Asian population and patients from western
countries. It is notable that natural killer/T-cell lymphoma and
T-cell lymphoma are rare in western countries but very common in
Asian countries (6). In addition,
the most common pathological type in Asian populations is NK/T-cell
LAHS (NK/T-LAHS), which accounts for 35% of patients with LAHS
(8).
There is no unified understanding of the clinical
features and prognostic factors of LAHS. As the clinical
manifestations and treatment response of LAHS are not only affected
by HPS itself but are also associated with lymphoma, the prognosis
is often poor (9,10). At present, there is no standard and
effective first-line treatment for LAHS, although the HLH-1994
treatment protocol is the most widely used. However, a clinical
study revealed that 30% of patients with HPS did not respond to the
standard HLH-1994 regimen and died during the first few weeks of
treatment, and the efficacy of this regimen in patients with LAHS
was particularly poor (11). A
study of 137 patients with HPS in Germany showed that the median
survival duration of patients with idiopathic HPS was 248 days and
that of patients with infection-associated HPS was 641 days
(12). Chang et al
(13) studied the prognosis of
patients with LAHS and observed a median survival time of 43 days
for all patients, with B-cell LAHS (B-LAHS) and NK/T-LAHS having
median survival times of 55 and 40 days, respectively. Furthermore,
Liu et al (14) also found
that the prognosis of patients with NK/T-LAHS was poor, with a
mortality rate of 96.4% and median survival time of only 15 days;
its prognosis is worse than that of other types of HPS (3,6,9,15).
In addition, a study of 567 patients with HPS in Japan between 2001
and 2005 showed that patients with NK/T-LAHS had the worst
prognosis, with a 5-year survival rate of <15%, followed by
familial HPS and B-LAHS (3).
Numerous studies have confirmed that the prognosis of patients with
LAHS is relatively poor (3–5,16,17).
Due to its rarity and the heterogeneity of inducing
factors and clinical outcomes, the management of LAHS remains
challenging. Few reports have focused on LAHS due to its low
incidence rate. Therefore, the present study retrospectively
analyzed the clinical features, treatment and prognosis of LAHS
with the goal of providing valuable guidance for the diagnosis and
treatment of patients with LAHS.
Patients and methods
Patients
The 139 patients with lymphoma and LAHS (LAHS group)
and 185 patients with HPS that was not associated with lymphoma
(non-LAHS group) who were admitted to The First Affiliated Hospital
of Medical School of Zhejiang University (Hangzhou, China) and the
Cancer Hospital of the University of Chinese Academy of Sciences
(Hangzhou, China) between January 2014 and February 2021 were
enrolled in the present study. These patients were retrospectively
analyzed in a clinical study designated as ‘Lymphoma-Associated
Hemophagocytic Syndrome’. All patients met the following inclusion
criteria: Adolescent and adult patients aged ≥14 years; met the
HLH-2004 diagnostic criteria (18); and evidence of malignant lymphoma
for the LAHS group. Exclusion criteria: Did not meet the HLH-2004
diagnostic criteria or met the diagnostic criteria but had not
received treatment; and no evidence of malignant lymphoma for the
non-LAHS group. The diagnosis of malignant lymphoma was based on
the 2008 World Health Organization classification. The medical
records of all the diagnosed cases at their presentation to
hospital were reviewed. The study was reviewed and approved by the
ethics committees of The First Affiliated Hospital of Medical
School of Zhejiang University and the Cancer Hospital Affiliated
with the University of Chinese Academy of Sciences (ethical
approval no.: IRB-2022-55). All patients provided written informed
consent. The study was performed in accordance with The Declaration
of Helsinki.
Diagnostic criteria
The diagnosis of HPS was based on the HLH-2004
protocol standard (Group 2004) (18). The patients were diagnosed with
LAHS if they were pathologically diagnosed with lymphoma and met at
least five of the following eight criteria: i) fever; ii)
splenomegaly; iii) cytopenia affecting at least two lineages in the
peripheral blood, defined as hemoglobin <90 g/l, platelets
<100×109/l and neutrophils <1.0×109/l;
iv) hypertriglyceridemia (triglycerides >3 mmol/l) and/or
hypofibrinogenemia (fibrinogen <1.5 g/l); v) ferritin ≥500 µg/l;
vi) soluble interleukin-2 receptor (sCD25) ≥2,400 U/ml; vii)
decreased or absent NK-cell activity; and viii) hemophagocytosis in
the bone marrow, spleen, liver or lymph nodes. NK cell activity and
the level of sCD25 were not tested in the patients because suitable
test methods were unavailable at the time of diagnosis. Most
patients in the LAHS group presented with HPS at the diagnosis of
lymphoma and a small number of patients developed HPS during the
clinical course of lymphoma, typically at the advanced stage.
Laboratory examination
All patients underwent a physical examination and
hematological tests, including a complete blood count, blood
biochemical function test and coagulation function test. The
percentage of macrophages in the bone marrow was also measured.
Biopsy specimens from involved sites were assessed, as well as bone
marrow smears and biopsies.
Therapeutic regimens
The patients received induction treatment with the
HLH-1994 regimen. For patients with LAHS, once the HPS was
controlled, the chemotherapy regimen appropriate for the type of
lymphoma that was present was administered to treat the primary
disease. The COPE or ECHOP (etoposide + dexamethasone + vindesine +
cyclophosphamide + nordoxorubicin) regimen was mainly used for
NK/T-cell lymphoma or T-cell lymphoma, the R-CHOP (rituximab +
dexamethasone + vindesine + cyclophosphamide + nordoxorubicin)
regimen was used for non-Hodgkin's B-cell lymphoma and the ABVD
(epirubicin + bleomycin + vindesin + dacarbazine) regimen was used
for Hodgkin's lymphoma. Patients with infections were treated with
antibiotics and/or antiviral drugs. Blood transfusions included the
transfusion of erythrocyte suspensions, platelets, fresh plasma and
blood products.
Study endpoints and definitions
Early death was defined as mortality due to any
cause within 30 days after the diagnosis of HPS. Overall survival
(OS) and recurrence free survival (RFS) refer to the time from the
diagnosis of HPS to patient death and diagnosis of HPS recurrence,
respectively. All cases were followed up through outpatient
re-examination or telephone interviews, and the follow-up was
performed up to January 31, 2022.
Statistical analysis
Clinical and laboratory data were compared between
the LAHS and non-LAHS groups using χ2 and Fisher's exact
probability tests for frequency data and Mann-Whitney U test for
age. Kaplan-Meier curves were used to calculate and analyze the OS
rate. The differences in survival rates between groups were
compared using the log-rank method. The Cox risk regression model
was used to analyze the factors associated with LAHS via univariate
analysis. The factors shown to be statistically significant by the
univariate analysis were incorporated into a Cox risk regression
model for multivariate analysis. P<0.05 was considered to
indicate a statistically significant result. Statistical analysis
was performed using SPSS 19 software (IBM Corp.).
Results
Patient characteristics
A total of 324 patients were included in the study.
The clinical parameters of the patients are shown in Tables I and II. The male to female ratio was 1.4:1
and the median age was 50 years (range, 14–90 years). In the LAHS
group, 119 patients developed HPS at the initial diagnosis of
lymphoma and 20 patients developed HPS at the recurrence of
lymphoma. Total bilirubin, indirect bilirubin, hepatomegaly and
lymphadenopathy in the LAHS group were significantly higher
compared with those in the non-LAHS group (P=0.005, P=0.006,
P=0.009 and P=0.003, respectively; Table I). Age, sex, white blood cell
count, hemoglobin and platelet level were not found to be
significantly different between the LAHS and non-LAHS groups.
During the follow-up, 134/324 (41.4%) patients died within 30 days
after diagnosis. These early deaths were of 70/139 (50.4%) patients
in the LAHS group and 64/185 (34.6%) patients in the non-LAHS
group. In addition, 70/324 (21.6%) patients experienced a
recurrence, including 42/139 (30.2%) patients in the LAHS group and
28/185 (15.1%) patients in the non-LAHS group. In the LAHS group,
the most frequent histopathological subtypes were T-cell lymphoma
in 100 patients (71.9%) and extranodal NK/T-cell lymphoma in 63
patients (45.3%), followed by diffuse large B-cell lymphoma in 27
patients (19.4%) and peripheral T-cell lymphoma in 15 patients
(10.8%) (Table II). In the
non-LAHS group, 17 (9.2%) patients had autoimmune disease and 32
(17.3%) had no confirmed basic diseases. There were also 33 (17.8%)
patients with Epstein-Barr virus infection, 4 (2.2%) with
cytomegalovirus, 86 (46.5%) with infection and 13 (7.0%) with
potential hematological diseases.
 | Table I.Comparison of clinical features and
prognosis in the LAHS and non-LAHS groups. |
Table I.
Comparison of clinical features and
prognosis in the LAHS and non-LAHS groups.
| Variable | Total (n=324) | LAHS group
(n=139) | Non-LAHS group
(n=185) | P-value |
|---|
| Age, median
(range), years | 50 (14–90) | 53 (17–84) | 49 (14–90) | 0.059 |
| Sex, n (%) |
|
|
| 0.058 |
|
Male | 188 (58.0) | 89 (64.0) | 99 (53.5) |
|
|
Female | 136 (42.0) | 50 (36.0) | 86 (46.5) |
|
| T >40.0°C, n
(%) | 94 (29.1) | 42 (30.2) | 52 (28.1) | 0.679 |
| WBC
≤4.0×109/l, n (%) | 263 (81.2) | 117 (84.2) | 146 (78.9) | 0.231 |
| N
≤0.5×109/l, n (%) | 75 (23.1) | 32 (23.0) | 43 (23.2) | 0.963 |
| L
≤0.5×109/l, n (%) | 198 (61.1) | 93 (66.9) | 105 (56.8) | 0.064 |
| Hb ≤60 g/l, n
(%) | 82 (25.3) | 36 (25.9) | 46 (24.9) | 0.832 |
| Plt
≤20×109/l, n (%) | 128 (39.5) | 63 (45.3) | 65 (35.1) | 0.063 |
| Fib ≤1.5 g/l, n
(%) | 217 (66.9) | 92 (66.2) | 125 (67.6) | 0.794 |
| APTT >36 sec, n
(%) | 239 (73.8) | 100 (71.9) | 139 (75.1) | 0.518 |
| PT >13.5 sec, n
(%) | 201 (62.0) | 79 (56.8) | 122 (65.9) | 0.094 |
| D-dimer >700
µg/l, n (%) | 283 (87.3) | 127 (91.4) | 156 (84.3) | 0.052 |
| ALB ≤30 g/l, n
(%) | 246 (75.9) | 105 (75.5) | 141 (76.2) | 0.888 |
| GLB ≤20 g/l, n
(%) | 190 (58.6) | 90 (64.7) | 100 (54.1) | 0.053 |
| ALT >40 U/l, n
(%) | 255 (78.7) | 105 (75.5) | 150 (81.1) | 0.228 |
| AST >50 U/l, n
(%) | 262 (80.9) | 106 (76.3) | 156 (84.3) | 0.068 |
| TB>21 µmol/l, n
(%) | 183 (56.5) | 91 (65.5) | 92 (49.7) | 0.005 |
| DB >10 µmol/l, n
(%) | 187 (57.7) | 84 (60.4) | 103 (55.7) | 0.391 |
| IB >14 µmol/l, n
(%) | 142 (43.8) | 73 (52.5) | 69 (37.3) | 0.006 |
| Cr abnormal, n
(%) | 208 (64.2) | 93 (66.9) | 115 (62.2) | 0.378 |
| Urea >8.2 mmol/l
n (%) | 164 (50.6) | 79 (56.8) | 85 (45.9) | 0.052 |
| TG >3 mmol/l, n
(%) | 157 (48.5) | 60 (43.2) | 97 (52.4) | 0.099 |
| LDH >1,000 U/l,
n (%) | 141 (43.5) | 59 (42.4) | 82 (44.3) | 0.736 |
| Ferritin >10,000
µg/l, n (%) | 137 (42.3) | 59 (42.4) | 78 (42.2) | 0.959 |
| CRP >100 mg/l, n
(%) | 76 (23.5) | 31 (22.3) | 45 (24.3) | 0.671 |
| Pneumonia, n
(%) | 160 (49.4) | 72 (51.8) | 88 (47.6) | 0.451 |
| Splenomegaly, n
(%) | 226 (69.8) | 97 (69.8) | 129 (69.7) | 0.992 |
| Hepatomegaly, n
(%) | 48 (14.8) | 29 (20.9) | 19 (10.3) | 0.009 |
| Lymphadenopathy, n
(%) | 91 (28.1) | 51 (36.7) | 40 (21.6) | 0.003 |
| Early mortality, n
(%) | 134 (41.4) | 70 (50.4) | 64 (34.6) | 0.004 |
| Recurrence, n
(%) | 70 (21.6) | 42 (30.2) | 28 (15.1) | 0.001 |
| 5-year overall
survival (%) | - | 21.5 | 52.4 | <0.001 |
| 5-year relapse-free
survival (%) | - | 7.7 | 48.3 | <0.001 |
| 5-year overall
survival (excluding early mortality) (%) | - | 43.3 | 80.2 | 0.041 |
| 5-year relapse-free
survival (excluding early mortality) (%) | - | 22.9 | 73.8 | 0.002 |
| Table II.Pathological subtypes of lymphoma of
the 139 patients with lymphoma-associated hemophagocytic
syndrome. |
Table II.
Pathological subtypes of lymphoma of
the 139 patients with lymphoma-associated hemophagocytic
syndrome.
| Pathological
subtype | N (%) |
|---|
| Hodgkin's
lymphoma | 3 (2.2) |
| Non-Hodgkin's
lymphoma | 136 (97.8) |
| B-cell
lymphoma | 36 (25.9) |
| Diffuse large
B-cell lymphoma | 27 (19.4) |
| Burkitt
lymphoma | 6 (4.4) |
| Mantle cell
lymphoma | 1 (0.7) |
| Lymphoplasmacytic
lymphoma | 2 (1.4) |
| T-cell
lymphoma | 100 (71.9) |
| Extranodal
NK/T-cell lymphoma | 63 (45.3) |
| Angioimmunoblastic
T-cell lymphoma | 14 (10.1) |
| Anaplastic large
cell lymphoma, ALK (−) | 8 (5.8) |
| Peripheral T-cell
lymphoma, NOS | 15 (10.8) |
Comparison of therapeutic response in
the LAHS and non-LAHS groups
The patients in the LAHS group received five
different treatments. Two patients received intravenous
immunoglobulin (IVIG) alone (1.4%), 41 patients received
glucocorticoid (GC) alone (29.5%), 10 patients received GC combined
with IVIG (7.2%), 72 patients received GC combined with
chemotherapy (51.8%), and 14 patients received GC combined with
IVIG and chemotherapy (10.1%; Fig.
1). The patients in the non-LAHS group received HLH-2004
treatments.
Association between disease
characteristics and early death in LASH
There were 70 early deaths of patients in the LAHS
group. The analysis of various disease characteristics revealed
that the early death rate was significantly higher in patients with
a platelet count of ≤20.0×109/l, fibrinogen level of
≤1.5 g/l, activated partial thromboplastin time (APTT) of >36.0
sec, prothrombin time of >13.5 sec, lactate dehydrogenase (LDH)
level of >1,000 U/l, ferritin level of >10,000 µg/l and
splenomegaly (Table III).
| Table III.Analysis of early mortality in
patients with lymphoma-associated hemophagocytic syndrome. |
Table III.
Analysis of early mortality in
patients with lymphoma-associated hemophagocytic syndrome.
| Variable | N | Early death, n
(%) | P-value |
|---|
| Age, years |
|
| 0.238 |
|
≤60 | 90 | 42 (46.7) |
|
|
>60 | 49 | 28 (57.1) |
|
| WBC, cells/l |
|
| 0.175 |
|
≤4.0×109 | 117 | 56 (47.9) |
|
|
>4.0×109 | 22 | 14 (63.6) |
|
| Hb, g/l |
|
| 0.662 |
| ≤60.0
g | 36 | 17 (47.2) |
|
|
>60.0 | 103 | 53 (51.5) |
|
| Plt, cells/l |
|
| 0.005 |
|
≤20.0×109 | 63 | 40 (63.5) |
|
|
>20.0×109 | 76 | 30 (39.5) |
|
| Fib, g/l |
|
| 0.002 |
|
≤1.5 | 92 | 55 (59.8) |
|
|
>1.5 | 47 | 15 (31.9) |
|
| APTT, sec |
|
| <0.001 |
|
≤36.0 | 39 | 10 (25.6) |
|
|
>36.0 | 100 | 60 (60.0) |
|
| PT, sec |
|
| 0.002 |
|
≤13.5 | 60 | 21 (35.0) |
|
|
>13.5 | 79 | 49 (62.0) |
|
| ALB, g/l |
|
| 0.104 |
|
≤30.0 | 105 | 57 (54.3) |
|
|
>30 | 34 | 13 (38.2) |
|
| GLB, g/l |
|
| 0.342 |
|
≤20.0 | 90 | 48 (53.3) |
|
|
>20.0 | 49 | 22 (44.9) |
|
| ALT, U/l |
|
| 0.218 |
|
≤40.0 | 34 | 14 (41.2) |
|
|
>40.0 | 105 | 56 (53.3) |
|
| AST, U/l |
|
| 0.149 |
|
≤50.0 | 33 | 13 (39.4) |
|
|
>50.0 | 106 | 57 (53.8) |
|
| TB, µmol/l |
|
| 0.676 |
|
≤21.0 | 48 | 23 (47.9) |
|
|
>21.0 | 91 | 47 (51.6) |
|
| DB, µmol/l |
|
| 0.775 |
|
≤10.0 | 55 | 25 (45.5) |
|
|
>10.0 | 84 | 45 (53.6) |
|
| IB, µmol/l |
|
| 0.674 |
|
≤14.0 | 66 | 32 (48.5) |
|
|
>14.0 | 73 | 38 (52.1) |
|
| Cr |
|
| 0.254 |
|
Abnormal | 93 | 50 (53.8) |
|
|
Normal | 46 | 20 (43.5) |
|
| TG, mmol/l |
|
| 0.340 |
|
≤3.0 | 79 | 37 (46.8) |
|
|
>3.0 | 60 | 33 (55.0) |
|
| LDH, U/l |
|
| <0.001 |
|
≤1,000 | 80 | 28 (35.0) |
|
|
>1,000 | 59 | 42 (71.2) |
|
| Ferritin, µg/l |
|
| 0.012 |
|
≤10,000 | 80 | 33 (41.2) |
|
|
>10,000 | 59 | 37 (62.7) |
|
| Spleen |
|
| 0.017 |
|
Splenomegaly | 97 | 55 (56.7) |
|
|
Normal | 42 | 15 (35.7) |
|
Comparison of recurrence and survival
between the LAHS and non-LAHS groups
The median follow-up time for the 324 patients was
12 months (interquartile range, 1–65 months). By the end of
follow-up, 90 (64.7%) patients in the LAHS group had died and 70
(50.4%) of these deaths were defined as early mortality. The 5-year
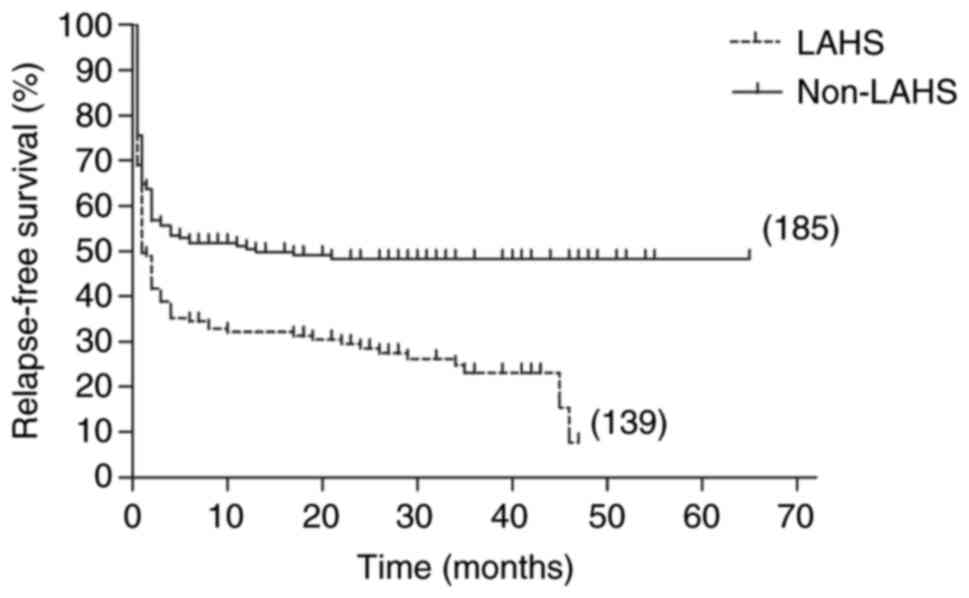
RFS and OS rates in the LAHS group were low at 7.7 and 21.5%,
respectively. By contrast, the 5-year RFS and OS rates in the
non-LAHS group were high at 48.3 and 52.4%, respectively. There was
a significant difference in OS between the two groups as indicated
by the log-rank test (P<0.001). In addition, the 5-year RFS in
the LAHS group was significantly lower than that in the non-LAHS
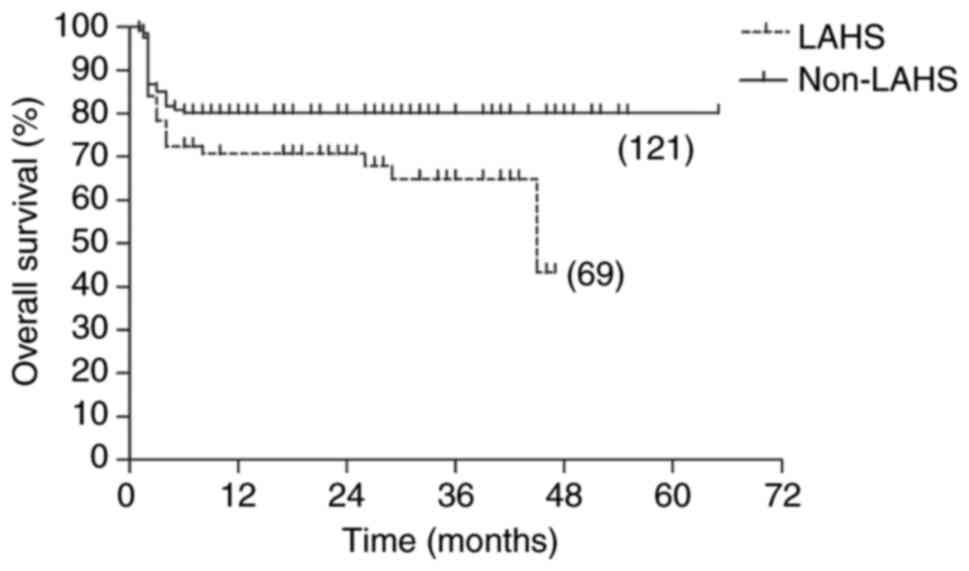
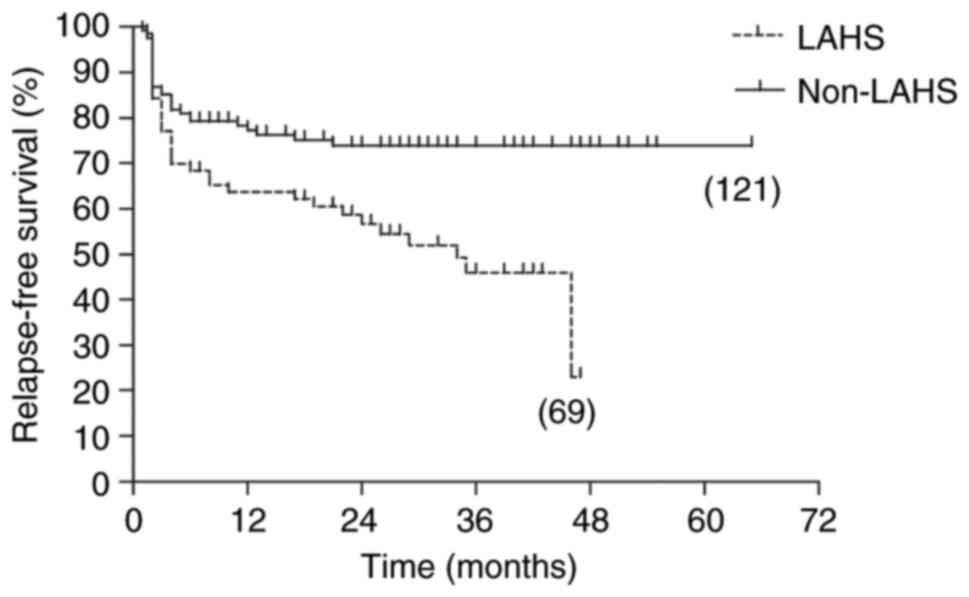
group (P<0.001). When patients who died early were excluded from
the two groups, the 5-year OS rates were improved and significantly
different (43.3 vs. 80.2; P=0.041), and the 5-year RFS rates also
remained significantly different (22.9 vs. 73.8; P=0.002; Fig. 2, Fig.
3, Fig. 4, Fig. 5).
Univariate and multivariate analysis
of early death in LAHS
Univariate analysis indicated that OS was
significantly associated with coagulation dysfunction (APTT
>36.0 sec), abnormal LDH (>1,000 U/l) and abnormal ferritin
(>10,000 µg/l). The subsequent multivariate analysis revealed
that coagulation dysfunction and abnormal LDH were independent
prognostic indicators of reduced OS (Table IV).
| Table IV.Univariate and multivariate analysis
of the overall survival of patients with lymphoma-associated
hemophagocytic syndrome. |
Table IV.
Univariate and multivariate analysis
of the overall survival of patients with lymphoma-associated
hemophagocytic syndrome.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Variable | HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| Plt
≤20.0×109/l | 1.556 | 0.971-2.493 | 0.066 | - | - | - |
| Fib ≤1.5 g/l | 1.573 | 0.920-2.689 | 0.098 | - | - | - |
| APTT >36.0
sec | 2.641 | 1.351-5.164 | 0.005 | 2.250 | 1.134-4.463 | 0.020 |
| PT >13.5
sec | 1.518 | 0.927-2.487 | 0.097 | - | - | - |
| LDH >1,000.0
U/l | 2.244 | 1.390-3.623 | 0.001 | 1.703 | 1.013-2.863 | 0.045 |
| Ferritin
>10,000.0 µg/l | 1.800 | 1.123-2.886 | 0.015 | 1.463 | 0.885-2.421 | 0.138 |
| Splenomegaly | 1.184 | 0.699-2.006 | 0.529 | - | - | - |
Survival rate comparison between LAHS
patients with and without chemotherapy
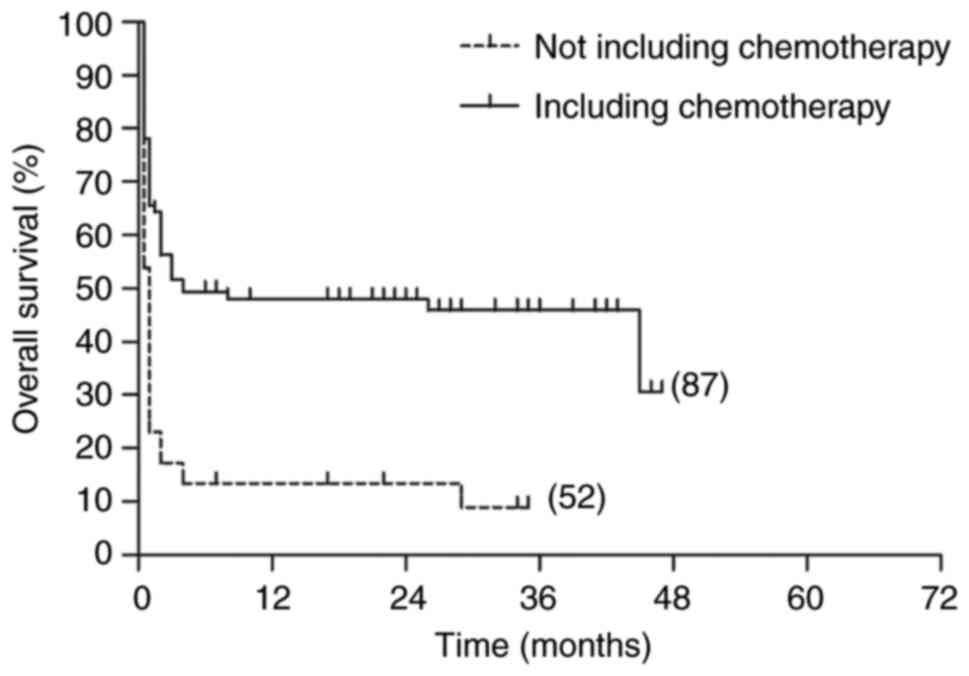
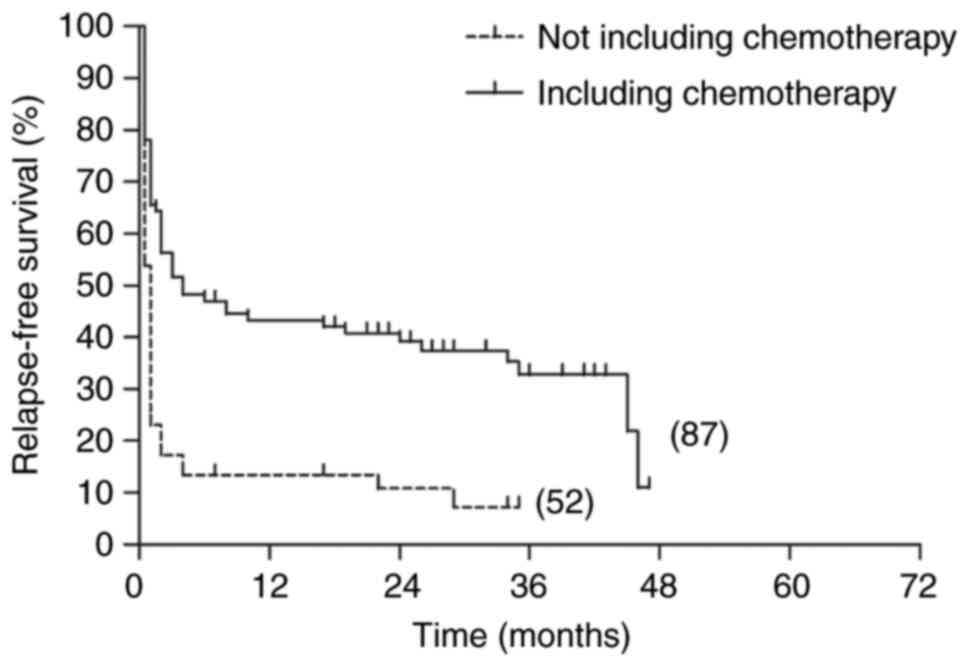
The 5-year OS and RFS rates in the patients with
LAHS who had received chemotherapy were 30.7 and 10.9%,
respectively. By comparison, the 5-year OS and RFS rates in the
patients with LAHS who had not received chemotherapy were very low
at 8.9 and 7.2%, respectively. Log-rank tests showed that there was
a significant difference in OS between the two groups (P<0.001),
and that the 5-year RFS of patients who had not received
chemotherapy was significantly lower than that of patients who had
undergone chemotherapy (P<0.001; Figs. 6 and 7).
Survival rate comparison between
patients with LAHS of different pathological subtypes
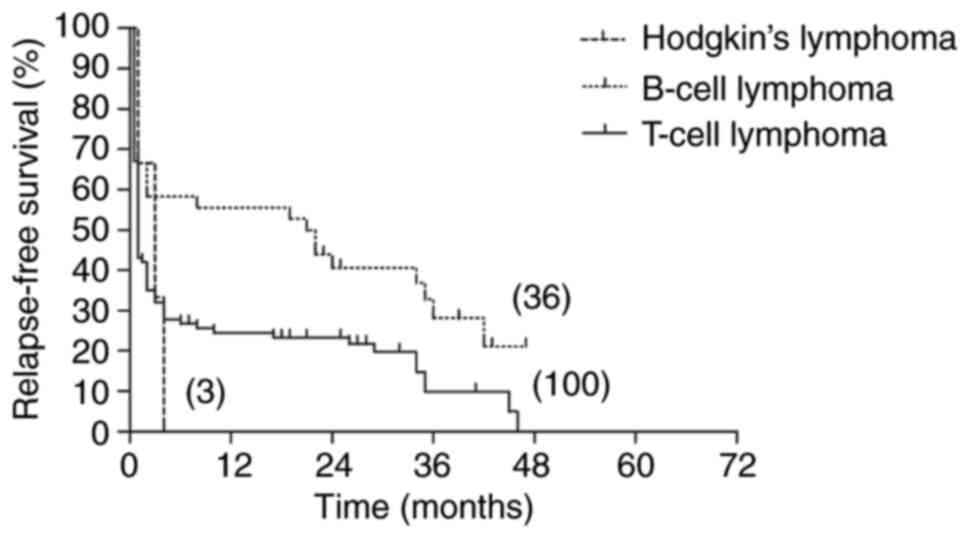
The 5-year OS and RFS rates in the B-cell lymphoma
group were high at 58.3 and 21.1%, respectively. The 5-year OS and
RFS rates in the T-cell lymphoma group were much lower, at 11.7 and
0% respectively. The 5-year OS and RFS rates in the Hodgkin's
lymphoma group were both 0%. Log-rank tests identified a
significant difference in OS among the three groups (P=0.022) and
indicated that the 5-year RFS of patients in the B-cell lymphoma
group was significantly higher than that of patients in the
chemotherapy and Hodgkin's lymphoma groups (P=0.047; Figs. 8 and 9).
Discussion
Tumor-associated HPS most frequently occurs
secondary to hematological malignancies, such as lymphoma (18). LAHS is a major subtype of secondary
HPS with a poor outcome (9,19,20).
Therefore, the identification of means for improving the survival
rate of patients with LAHS is a key and challenging issue in the
treatment of HPS, for which clinical data are lacking. The present
study sought to investigate the clinical features and prognostic
factors of patients with LAHS, with the aim of exploring
individualized therapeutic strategies to reduce the recurrence rate
and early mortality in patients with LAHS and to improve their
long-term survival rate.
The present study retrospectively analyzed 324
patients who were diagnosed with HPS between January 2014 and
February 2021. A total of 139 patients with LAHS were evaluated,
accounting for 42.9% of the cohort, which was consistent with
previous reports (20,21). Studies have shown that LAHS is most
commonly secondary to diffuse large B-cell lymphoma in the Europe
and Japan, but predominantly secondary to T-cell lymphoma in China
and Korea (3,6,20).
In the present study, LAHS was associated with T-LAHS in~71.9% of
patients, which is in line with previous reports of 50.0-75.4%
(13,21,22).
No significant differences in age, sex, white blood
cell count, hemoglobin or platelet levels were detected between the
LAHS and non-LAHS groups. The percentages of patients with a total
bilirubin level of >21 µmol/l and indirect bilirubin level of
>14 µmol/l in the LAHS group were significantly higher compared
with those in the non-LAHS group. Previous studies have
demonstrated that there is a significant difference in total
bilirubin indicators between groups of patients with different
prognostic factors, and the evident anomaly of various indicators
is associated with disease-induced multiple organ function
impairment (23,24). Therefore, these may be among the
causes of the high early mortality rate in patients with LAHS.
Hyperbilirubinemia is considered to be a risk factor of early death
(25). Hepatomegaly and
lymphadenopathy were also significantly different between the LAHS
and non-LAHS groups in the present study, which was likely
associated with secondary lymphoma in patients with LAHS. Notably,
hepatomegaly is a risk factor for NK/T-LAHS (26).
In a previous study, Ishii et al (3) analyzed the outcomes of distinct
subtypes of HPS, which revealed that the 5-year OS rates were 89.6%
for autoimmune-associated HPS, 89.0% for other infection-associated
HPS, 82.7% for Epstein-Barr virus-HPS, 48.2% for B-LAHS and 12.2%
for NK/T-LAHS. The overall mortality associated with LAHS has been
reported to be 79.6% (26). A
study of 28 cases of T-LAHS reported that all patients died, with
89.0% deaths occurring within 6 months of diagnosis (9). The present study retrospectively
analyzed the prognosis for 139 patients with LAHS. The OS of
patients with LAHS was significantly lower than that in patients
with non-LAHS (21.5 vs. 52.4%; P<0.001). However, when patients
with early mortality were excluded, the difference in OS between
the patients with and without LAHS appeared to decrease, as the
difference was less significant (P=0.041). This shows that if LAHS
patients can overcome early death, their OS improves significantly.
Therefore, it is essential to reduce early mortality in order to
improve the prognosis of patients with LAHS. At present, there is
no unified standard treatment for LAHS, and the optimal therapeutic
option remains largely undefined. Various therapeutic strategies
have been reported, such as steroid pulse therapy, IVIG, the CHOP
regimen, the HPS-2004 protocol, the HPS-94 protocol and CD25
monoclonal antibody treatment (6).
However, the complexity of LAHS may delay the timing of the primary
disease treatment, which is not conducive to a favorable outcome
and the prognosis of the disease (27,28).
Chemotherapy-resistant LAHS requires hematopoietic stem cell
transplantation (7,9,13,29).
The median survival time for patients with LAHS who do not receive
chemotherapy has been reported to be only 11 days (30). In the present study, 61.9% of the
patients with LAHS received a chemotherapy regimen. The 5-year OS
and RFS of patients who received chemotherapy were significantly
higher than those in the patients who did not receive chemotherapy.
The aggressive treatment of primary tumors in cases of
tumor-associated HPS has been recommended (13,27,31).
Although the current study focused on a small number of patients,
it indicates that chemotherapy for lymphoma has great potential in
the treatment of LAHS, and can improve patient prognosis.
Early mortality continues to be the main factor
affecting the efficacy of HPS. The mortality associated with LAHS
is notably high. Therefore, early treatment with intensive
chemotherapy is critical for improving OS. In the present study,
the early mortality of patients with LAHS was significantly higher
than that of the patients in the non-LAHS group (50.4 vs. 34.6%;
P=0.004). Jin et al (27)
reported an early (30-day) mortality rate of 51.1% for patients
with NK/T-LAHS. Other studies have reported a median survival time
for patients with T-LAHS ranging from 11 to 40 days (9,30,32).
Hence, it is clear that LAHS is dangerous and the early stage of
the disease is the time at which the risk of death is greatest. The
present study further analyzed the risk factors associated with
early death. Univariate analysis indicated that OS was
significantly associated with coagulation dysfunction (>36.0
sec), abnormal LDH (>1,000 U/l) and abnormal ferritin
(>10,000 µg/l). Multivariate analysis indicated that coagulation
dysfunction and abnormal LDH were independent prognostic indicators
of reduced OS, which was consistent with previous research
(27,33–35).
Disseminated intravascular coagulation (DIC) caused by coagulation
dysfunction is one of the main causes of HPS-associated death
(9,36–38).
Patients with T-LAHS are prone to DIC, while the association of
B-LAHS with DIC is less common (39). Li et al (26) found that 10 cases of early death
among 103 patients with HPS were complicated by DIC. Therefore, the
early detection of coagulation abnormalities, the timely correction
and prevention of DIC and thus the reduction of early mortality are
critical (26). For this group of
patients, early use of the CHOP regimen to treat LAHS is
recommended, in order to suppress inflammation, prevent severe
coagulopathy and severe infections and minimize early mortality,
thereby achieving better results.
The present study has some limitations. Firstly, it
is a retrospective study that is based on the analysis of medical
records, which may have some bias. Secondly, the detection of sCD25
and NK cell activity were not performed. Another limitation is that
immortal time bias was not accounted for.
In summary, the onset of LAHS is dangerous. LAHS has
a high early mortality rate and the prognosis of patients with LAHS
is significantly worse than that of patients with other types of
HPS. Once LAHS has been diagnosed, the treatment of lymphoma is
particularly important. Since the mortality associated with LAHS is
high, the early recognition and treatment of DIC and reduction of
early mortality rate are critical for improving OS.
Acknowledgements
Not applicable.
Funding
This study was partly supported by Zhejiang Provincial Public
Welfare Technology Application Research and Subsidy Project (grant
no. LGF19H080003) and Zhejiang Medical and Health Science and
Technology Program (grant no. 2020ky1071).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QZ collected, analyzed and interpreted the data and
drafted the manuscript. LW collected, analyzed and interpreted the
data and revised the manuscript. DZ performed statistical analyses,
interpreted the data and revised the manuscript. LZ, LL and WX
obtained the samples and added clinical data in the process of
article repair. YT obtained the samples, added clinical data in the
process of article repair and reviewed the manuscript. XY conceived
and coordinated the study, designed and analyzed the experiments,
reviewed the paper and gave final approval for the submitted
version. QZ and XY confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Cancer Hospital of
the University of Chinese Academy of Sciences (Hangzhou, China).
All patients provided written informed consent to participate.
Patient consent for publication
All patients consented to publication of their
data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Canna SW and Marsh RA: Pediatric
hemophagocytic lymphohistiocytosis. Blood. 135:1332–1343. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu P, Pan X, Chen C, Niu T, Shuai X, Wang
J, Chen X, Liu J, Guo Y, Xie L, et al: Nivolumab treatment of
relapsed/refractory Epstein-Barr virus-associated hemophagocytic
lymphohistiocytosis in adults. Blood. 135:826–833. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ishii E, Ohga S, Imashuku S, Yasukawa M,
Tsuda H, Miura I, Yamamoto K, Horiuchi H, Takada K, Ohshima K, et
al: Nationwide survey of hemophagocytic lymphohistiocytosis in
Japan. Int J Hematol. 86:58–65. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yao S, Jin Z, He L, Zhang R, Liu M, Hua Z,
Wang Z and Wang Y: Clinical features and prognostic risk prediction
of non-Hodgkin lymphoma-associated hemophagocytic syndrome. Front
Onco. 11:7880562021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin CH, Shih YH, Chen TC, Chou CW, Hsu CY
and Teng CJ: A decade of lymphoma-associated hemophagocytic
lymphohistiocytosis: Does the outcome improve? J Clin Med.
10:51142021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bigenwald C, Fardet L, Coppo P, Meignin V,
Lazure T, Fabiani B, Kohn M, Oksenhendler E, Boutboul D, Uzzan M,
et al: A comprehensive analysis of Lymphoma-associated
haemophagocytic syndrome in a large French multicentre cohort
detects some clues to improve prognosis. Br J Haematol. 183:68–75.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Giza A, Gałązka K, Jońca M, Raźny M,
Zimowska-Curyło D, Wilk M, Goldman-Mazur S, Piątkowska-Jakubas B
and Sacha T: Subcutaneous panniculitis-like T-cell lymphoma (SPTCL)
with probable mesentery involvement with associated hemophagocytic
syndrome (HPS)-how to treat it? J Dermatolog Trea. 33:2674–2676.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lehmberg K, Nichols KE, Henter JI,
Girschikofsky M, Greenwood T, Jordan M, Kumar A, Minkov M, La Rosée
P and Weitzman S; Study Group on Hemophagocytic Lymphohistiocytosis
Subtypes of the Histiocyte Society, : Consensus recommendations for
the diagnosis and management of hemophagocytic lymphohistiocytosis
associated with malignancies. Haematologic. 100:997–1004.
2015.PubMed/NCBI
|
|
9
|
Tong H, Ren Y, Liu H, Xiao F, Mai W, Meng
H, Qian W, Huang J, Mao L, Tong Y, et al: Clinical characteristics
of T-cell lymphoma associated with hemophagocytic syndrome:
Comparison of T-cell lymphoma with and without hemophagocytic
syndrome. Leuk Lymphoma. 49:81–87. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song Y, Yin Q, Wang J and Wang Z:
Autologous hematopoietic stem cell transplantation for patients
with lymphoma-associated hemophagocytic lymphohistiocytosis. Cell
Transplant. 30:96368972110570772021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Henter JL, Samuelsson-Horne AC, Aricò M,
Egeler RM, Elinder G, Filipovich AH, Gadner H, Imashuku S, Komp D,
Ladisch S, et al: Treatment of hemophagocytic lymphohistiocytosis
with HLH-94 immunochemotherapy and bone marrow transplantation.
Blood. 100:2367–2373. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Birndt S, Schenk T, Heinevetter B,
Brunkhorst FM, Maschmeyer G, Rothmann F, Weber T, Müller M, Panse
J, Penack O, et al: Hemophagocytic lymphohistiocytosis in adults:
Collaborative analysis of 137 cases of a nationwide German
registry. J Cancer Res Clin Onco. 146:1065–1077. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang Y, Cui M, Fu X, Han L, Zhang L, Li
L, Li X, Sun Z, Wu J, Zhang X, et al: Lymphoma associated
hemophagocytic syndrome: A single-center retrospective study. Oncol
Lett. 16:1275–1284. 2018.PubMed/NCBI
|
|
14
|
Liu YZ, Bi LQ, Chang GL, Guo Y and Sun S:
Clinical characteristics of extranodal NK/T-cell
lymphoma-associated hemophagocytic lymphohistiocytosis. Cancer
Manag Res. 11:997–1002. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Daver N, McClain K, Allen CE, Parikh SA,
Otrock Z, Rojas-Hernandez C, Blechacz B, Wang S, Minkov M, Jordan
MB, et al: A consensus review on malignancy-associated
hemophagocytic lymphohistiocytosis in adults. Cancer.
123:3229–3240. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ren Q, Chan KW, Huang H, Wang Z, Fang X,
Guo C, Li F, Zhang L, Yao Y, Chen Z, et al: Platelet-derived
alpha-granules are associated with inflammation in patients with
NK/T-cell lymphoma-associated hemophagocytic syndrome. Cytokine.
126:1548782020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meng G, Wang Y, Wang J and Wang Z: The DEP
regimen is superior to the HLH-1994 regimen as first-line therapy
for lymphoma-associated haemophagocytic lymphohistiocytosis. Leuk
Lymphoma. 62:854–860. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Henter JI, Horne A, Aricó M, Egeler RM,
Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski
J and Janka G: HLH-2004: Diagnostic and therapeutic guidelines for
hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer.
48:124–131. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jin Z, Wang Y, Wei N and Wang Z: Hodgkin
lymphoma-associated hemophagocytic lymphohistiocytosis-a dangerous
disease. Ann Hematol. 99:1575–1581. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pasvolsky O, Zoref-Lorenz A, Abadi U,
Geiger KR, Hayman L, Vaxman I, Raanani P and Leader A:
Hemophagocytic lymphohistiocytosis as a harbinger of aggressive
lymphoma: A case series. Int J Hematol. 109:553–562. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li W, Zhong Y, Shuang Y, Huang H, Huang Y,
Yu L and Huang X: High concentration of miR-133 is a useful marker
for the diagnosis of lymphoma-associated hemophagocytic syndrome.
Cancer Biomark. 20:159–164. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yuan S, Wang Y, Luo H, Jiang Z, Qiao B,
Jiang Y, Hu Y, Cheng Y, Chen X, Gong W, et al: Serum soluble VSIG4
as a surrogate marker for the diagnosis of lymphoma-associated
hemophagocytic lymphohistiocytosis. Br J Haematol. 189:72–83. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bin Q, Gao JH and Luo JM: Prognostic
factors of early outcome in pediatric hemophagocytic
lymphohistiocytosis: An analysis of 116 cases. Ann Hematol.
95:1411–1418. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
George MR: Hemophagocytic
lymphohistiocytosis: Review of etiologies and management. J Blood
Med. 5:69–86. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Prokesch BC, Nagalla S, Ezzati F, Tujios
SR, Dominguez A, Chen W, Kershaw C, Patel P, de la Flor C, Foster
J, et al: What's in a name? The heterogeneous clinical spectrum and
prognostic factors in a cohort of adults with hemophagocytic
lymphohistiocytosis. Transfus Apher Sci. 57:779–784. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li J, Wang Q, Zheng W, Ma J, Zhang W, Wang
W and Tian X: Hemophagocytic lymphohistiocytosis: Clinical analysis
of 103 adult patients. Medicine (Baltimore). 93:100–105. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin Z, Wang Y, Wang J, Wu L, Pei R, Lai W
and Wang Z: Multivariate analysis of prognosis for patients with
natural killer/T cell lymphoma-associated hemophagocytic
lymphohistiocytosis. Hematology. 23:228–234. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bhatt NS, Oshrine B and An Talano J:
Hemophagocytic lymphohistiocytosis in adults. Leuk Lymphoma.
60:19–28. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee DE, Martinez-Escala ME, Serrano LM,
Zhou XA, Kaplan JB, Pro B, Choi J and Guitart J: Hemophagocytic
lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol.
154:828–831. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takahashi N, Chubachi A, Miura I, Nakamura
S and Miura AB: Lymphoma-associated hemophagocytic syndrome in
Japan. Rinsho Ketsueki. 40:542–549. 1999.(In Japanese). PubMed/NCBI
|
|
31
|
Malkan UY, Albayrak M, Yildiz M, Maral S,
Afacan Ozturk HB and Comert P: A rare case of diffuse large B-cell
lymphoma-associated hemophagocytic lymphohistiocytosis. J Oncol
Pharm Pract. 27:250–252. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Han L, Li L, Wu J, Li X, Zhang L, Wang X,
Fu X, Ma W, Sun Z, Zhang X, et al: Clinical features and treatment
of natural killer/T cell lymphoma associated with hemophagocytic
syndrome: Comparison with other T cell lymphoma associated with
hemophagocytic syndrome. Leuk Lymphoma. 55:2048–2055. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Signoff JK, Fitzgerald JC, Teachey DT,
Balamuth F and Weiss SL: Hypofibrinogenemia is associated with poor
outcome and secondary hemophagocytic lymphohistiocytosis/macrophage
activation syndrome in pediatric severe sepsis. Pediatr Crit Care
Med. 19:397–405. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li N, Zhang L, Liu J, Zhang J, Weng HW,
Zhuo HY and Zou LQ: A clinical study of 21 patients with
hemophagocytic syndrome in 295 cases diagnosed with nasal type,
extranodal nature killer/T cell lymphoma. Cancer Biol Ther.
18:252–256. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang T, Ding X and Lu W: The prognostic
significance of beta2 microglobulin in patients with hemophagocytic
lymphohistiocytosis. Dis Markers. 2016:15239592016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pan H, Huo Y and Sun L: Comparison between
clinical features and prognosis of malignancy- and
non-malignancy-associated pediatric hemophagocytic
lymphohistiocytosis. BMC Pediatr. 19:4682019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Valade S, Mariotte E and Azoulay E:
Coagulation disorders in hemophagocytic
lymphohistiocytosis/macrophage activation syndrome. Crit Care Clin.
36:415–426. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jia J, Song Y, Lin N, Liu W, Ping L, Zheng
W, Wang X, Xie Y, Tu M, Zhang C, et al: Clinical features and
survival of extranodal natural killer/T cell lymphoma with and
without hemophagocytic syndrome. Ann Hematol. 95:2023–2031. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Uni M, Yoshimi A, Maki H, Maeda D,
Nakazaki K, Nakamura F, Fukayama M and Kurokawa M: Successful
treatment with recombinant thrombomodulin for B-cell
lymphoma-associated hemophagocytic syndrome complicated by
disseminated intravascular coagulation. Int J Clin Exp Pathol.
6:1190–1194. 2013.PubMed/NCBI
|