Introduction
Tumors of the brain and Central Nervous System (CNS)
are uncommon. Of these tumors, gliomas represent approximately
one-third of all primary CNS tumors and the majority of malignant
CNS tumors with a yearly incidence of about 6/100000 (1). Patients with gliomas have a varying
5-year survival rate, a high mortality rate, and a poor prognosis
depending on histology and grading. The poorest prognosis is for
glioblastoma, which has a 5-year relative survival rate of 6.8
(2,3).
Traditionally, the classification of tumors has been
based on histopathological features and immunochemistry, but in
2016 the WHO (World Health Organization) took molecular
characteristics into account in its reclassification of tumors, and
in 2021 it underlined molecular markers after additional update
classifications (4–6).
Gliomas have been found to have several genetic
changes that maybe used as diagnostic, prognostic, predictive, and
potentially therapeutic biomarkers. The most commonly reported
genetic variations include mutations in ATRX, BRAF, CDKN2A/B,
IDH, NF1, RB1, TERT, and TP53 genes as well as
MGMT promoter methylation status, copy number alterations
such as 1p/19q codeletion, EGFR amplification and combined
gain of chromosome 7 and loss of chromosome 10 (7+/10-), and
rearrangements (7–9).
Subsequently, the advent of next-generation
sequencing (NGS) technologies over the past ten years, has
significantly influenced the field of molecular oncology in
gliomas, demonstrating a significant variability by age, gender,
and ethnicity (10,11).
Implementation and broad use of multigene panels on
Formalin-Fixed, Paraffin-Embedded (FFPE) tissues enable the
cost-effective study of many genes and detection of genetic
abnormalities by massively parallel sequencing.
To date, a large number of investigations
encompassing commonly affected and new candidate genes have been
performed to more precisely categorize gliomas and identify
distinct prognostic categories that may have treatment
implications, with varied results being obtained (12,13).
Therefore, the aim of the present study is to further elucidate and
determine the prevalence of somatic mutations by implementing a
broad 80-gene panel among 32 patients with gliomas, while reporting
their clinicopathological features and outcomes. Subsequently, 14
of 32 tumor samples were examined using another multigene panel to
evaluate the accuracy and clinical utility of the broad panel.
Materials and methods
This study initially included 48 patients diagnosed
with high-grade gliomas and 48 available FFPE tissue samples
selected in the clinical centers affiliated with Hellenic
Cooperative Oncology Group (HeCOG). Sixteen out of these 48 tumor
samples did not meet the quality control criteria for further
analysis. Subsequently, thirty-two tumor paraffin blocks from 32
patients were examined. The median age of cancer diagnosis among
the patients was 54.9 years (range: 25.2-77.8 years). Clinical data
were obtained from the HeCOG data office. Written informed consent
was obtained from all individuals prior to the use of their
biological material for research purposes as specified in the
Declaration of Helsinki. Tumor blocks were stored and centrally
processed in the Laboratory of Molecular Oncology (MOL; Hellenic
Foundation for Cancer Research/Aristotle University of
Thessaloniki) for histology review, including confirmation of tumor
tissue on the section; comparison to local typing; histologic
grade; areas with necrosis; microvascular proliferation; assessment
of tumor areas for macrodissection. The study was approved by the
Bioethics Committee of the Aristotle University of Thessaloniki
School of Medicine (AUTH #No 2/February 4, 2015) and by Cyprus
National Bioethics Committee (EEBK/EΠ/2016/54, 22/4/2021).
Tissue processing, DNA extraction and
NGS genotyping
Tumor dense areas were marked on H&Es and
microdissected manually from 10 µm unstained FFPE sections prior to
DNA extraction. FFPE samples from 48 patients diagnosed with
gliomas were processed. DNA was isolated from FFPE tissue sections
using the GeneRead DNA FFPE Kit (Qiagen GmbH, Hilden, Germany) in
accordance with manufacturer's instructions. DNA was quantified
with a fluorometric-based assay for FFPE tissue-derived DNA (Qubit
flex fluorometer, Qubit dsDNA high sensitivity assay, ThermoFisher
Scientific, Carlsbad, CA, USA). Sixteen samples that did not meet
the minimum acceptable quality control (QC) criteria (a minimum of
10 ng of DNA and a minimum DNA concentration of 1 ng/µl) were
excluded from the analysis. Therefore, 32 FFPE samples from 32
patients were included and analysed.
Tumor genotyping was performed in the Clinical
Laboratory Improvement Amendments (CLIA)-certified laboratory,
using a comprehensive, commercially available, pan-cancer tumor
profile panel of 80 genes (NIPD Genetics, Nicosia, Cyprus,
http://nipd.com/products/oncology/foresentia/), from
which Single Nucleotide Variants (SNVs), Insertions and Deletions
(InDels), Copy Number Alterations (CNAs) and Rearrangements were
detected in accordance with manufacturer's instructions (Table SI). Targeted genomic loci were
captured using an in-solution hybridization method (NIPD Genetics,
Nicosia, Cyprus) (Supplementary materials and methods). Notably, 14
of 32 tumor samples were also genotyped on Ion Personal Genome
Machine (Thermo Fisher/IonTorrent) at the MOL with a custom
Ampliseq panel (IAD68363_167), targeting mutations in 55 genes, as
previously described (14).
Library construction with the MOL panel was performed with standard
protocols, using 20 ng DNA per sample and the Ampliseq primers
along with the Ampliseq Library Kit v.2.0 and Ion Xpress barcodes
(Life Technologies, Carlsbad, CA). Resulting libraries, once
normalized to 15 ng/ml, were clonally amplified on the One-Touch-2
instrument, enriched on the OneTouch ES station and sequenced on
the Ion Personal Genome Machine sequencer. For data retrieval, base
calling was performed on the Torrent Server with Torrent Suite
v.4.4.2. Consequently, variants were annotated with Ion Reporter
v.4 and accepted for analysis if they had P-value <0.0001;
>100 amplicon reads; position coverage >100; variant coverage
>40 for position coverage of 100; variant allele frequency (VAF)
>5% and Indels without GC stretches. Finally, only tumor samples
with mean depth >150 and at least 5 variants were considered
eligible and included in the study (14). The study design is presented in
Fig. 1.
Bioinformatics analysis
Sequencing data were de-multiplexed with bcl2fastq
(v.2.16.0) and paired-end DNA sequencing reads were processed to
remove adapter sequences and poor-quality reads. The remaining
sequences were aligned to the human reference genome build (hg19)
using the Burrows-Wheeler alignment algorithm (15). Duplicate read entries were removed
and aligned reads files were converted to a binary (BAM) format
(16). Variant calling was
performed using a versatile somatic variant caller (17) and variant annotation was performed
using the ENSEMBLE Variant Effect Predictor (VEP) (18). Subsequently, variants were filtered
on the basis of variant mapping and read quality (amplicon coverage
>100; and base coverage >50). Moreover, variants that
fulfilled selection criteria for rarity (minor allele frequency
<0.1% based on dbSNP, 5000 Exomes and ExAC) and were identified
in coding regions and splice junctions were included in the study.
The identified variants were classified and interpreted in
accordance with ClinVar and COSMIC databases (19,20).
Gene-level CNAs were detected using an in-house bioinformatics
pipeline that implements a circular binary segmentation method
(21). Rearrangement calling was
performed by utilizing discordant pair and split-read alignments
following local assembly, realignment, and an in-house filtering
pipeline to refine the set of candidate events (22–24).
Statistical analysis
The objectives of the study were descriptive.
Categorical variables were summarized using frequencies and
percentages while continuous variables using median (min, max).
Since Greece lacks a national Tumor Registry, we proportionally
estimated the number of glioma patients to the country's
approximate 10 million residents using information from
glioblastoma (GBM) patients in Malta, a country that is
geographically close to Greece. More specifically, a recent study
reported that glioblastoma has an incidence rate of 4.5 cases per
100,000 population in Malta (25).
Therefore, we assumed that there were 450 glioma patients in Greece
and 48 patients would generate a confidence interval of maximum
width +/- 14.07% for the percentage estimates. Associations between
categorical and continuous variables were examined using the
Kruskal-Wallis test whilst associations between categorical
variables were examined using the chi-squared test. Overall
survival was defined as the time elapsed from cancer diagnosis
until death or last contact. The Kaplan-Meier method was used to
estimate the survival rate since diagnosis. Univariable Cox
regression was used to generate hazard ratios which were presented
alongside 95% C.I. The long-rank test was used to examine
differences in event-free probability between patient subgroups.
Significance level was set to 0.05 for all significance tests. The
data management and statistical analysis were mainly performed
using the SAS software (version 9.4). The Python 3 and the R 4.2.0
programming languages were employed to build different types of
charts for data visualization. The R packages readxl and GenVisR
were used to produce the genetic variants' map.
Results
Patient characteristics
A total of 32 patients with high-grade gliomas were
involved in the present study, of which 10 (31.3%) were female and
22 (68.7%) were male. The median age of cancer diagnosis was 54.9
years of age, with ages ranging from 25.2 to 77.8 years of age.
Among the 31 patients who had a known type of surgery, biopsy was
carried out on 16.1% (5/31) while the vast majority of patients
underwent subtotal tumor excision (67.8%; 21/31). In our cohort,
tumors were predominantly glioblastomas (81.2%; 26/32) whereas
12.5% (4/32) were grade 3 astrocytomas. Of 30 cancer patients with
information on the tumor side, tumors were located in the right or
in the left hemisphere with the same ratio (each 46.7%; 14/30) and
two tumors in both hemispheres (6.6%; 2/30). All information is
summarized in Table I.
 | Table I.Clinical and histopathological
characteristics of patients diagnosed with gliomas. |
Table I.
Clinical and histopathological
characteristics of patients diagnosed with gliomas.
| Characteristic | Value |
|---|
| Age at diagnosis,
years |
|
| Median
(minimum-maximum) | 54.9
(25.2-77.8) |
| Sex |
|
|
Female | 31.3% (10/32) |
|
Male | 68.7% (22/32) |
| Type of
surgery |
|
| Biopsy
(<75% of the tumor) | 16.1% (5/31) |
|
Subtotal (75–99% of the
tumor) | 67.8% (21/31) |
| Total
excision | 16.1% (5/31) |
|
Unknown | 1 |
| Histology |
|
| Grade 3
astrocytoma | 12.5% (4/32) |
|
Glioblastoma | 81.2% (26/32) |
|
Othera | 6.3% (2/32) |
| Hemisphere |
|
|
Bilateral | 6.6% (2/30) |
|
Left | 46.7% (14/30) |
|
Right | 46.7% (14/30) |
|
Unknown | 2 |
| Necrosis |
|
| No | 21.9% (7/32) |
|
Yes | 78.1% (25/32) |
| Hemorrhage |
|
| No | 56.3% (18/32) |
|
Yes | 43.7% (14/32) |
| Endothelial
hyperplasia |
|
| No | 21.9% (7/32) |
|
Yes | 78.1% (25/32) |
Genomic landscape of somatic variants
in gliomas
In total, 96 variants (SNVs and InDels) were
identified in 30 of the 32 (93.75%) tumors in 34 genes, namely
APC, AR, ATM, ATRX, BARD1, BRCA1, CDKN2A, CHEK2, DICER1, EGFR,
ERBB2, ERBB3, FANCA, FGFR1, FGFR2, IDH1, KEAP1, MLH1, MRE11, MSH2,
NF1, NTRK1, NTRK3, RB1, PALB2, PIK3CA, POLD1, POLE, PTEN, RAD51D,
STK11, TMPRSS2, TP53 and ZNF276. The number of
identified variants per sample ranged from 1 to 14. Among the 30
tumors with variants, 6 (20%) had only one, 9 (30%) had two and 15
(50%) had three or more variants. Two tumor samples (6.25%; 2/32)
demonstrated no variants in the targeted genomic regions. In total,
the analysis revealed 38 pathogenic variants and 58 variants of
unknown clinical significance (VUS) of which 5 were in-frame
insertions/deletions. Among 38 pathogenic variants in 23 tumor
samples in 13 genes, 19 were missense, 7 were frameshift, 6 were
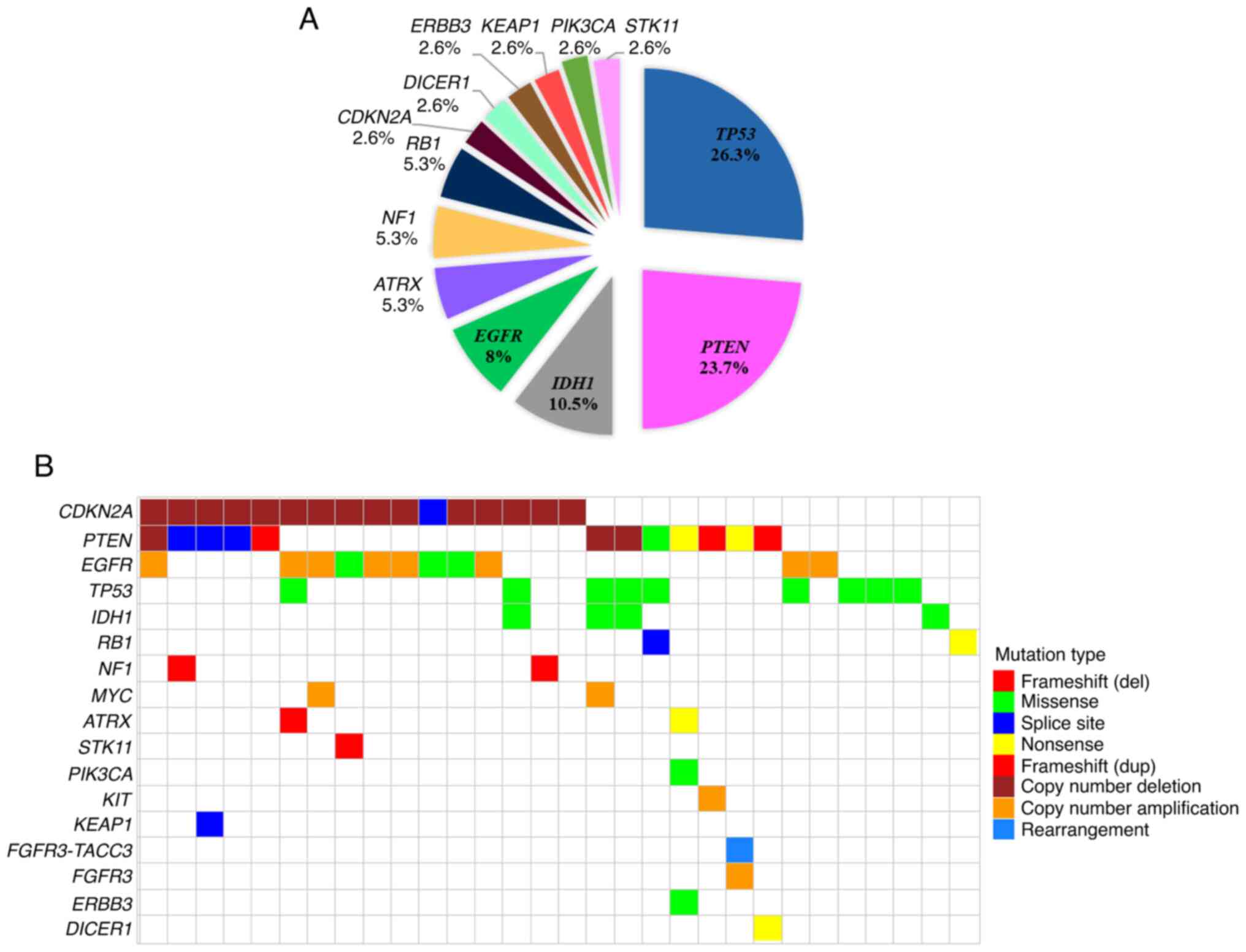
nonsense and 6 affected a conserved splice-site. TP53
mutations were the most prevalent (10/38; 26.3%), followed by
PTEN (9) and IDH1
(4) (23.7 and 10.5% respectively).
Four out of ten TP53 pathogenic variants were located at
mutational hotspot codons 248 and 273. Notably, all the IDH1
mutations involved the p.(R132H) one. In EGFR, all
pathogenic variants were located in the extracellular domain. The
distribution of pathogenic variants per gene is shown in Fig. 2A.
Twenty-two out of 32 tumor samples (68.75%) showed
evidence of structural variants (SVs) (Table SII). Overall, 33 SVs were
identified of which the copy number deletions (53.2%; 17 out of 32
patients) were more frequent. Copy number amplifications involved
40.7% (13 out of 32 patients), followed by rearrangements (1 out of
32 patients; 3.1%) (Fig. S1A).
Copy number deletions were identified in CDKN2A (46.9%; 15
out of 32 patients) and PTEN (9.4%; 3 out of 32 patients)
whereas gene amplifications were identified most frequently in
EGFR (31.3%; 10 out of 32 patients) followed by MYC
(6.3%; 2 out of 32 patients), FGFR3 and KIT (3.1%; 1
out of 32 patients for each gene). Interestingly, a
FGFR3-TACC3 rearrangement was also identified in one patient
(1/32; 3.1%) concurrently with FGFR3 amplification (Fig. S1B). And also interestingly, 8 out
of 22 patients with copy number alterations had concurrent CNAs,
predominately CDKN2A deletions with EGFR
amplifications (Fig. S1C).
Finally, it was noteworthy to mention that 15 out of
the 32 tumors had both SNVs/Indels and SVs. The distribution of
genetic alterations encompassing SNVs, InDels, and SVs per gene and
per tumor is demonstrated in Fig.
2B.
Comparison of mutational profiles
derived from a comprehensive pan-cancer gene panel and EMO
panel
To evaluate the newly implemented targeted
sequencing panel, we compared the results from analysis of 14 tumor
blocks genotyped with both the comprehensive pan-cancer and the MOL
panels. Implementation of the comprehensive pan-cancer and the MOL
panels resulted in the identification of 37 and 15 variants,
respectively. Of those, 13 were common. Comprehensive pan-cancer
panel identified 24 additional variants, 22 of which located in
regions that were not targeted by the MOL panel. On the contrary,
the MOL panel identified two additional variants. Of these, one was
located in a non-targeted region by a comprehensive pan-cancer
panel. Interestingly, six variants that were identified only by the
comprehensive pan-cancer panel were pathogenic, whereas two
variants identified only by the MOL panel were pathogenic.
Subsequently, 18 tumor blocks were genotyped with a
comprehensive pan-cancer panel only. A total of 59 genetic variants
were identified, of which 43 were located in regions targeted by
the comprehensive pan-cancer panel only, while the remaining 16
variants were detected in regions by both panels. Notably,
twenty-one of all detected variants were pathogenic.
Associations between clinicopathological
features and mutations in IDH1, PTEN and TP53 genes
IDH1 mutational status
Patients with IDH1 positive tumors were
diagnosed with brain tumors at a statistically significant younger
age than those with IDH1 wild-type tumors (34.4 vs. 56.3
years, P<0.05). Moreover, IDH1 mutational status was
significantly associated with specific type of surgery
(x2 P<0.05). The majority (75%; 3/4) of patients with
IDH1 positive tumors carried out biopsy whereas 25% (1/4)
underwent subtotal excision. In contrast, the majority (74.1%;
20/27) of patients with IDH1 wild-type tumors underwent
subtotal surgery, while 18.5% (5/27) performed total excision and
7.4% (2/27) carried out biopsy. Additionally, half of the
IDH1 positive tumors were glioblastomas, one was grade 3
astrocytomas and one was anaplastic oligodendroglioma. IDH1
mutant tumors were located either in the right or left hemisphere
with the same proportion (50% each). This was also the case with
the IDH1 wild-type tumors (46.2% each). All data are
summarized in Table II.
| Table II.Associations between
clinicopathological characteristics and the mutational status of
several genes. |
Table II.
Associations between
clinicopathological characteristics and the mutational status of
several genes.
|
|
| IDH1 mutation | PTEN mutation | TP53 mutation |
|---|
|
|
|
|
|
|
|---|
| Characteristic | Eligible | No | Yes | P-value | No | Yes | P-value | No | Yes | P-value |
|---|
| Age at diagnosis,
years | 32 | 56.3 | 34.4 | 0.003a | 52.1 | 59.6 | 0.167 | 58.7 | 42.6 | 0.065 |
|
|
| (40.3,77.8) | (25.2,42.1) |
| (25.2,77.8) | (40.3,69.2) |
| (40.3,77.8) | (25.2,59.1) |
|
| Sex | 32 |
|
| 0.28 |
|
| 0.87 |
|
| 0.66 |
|
Female |
| 10 (35.7) | 0 (0.0) |
| 7 (30.4) | 3 (33.3) |
| 7 (30.4) | 3 (33.3) |
|
|
Male |
| 18 (64.3) | 4 (100.0) |
| 16 (69.6) | 6 (66.7) |
| 16 (69.6) | 6 (66.7) |
|
| Surgery
specify | 31 |
|
| 0.02a |
|
| 0.49 |
|
| 0.57 |
| Biopsy
(<75% of the tumor) |
| 2 (7.4) | 3 (75.0) |
| 3 (13.6) | 2 (22.2) |
| 2 (8.7) | 3 (33.3) |
|
|
Subtotal (75–99% of the
tumor) |
| 20 (74.1) | 1 (25.0) |
| 14 (63.6) | 7 (77.8) |
| 17 (73.9) | 4 (44.4) |
|
| Total
excision |
| 5 (18.5) | 0 (0.0) |
| 5 (22.7) | 0 (0.0) |
| 3 (13.0) | 2 (22.2) |
|
|
Unknown |
|
|
|
|
|
|
| 1 (4.3) | 0 (0.0) |
|
| Histology | 32 |
|
| 0.15 |
|
| 0.45 |
|
| 0.99 |
| Grade 3
astrocytoma |
| 3 (10.7) | 1 (25.0) |
| 4 (17.4) | 0 (0.0) |
| 3 (13.0) | 1 (11.1) |
|
|
Glioblastoma |
| 24 (85.7) | 2 (50.0) |
| 17 (73.9) | 9 (100.0) |
| 18 (78.3) | 8 (88.9) |
|
|
Other/Unknown |
| 1 (3.6) | 1 (25.0) |
| 2 (8.7) | 0 (0.0) |
| 2 (8.7) | 0 (0.0) |
|
| Hemisphere | 30 |
|
| 0.99 |
|
| 0.83 |
|
| 0.83 |
|
Bilateral |
| 2 (7.7) | 0 (0.0) |
| 2 (9.1) | 0 (0.0) |
| 2 (9.5) | 0 (0.0) |
|
|
Left |
| 12 (46.2) | 2 (50.0) |
| 9 (40.9) | 5 (62.5) |
| 9 (42.9) | 5 (55.6) |
|
|
Right |
| 12 (46.2) | 2 (50.0) |
| 11 (50) | 3 (37.5) |
| 10 (47.6) | 4 (44.4) |
|
| Necrosis | 32 |
|
| 0.20 |
|
| 0.36 |
|
| 0.46 |
| No |
| 5 (17.9) | 2 (50.0) |
| 6 (26.1) | 1 (11.1) |
| 6 (26.1) | 1 (11.1) |
|
|
Yes |
| 23 (82.1) | 2 (50.0) |
| 17 (73.9) | 8 (88.9) |
| 17 (73.9) | 8 (88.9) |
|
| Hemorrhage | 32 |
|
| 0.61 |
|
| 0.40 |
|
| 0.99 |
| No |
| 15 (53.6) | 3 (75.0) |
| 14 (60.9) | 4 (44.4) |
| 13 (56.5) | 5 (55.6) |
|
|
Yes |
| 13 (46.4) | 1 (25.0) |
| 9 (39.1) | 5 (55.6) |
| 10 (43.5) | 4 (44.4) |
|
| Endothelial
hyperplasia | 32 |
|
| 0.20 |
|
| 0.36 |
|
| 0.46 |
| No |
| 5 (17.9) | 2 (50.0) |
| 6 (26.1) | 1 (11.1) |
| 6 (26.1) | 1 (11.1) |
|
|
Yes |
| 23 (82.1) | 2 (50.0) |
| 17 (73.9) | 8 (88.9) |
| 17 (73.9) | 8 (88.9) |
|
PTEN mutational status
Tumors with PTEN mutations (SNVs and InDels)
were identified in older patients when compared to PTEN
wild-type tumors (59.6 vs. 52.1 years) although this finding was
not statistically significant. The vast majority (77.8%; 7/9) of
patients with PTEN mutant tumors underwent subtotal surgery,
while all involved glioblastomas. No associations were observed
between PTEN mutational status and the side of the tumor
(Table II).
TP53 mutational status
Tumors with TP53 mutations were more
frequently detected in younger patients, when compared to
TP53 wild-type tumors (42.6 vs. 58.7 years). One third of
patients (3/9; 33.3%) with TP53 positive tumors carried out
biopsy. Most ofTP53 mutant tumors were glioblastomas (8/9;
88.9%). According to available information (n=30), five patients
with TP53 mutant tumors developed left-sided and four
right-sided tumors, respectively (Table II).
Associations of the mutational status
between IDH1, PTEN and TP53 genes
Wild-type TP53 was present in IDH1
wild-type tumors only (P<0.05). Similarly, wild-type TP53
was more frequently identified in PTEN wild-type than
PTEN mutated tumors. Additionally, PTEN mutations did
not co-occur with IDH1 mutations. All the data are
summarized in Table III.
| Table III.Associations of the mutational status
between IDH1, PTEN and TP53. |
Table III.
Associations of the mutational status
between IDH1, PTEN and TP53.
| A, IDH1
mutation |
|---|
|
|---|
| Mutation | No | Yes | P-value |
|---|
| TP53
mutation |
|
| 0.25 |
| No | 22 (78.6) | 2 (50.0) |
|
|
Yes | 6 (21.4) | 2 (50.0) |
|
| PTEN
mutation |
|
| 0.55 |
| No | 20 (71.4) | 4 (100.0) |
|
|
Yes | 8 (28.6) | 0 (0.0) |
|
|
| B, PTEN
mutation |
|
|
Mutation | No | Yes | P-value |
|
| TP53
mutation |
|
| 0.18 |
| No | 15 (65.2) | 8 (88.9) |
|
|
Yes | 8 (34.8) | 1 (11.1) |
|
| IDH1
mutation |
|
| 0.54 |
| No | 20 (87.0) | 9 (100.0) |
|
|
Yes | 3 (13.0) | 0 (0.0) |
|
|
| C, TP53
mutation |
|
|
Mutation | No | Yes | P-value |
|
| PTEN
mutation |
|
| 0.18 |
| No | 15 (65.2) | 8 (88.9) |
|
|
Yes | 8 (34.8) | 1 (11.1) |
|
| IDH1
mutation |
|
| 0.017a |
| No | 23 (100.0) | 6 (66.7) |
|
|
Yes | 0 (0.0) | 3 (33.3) |
|
Patient outcomes
Herein, was assessed whether having a mutant tumor
on a specific gene had an impact on patients' overall survival
(OS). Notably, all the patients received standard of care
treatment. The median follow-up of patients from diagnosis until
death or last contact was 19.2 months and during this time period
29 deaths (90.6%; 29/32) occurred and three patients were alive
(9.4%; 3/32). Our analysis showed that patients with IDH1
mutant tumors had a statistically significant longer survival when
compared to patients with IDH1 wild-type tumors, although
due to small size, a confidence interval was not available
(Fig. 3).
Discussion
In the present study, we explored the genomic
landscape of 32 tumor tissues from patients with high-grade gliomas
by massively parallel sequencing using an 80 multigene panel.
Subsequently, we examined the correlation between our results and
patient outcomes as well as histopathological characteristics.
Additionally, implementing two distinct panels on 14 common
samples, we evaluated the molecular profiles of tumors.
Our findings showed that 96 genetic alterations
(SNVS and InDels) were identified in the tumor samples. Of these,
38 were pathogenic and scattered among 13 distinct genes. IDH1,
PTEN, and TP53 pathogenic variants exhibited high
incidence in mutational spectrum, with TP53 being the most
prevalent. These results were in line with those of other studies
since the p53 pathway was deregulated in many cancers and these
three genes, along with EGFR, were the most frequently
altered genes in gliomas (26–29).
In the present study, the most frequently detected
mutations at codons 248 and 273 of the TP53 gene were also
found in TP53 positive tumors, but at a decreased frequency
compared to other series (30,31).
Additionally, among IDH1 mutated tumors, the p.(R132H)
variant was solely found. Given that more than 90% of all
IDH1 mutants have the present variant, this finding is
consistent with other reports (32). Genetic alterations were also
detected in genes that were involved in cell cycle regulation, DNA
damage response pathways, the MAPK/PI3K pathway, the receptor
tyrosine kinase pathway and telomere maintenance including ATRX,
CDKN2A, NF1 andRB1. These genes could also serve as
therapeutic targets for the design of a more individualized
treatment protocol (33).
Furthermore, the use of the comprehensive pan-cancer
panel allowed for the detection of structural variations. The
identification of CDKN2A deletions and EGFR
amplifications is consistent with previous research since both
genetic changes are frequent in glioblastomas and have previously
been linked to a poor prognosis (34). Additionally, an FGFR-TACC3
rearrangement was identified; these genomic events are present in
approximately 2–3% of glioblastomas. Clinical studies and case
reports have provided some preliminary evidence that suggests that
this result could serve as an actionable therapeutic target in
advanced solid tumors. Erdafitinib, a pan-FGFR tyrosine kinase
inhibitor, produced one stable disease and two partial responses in
three glioblastoma patients with FGFR-TACC3 positive tumors
in two phase I studies (35,36).
We also assessed the extended panel's clinical
usefulness and accuracy. We presented a comparison of the results
from the tumor analysis using both the comprehensive pan-cancer
panel and the MOL panel. We observed that there was a high
concordance (86.7%; 13/15) between the two panels since 13 variants
were common. It's noteworthy that a significant portion (22/24;
91.6%) of additional variants that were identified by the
comprehensive pan-cancer panel occurred in regions that were not
targeted by the MOL panel, emphasizing the necessity of
implementing a broader panel with additional targeted regions.
In this study, a mutation in exon 14 of the
EGFR gene was found in the tumor of a glioblastoma patient,
using the comprehensive pan-cancer panel; this exon was not
included in the MOL panel. Although numerous prior studies
involving EGFR tyrosine kinase inhibitors have failed to
demonstrate anti-tumor efficacy in tumors with EGFR
mutations, there is still continuing research using anti
EGFR-immunotherapy approaches, such as antibody-based strategies
and vaccines (37,38).
Subsequently, variant calling parameters; such as
relatively low variant allele frequency (VAF; ≤5%) and/or low base
coverage (≤50%), explained why despite the fact that two genetic
alterations although were targeted by the MOL panel, they were not
detected and reported.
Moreover, a mutation in the LZTR1 gene's
Kelch domain was only detected by the MOL panel due to the absence
of the LZTR1 gene from the extended panel. This could be
considered a limitation of the comprehensive pan-cancer panel,
because it has been demonstrated that LZTR1 mutations
facilitate glioma sphere development and self-renewal, these
mutations could be considered as a potential therapeutic target for
glioma (39,40). Additionally, the IDH1
p.(R132H) mutation was identified by the MOL panel but not by the
comprehensive pan-cancer panel, although it was targeted by the
latter, and sufficient quality control parameters were reached,
ruling out the likelihood of decreased assay sensitivity. This
disagreement may be explained by the fact that both assays used DNA
that was isolated from two separate sets of FFPE sections from the
same FFPE block, demonstrating intra-tumor heterogeneity (41,42).
Finally, our findings showed that SVs could be identified using NGS
data if the suitable NGS platform and multigene panel were
implemented. This is essential for the comprehensive molecular
characterization of tumor profiles.
Therefore, the implementation of a pan-cancer panel
enables the identification of a larger number of mutations as well
as rare genomic events, thus providing more therapeutic choices for
patients with gliomas. Consequently, these findings may help with
glioma patient diagnosis, prognosis, eligibility for clinical trial
enrollment, and treatment as previously described (12,28,43).
The application of broad panel sequencing also
provided insightful information about clinicopathological traits
and patient outcomes. We found that IDH1 positive tumors
were more frequently detected in younger patients, when compared to
IDH1 wild-type tumors. Patients with IDH1 mutated
tumors were characterized by better prognosis compared to patients
with IDH1 wild-type tumors which is consistent with previous
studies (44–46). Patients with IDH1 mutant
tumors mainly underwent biopsies rather than tumor resections; that
were more frequently diffused and cannot be completely removed.
Moreover, we observed that patients with TP53
positive tumors were more likely to be diagnosed with cancer at a
younger age than patients with wild-type TP53 tumors, as
expected (47). Interestingly,
PTEN mutations were mutually exclusive with IDH1
mutations and wild-type TP53 tumors were presented in
IDH1 wild-type tumors only (14,26,48).
However, these data should be regarded with caution due to the
small examined number of tumors.
All patients in our study have received
standard-of-care and have long term follow up that reflects actual
survival. This, gives our study a benefit in identifying prognostic
molecular biomarkers for glioma patients.
These observations are consistent with other reports
and established findings as mentioned above.
Furthermore, both platforms verified the tumor
genotyping results for the common samples. This study also
demonstrates the importance of collaboration between many
institutions and clinics in the acquisition of tumor blocks, along
with comprehensive clinical information, histology reports, and
patient outcomes. On the other hand, there are certain restrictions
with the present study.
First of all, the small sample size restricts the
statistical power of our research, necessitating careful
interpretation of the results. Second, tumors were chosen based on
the quantity and quality of available tissue, thereby bringing
selection bias into the study.
Overall, utilizing FFPE samples in clinical and
research contexts, the use of a broader sequencing panel enables
the simultaneous identification of several genetic alterations in a
wide range of genes across gliomas, improving tumor molecular
characterization and classification. The application of the new
technology improves clinical outcomes by facilitating accurate
molecular diagnosis as well as the identification of known and
candidate prognostic biomarkers that could serve as possible
therapeutic targets for the personalized treatment of glioma
patients.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by an internal HeCOG research grant
(grant no. HE R_17/15), by NIPD Genetics Limited and by a HeSMO
Grant.
Availability of data and materials
We have submitted our data to the ‘Figshare’
Repository. They are available from Romanidou, Ourania (2022):
Accession no. figshare. Dataset. Version 1 25.10.22, 13:15 first
online date https://doi.org/10.6084/m9.figshare.21394077.v1 and
the link https://doi.org/10.6084/m9.figshare.21394077.
Authors' contributions
OR, PA, VK, GF and PCP conceptualized the study. KK,
AA, CLo, CLe, AP and FF analyzed the data. KT, AE, KP and VK
performed the experiments. OR, MI, EK, GK, GR, IX, GF and PCP
acquired the data. KT, AP and FF validated the reproducibility of
results. KT and KP designed the methodology of next generation
sequencing. AA, ChrL and AP software development. AE, AA and ChaL
curated the data. MI, EK and GK supervised the study. PCP was
project administrator and acquired funding. OR, PA, AE and GF wrote
the original draft. PA, KK, KT, AA, CLo, CLe, MI, EK, GK, KP, AP,
GR, IX, FF, VK and PCP wrote or revised critically the manuscript.
KP and AE confirm the authenticity of all the raw data. All authors
read and approved the final manuscript and agreed to be accountable
for all aspects of the work in ensuring that questions related to
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The study was approved by the Bioethics Committee of
the Aristotle University of Thessaloniki School of Medicine
(approval no. 2/February 4, 2015) and by Cyprus National Bioethics
Committee (approval no. EEBK/EΠ/2016/54; 22/4/2021).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CNAs
|
copy number alterations
|
|
CNS
|
central nervous system
|
|
FFPE
|
formalin-fixed, paraffin-embedded
|
|
HeCOG
|
Hellenic cooperative oncology
group
|
|
InDels
|
insertions and deletions
|
|
MOL
|
laboratory of molecular oncology
|
|
NGS
|
next generation sequencing
|
|
OS
|
overall survival
|
|
P/LP
|
pathogenic/likely pathogenic
|
|
SNVs
|
single nucleotide variants
|
|
SVs
|
structural variants
|
|
VEP
|
variant effect predictor
|
|
VUS
|
variants of unknown clinical
significance
|
|
WHO
|
World Health Organization
|
References
|
1
|
Schwartzbaum JA, Fisher JL, Aldape KD and
Wrensch M: Epidemiology and molecular pathology of glioma. Nat Clin
Pract Neurol. 2:494–503. 15162006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Brada M, van den Bent MJ, Tonn JC
and Pentheroudakis G; ESMO Guidelines Working Group, : High-grade
glioma: ESMO clinical practice guidelines for diagnosis, treatment
and follow-up. Ann Oncol. 25 (Suppl 3):iii93–iii101. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ostrom QT, Cioffi G, Gittleman H, Patil N,
Waite K, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical
report: Primary brain and other central nervous system tumors
diagnosed in the United States in 2012–2016. Neuro Oncol. 21 (Suppl
5):v1–v100. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wesseling P and Capper D: WHO 2016
classification of gliomas. Neuropathol Appl Neurobiol. 44:139–150.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Śledzińska P, Bebyn MG, Furtak J,
Kowalewski J and Lewandowska MA: Prognostic and predictive
biomarkers in gliomas. Int J Mol Sci. 22:103732021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Weller M and Reifenberger G: Beyond the
World Health Organization classification of central nervous system
tumors 2016: What are the new developments for gliomas from a
clinician' perspective? Curr Opin Neurol. 33:701–706. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kristensen BW, Priesterbach-Ackley LP,
Petersen JK and Wesseling P: Molecular pathology of tumors of the
central nervous system. Ann Oncol. 30:1265–1278. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reinhardt A, Stichel D, Schrimpf D, Sahm
F, Korshunov A, Reuss DE, Koelsche C, Huang K, Wefers AK, Hovestadt
V, et al: Anaplastic astrocytoma with piloid features, a novel
molecular class of IDH wildtype glioma with recurrent MAPK pathway,
CDKN2A/B and ATRX alterations. Acta Neuropathol. 136:273–291. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ostrom QT, Kinnersley B, Wrensch MR,
Eckel-Passow JE, Armstrong G, Rice T, Chen Y, Wiencke JK, McCoy LS,
Hansen HM, et al: Sex-specific glioma genome-wide association study
identifies new risk locus at 3p21.31 in females, and finds
sex-differences in risk at 8q24.21. Sci Rep. 8:73522018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ostrom QT, Cote DJ, Ascha M, Kruchko C and
Barnholtz-Sloan JS: Adult glioma incidence and survival by race or
ethnicity in the united states from 2000 to 2014. JAMA Oncol.
4:1254–1262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zacher A, Kaulich K, Stepanow S, Wolter M,
Köhrer K, Felsberg J, Malzkorn B and Reifenberger G: Molecular
diagnostics of gliomas using next generation sequencing of a
glioma-tailored gene panel. Brain Pathol. 27:146–159. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim HS, Kwon MJ, Song JH, Kim ES, Kim HY
and Min KW: Clinical implications of TERT promoter mutation on IDH
mutation and MGMT promoter methylation in diffuse gliomas. Pathol
Res Pract. 214:881–888. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Razis E, Kotoula V, Koliou GA,
Papadopoulou K, Vrettou E, Giannoulatou E, Tikas I, Labropoulos SV,
Rigakos G, Papaemmanoyil S, et al: Is there an independent role of
TERT and NF1 in high grade gliomas? Transl Oncol. 13:346–354. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li H: Aligning sequence reads, clone
sequences and assembly contigs with BWA-MEM. arXiv.
3:130339972013.
|
|
16
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The sequence alignment/map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li MM, Datto M, Duncavage EJ, Kulkarni S,
Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ,
Younes A and Nikiforova MN: Standards and guidelines for the
interpretation and reporting of sequence variants in cancer: A
joint consensus recommendation of the association for molecular
pathology, American society of clinical oncology, and college of
American pathologists. J Mol Diagn. 19:4–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McLaren W, Gil L, Hunt SE, Riat HS,
Ritchie GR, Thormann A, Flicek P and Cunningham F: The ensembl
variant effect predictor. Genome Biol. 17:1222016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Landrum MJ, Lee JM, Benson M, Brown GR,
Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al:
ClinVar: Improving access to variant interpretations and supporting
evidence. Nucleic Acids Res. 46(D1): D1062–D1067. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tate JG, Bamford S, Jubb HC, Sondka Z,
Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E,
et al: COSMIC: The catalogue of somatic mutations in cancer.
Nucleic Acids Res. 47(D1): D941–D947. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Seshan VE and Olshen AB: DNAcopy: A
package for analyzing DNA copy data. Bioconductor Vignette. 1–7.
2014.
|
|
22
|
Chen X, Schulz-Trieglaff O, Shaw R, Barnes
B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S and Saunders CT:
Manta: Rapid detection of structural variants and indels for
germline and cancer sequencing applications. Bioinformatics.
32:1220–1222. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cameron DL, Schröder J, Penington JS, Do
H, Molania R, Dobrovic A, Speed TP and Papenfuss AT: GRIDSS:
Sensitive and specific genomic rearrangement detection using
positional de Bruijn graph assembly. Genome Res. 27:2050–2060.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Layer RM, Chiang C, Quinlan AR and Hall
IM: LUMPY: A probabilistic framework for structural variant
discovery. Genome Biol. 15:R842014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grech N, Dalli T, Mizzi S, Meilak L,
Calleja N and Zrinzo A: Rising incidence of glioblastoma multiforme
in a well-defined population. Cureus. 12:e81952020.PubMed/NCBI
|
|
26
|
Nie Q, Hsiao MC, Chandok H, Rowe S, Prego
M, Meyers B, Omerza G, Hesse A, Uvalic J, Soucy M, et al: Molecular
profiling of CNS tumors for the treatment and management of
disease. J Clin Neurosci. 71:311–315. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ballester LY, Fuller GN, Powell SZ, Sulman
EP, Patel KP, Luthra R and Routbort MJ: Retrospective analysis of
molecular and immunohistochemical characterization of 381 primary
brain tumors. J Neuropathol Exp Neurol. 76:179–188. 2017.PubMed/NCBI
|
|
28
|
Zeng C, Wang J, Li M, Wang H, Lou F, Cao S
and Lu C: Comprehensive molecular characterization of Chinese
patients with glioma by extensive next-generation sequencing panel
analysis. Cancer Manag Res. 13:3573–3588. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zheng S, Alfaro-Munoz K, Wei W, Wang X,
Wang F, Eterovic AK, Shaw KRM, Meric-Bernstam F, Fuller GN, Chen K,
et al: Prospective clinical sequencing of adult glioma. Mol Cancer
Ther. 18:991–1000. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gillet E, Alentorn A, Doukouré B,
Mundwiller E, van Thuijl HF, Reijneveld JC, Medina JA, Liou A,
Marie Y, Mokhtari K, et al: TP53 and p53 statuses and their
clinical impact in diffuse low grade gliomas. J Neurooncol.
118:131–139. 2014.PubMed/NCBI
|
|
31
|
Pessôa IA, Amorim CK, Ferreira WAS, Sagica
F, Brito JR, Othman M, Meyer B, Liehr T and de Oliveira EHC:
Detection and correlation of single and concomitant TP53, PTEN, and
CDKN2A alterations in gliomas. Int J Mol Sci. 20:26582019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park C, Kim TM, Bae JM, Yun H, Kim JW,
Choi SH, Lee ST, Lee JH, Park SH and Park CK: Clinical and genomic
characteristics of adult diffuse midline glioma. Cancer Res Treat.
53:389–398. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Reis GF, Pekmezci M, Hansen HM, Rice T,
Marshall RE, Molinaro AM, Phillips JJ, Vogel H, Wiencke JK, Wrensch
MR, et al: CDKN2A loss is associated with shortened overall
survival in lower-grade (World Health Organization grades II–III)
astrocytomas. J Neuropathol Exp Neurol. 74:442–452. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Loriot Y, Schuler MH, Iyer G, Witt O, Doi
T, Qin S, Tabernero J, Reardon DA, Massard C, Palmer D, et al:
Tumor agnostic efficacy and safety of erdafitinib in patients (pts)
with advanced solid tumors with prespecified fibroblast growth
factor receptor alterations (FGFRalt) in RAGNAR: Interim analysis
(IA) results. J Clin Oncol. 40 (16 Suppl):S30072022. View Article : Google Scholar
|
|
36
|
Costa R, Carneiro BA, Taxter T, Tavora FA,
Kalyan A, Pai SA, Chae YK and Giles FJ: FGFR3-TACC3 fusion in solid
tumors: Mini review. Oncotarget. 7:55924–55938. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang K, Wu Z, Zhang H, Zhang N, Wu W, Wang
Z, Dai Z, Zhang X, Zhang L, Peng Y, et al: Glioma targeted therapy:
Insight into future of molecular approaches. Mol Cancer. 21:392022.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Higa N, Akahane T, Hamada T, Yonezawa H,
Uchida H, Makino R, Watanabe S, Takajo T, Yokoyama S, Kirishima M,
et al: Detection of EGFR mutation distribution and transcriptional
variants in IDH-wildtype high-grade gliomas using a next-generation
sequencing oncopanel. Res Sq. 1–11. 2021.
|
|
39
|
Wang Y, Zhang J, Zhang P, Zhao Z, Huang Q,
Yun D, Chen J, Chen H, Wang C and Lu D: LZTR1 inactivation promotes
MAPK/ERK pathway activation in glioblastoma by stabilizing
oncoprotein RIT1. bioRxiv. 2020.
|
|
40
|
Frattini V, Trifonov V, Chan JM, Castano
A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al: The
integrated landscape of driver genomic alterations in glioblastoma.
Nat Genet. 45:1141–1149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Comba A, Faisal SM, Varela ML, Hollon T,
Al-Holou WN, Umemura Y, Nunez FJ, Motsch S, Castro MG and
Lowenstein PR: Uncovering spatiotemporal heterogeneity of
high-grade gliomas: From disease biology to therapeutic
implications. Front Oncol. 11:7037642021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sottoriva A, Spiteri I, Piccirillo SG,
Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C and Tavaré
S: Intratumor heterogeneity in human glioblastoma reflects cancer
evolutionary dynamics. Proc Natl Acad Sci USA. 110:4009–4014. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tirrò E, Massimino M, Broggi G, Romano C,
Minasi S, Gianno F, Antonelli M, Motta G, Certo F, Altieri R, et
al: A custom DNA-based NGS panel for the molecular characterization
of patients with diffuse gliomas: Diagnostic and therapeutic
applications. Front Oncol. 12:8610782022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zou P, Xu H, Chen P, Yan Q, Zhao L, Zhao P
and Gu A: IDH1/IDH2 mutations define the prognosis and molecular
profiles of patients with gliomas: A meta-analysis. PLoS One.
8:e687822013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yan H, Parsons DW, Jin G, McLendon R,
Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ,
et al: IDH1 and IDH2 mutations in gliomas. N Engl J Med.
360:765–773. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sun H, Yin L, Li S, Han S, Song G, Liu N
and Yan C: Prognostic significance of IDH mutation in adult
low-grade gliomas: A meta-analysis. J Neurooncol. 113:277–284.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Park Y, Park J, Ahn JW, Sim JM, Kang SJ,
Kim S, Hwang SJ, Han SH, Sung KS and Lim J: Transcriptomic
landscape of lower grade glioma based on age-related non-silent
somatic mutations. Curr Oncol. 28:2281–2295. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ichimura K, Pearson DM, Kocialkowski S,
Bäcklund LM, Chan R, Jones DT and Collins VP: IDH1 mutations are
present in the majority of common adult gliomas but rare in primary
glioblastomas. Neuro Oncol. 11:341–347. 2009. View Article : Google Scholar : PubMed/NCBI
|