Introduction
Immunotherapy has improved the options for cancer
therapy by enabling the immune cells to target cancer cells in a
patient. Immunotherapy is now clinically offered for melanoma and
lung cancer patients, while some cancers, including the majority of
colorectal cancer (CRC), remain recalcitrant (1–3).
Immunotherapy has been approved for microsatellite
instable (MSI) types of CRC (4),
which account for only 10–15% of the total CRC cases. This leaves
up to 85% of the cases, most of which display microsatellite
stability (MSS), to be managed through the conventional approach.
While early and localized cases of CRC may be surgically resected
with or without neoadjuvant therapy, advanced and metastatic cases
are treated with chemotherapy drugs or combinations of surgery,
radiation and drugs (2,5).
Studies on MSI CRC responsive to immunotherapy
suggest that the genomic instability in such tumors may enable
generation of neoantigens that are recognized by immune cells
(4,6,7).
Such instability combined with defects in mismatch repair would
enhance the visibility of altered cancer cells to T-cells which
could orchestrate cytotoxic responses. Despite such mechanistically
sound explanation for the visibility of MSI tumors, not all MSI
tumors respond to immunotherapy, and the reasons for this remain to
be uncovered.
Multiple theories have been proposed for the
non-responsiveness of most CRC tumors to immunotherapy (6). One of the possibilities is
immune-exclusion, where the lack of immune cells in the
microenvironment may be the primary factor. However, the abundance
of immune cells in the microenvironment has been associated with
either positive or negative outcomes (5,8–10).
In addition to the physical presence of immune cells in the tumor,
other factors including the sub-types and proportions of the immune
cells, their activation status, and the cytokine microenvironment
may modulate the effector functions and clinical outcomes (6,11,12).
Cancer cells are known to respond to replication
stress, chemotherapy, and radiation by activating DNA-damage
response (DDR) and nuclear factor-κB (NF-κB) pathways (13–18),
both of which may serve as survival mechanisms for the cells
damaged during therapy. Studies on CRC treatment to strategically
combine chemotherapy, radiation, and immunotherapy are ongoing,
with some encouraging preliminary results (19–26).
However, mechanistic studies on resistance mechanisms, and
experiments to rationalize combination strategies are urgently
needed (27). Combination
therapies could markedly benefit from studies that identify
effective strategies for patient selection, drugs to combine, and
timing and sequence of combination.
In this study, we examined the responses of CRC cell
lines treated with clinically used topoisomerase inhibitor in
combination with drugs that interfere with pathways initiated by
topoisomerase inhibition. We show that the cell surface expression
of immunoregulatory molecules on cancer cells may be modulated by
the combination of topoisomerase inhibitors and DDR pathway
blockers. This suggests that chemotherapy-activated pathways may be
identified and selectively targeted to enable the broader clinical
use of immunotherapy in CRC and to improve therapeutic
outcomes.
Materials and methods
Cell lines and culture
Colon cancer cells (SW620, RKO, HCT116) and the
colorectal cancer cell line (HT-29) were purchased from the
American Type Cell Culture (ATCC) collection. No further
authentication was done. Soon after receipt, multiple stock
cultures were prepared and stored in liquid nitrogen, and
periodically thawed and used for specific experiments. All parental
cell lines were grown in DMEM (Dulbecco's Modified Eagle's Medium,
Corning #10-013-CV) culture medium containing 10% fetal bovine
serum plus 10 µg/ml ciprofloxacin. SN-38-tolerant cell lines were
developed as previously described (28) using culture medium containing 10%
fetal bovine serum, penicillin (10,000 U/ml), and streptomycin (10
mg/ml). Briefly, parental cells were exposed to an initial SN-38
dose of 1 nM and cultured to a confluency of 80% for three passages
(~6 weeks). The cells that survived the initial SN-38 treatment
were then exposed to 5 nM SN-38 for three passages (~8 weeks) and
to 10 nM for three more passages (~eight weeks). Finally, the SN-38
concentration was increased to the clinically relevant plasma drug
concentration of 50 nM for three weeks.
Drugs and reagents
Stock concentrations of the compounds were prepared
in Dimethyl sulfoxide (DMSO) and stored at −20°C. SM-7368
(481411-5mg), CPT (C9911-250MG), KU60019 (SML1416-5MG), Irinotecan
(IRI) (SKU-I1406-50MG), and VE-822 (1232416-25-9) were purchased
from Sigma-Aldrich (St. Louis, MO). AZD7762 (Enzo, ENZ-CHM185-0005)
from ENZO. Flow Cytometry Staining Buffer (1X) (Cat# FC001), Mouse
PE-IgG2a (R&D IC003p), and Human HLA Class I (MHC I)
Phycoerythrin mAb (Clone W6/32) were purchased from R&D
Systems. Unless indicated otherwise, the drugs were used at the
following concentrations: CPT 0.5-1 µM; SM-7368 5 µM; irinotecan 25
µg/ml; VP-16 (Etoposide, LC laboratories) 25 µM; γ-IFN (Sigma) 50
ng/ml; VE822 (Selleckchem) 100 nM; AZD7762 100 nM, KU60019 at 1 µM;
erlotinib (LC laboratories) 20 µM; oxaliplatin (Sigma) 10 µM; taxol
(Sigma) 50 nM; 5-Fluorouracil (5-FU, Sigma) 10 µM. For experiments
in this study, single drug concentrations were pre-determined based
on the length of treatment (24–48 h) that induced the expression of
target proteins without visibly killing the treated cells.
Immunoblotting
Monolayer culture cells were treated in 6-cm culture
dishes and lysed in RIPA buffer containing a cocktail of protease
and phosphatase inhibitors. Samples containing equivalent protein
concentrations were resolved by SDS-PAGE and analyzed by
immunoblotting using pre-made 4–15% gradient gels. AI680 digital
imager (GE Lifesciences, Pittsburg, PA, USA) was used to scan the
chemiluminescent signals. Primary antibodies, at 1:1,000 dilutions,
used to target PD-L1 (catalog #13684, rabbit mAb) and were obtained
from Cell Signaling Technologies (Danvers, MA, USA).
Peroxidase-conjugated anti-rabbit and anti-mouse IgG secondary
antibodies were from Millipore (Temecula, CA, USA) and used at
1:3,000 dilutions.
MHC-I and PD-L1 detection by
fluorescence activated cell sorting (FACS)
Colon cancer cells (HCT116, RKO, SW620), colorectal
HT-29 cells and the SN-38-adapted derivatives were seeded and
treated with drugs (Irinotecan, VE822, KU60019, VE822, AZD) in
6-well plates for 24 h. Later, the culture medium was removed by
aspiration, and cells were harvested by brief trypsinization and
neutralized by adding 1 ml of medium for each well. The cells were
collected in a 2 ml tube on ice and centrifuged for 2 min at 3K
rpm. The medium was removed by aspiration, and the pellet was
washed two times in 1 ml cold staining buffer (FC001) (centrifuge
for 2 min at 3,000 rpm). Each pellet of about 106 cells
was resuspended in 100 µl staining buffer with the respective
antibody (100 µl X no. of tubes for Isotype control (Mouse PE-IgG2a
(IC003p) & 100 µl X no. of tubes for HLA I (MHC I) Ab (FAB7098P
PE conjugated anti-human HLA Class I). Then, the cells were
incubated for 1 h on ice in the dark and washed two times with 1 ml
staining buffer. The final pellet was resuspended with 500 µl
staining buffer and filtered (70 microns filter) into FACS tubes
(on ice) and analyzed. For PD-L1 FACS, each pellet of about
106 cells was resuspend in 100 µl staining buffer with
the respective antibody (100 µl with 2.5 µl Isotype Ab R&D
(AF488) & 100 µl with 2.5 µl PD-L1 Ab R&D (AF488) and
incubated for 1 h on ice in the dark and washed two times with 1 ml
staining buffer. The final pellet was resuspended in 500 µl
staining buffer and filtered (70 Microns filter) into FACS tubes
(on ice) before data collection on BD FACSCalibur, (Becton
Dickinson Biosciences, San Jose, CA, USA) using FlowJo Collectors'
Edition (FJCE) version 7.5.109 (Cytek Biosciences, Fremont, CA,
USA). Data analysis was done by using FlowJo versions 7.5 or 10.4
(BD Biosciences, Ashland, OR, USA).
Statistical analysis
Statistical comparisons of treatment effects were
performed using one-way ANOVA followed by Dunnett's multiple
comparisons test in GraphPad Prism software (version 9.4.0) for
Windows (GraphPad Software, Inc.). Median fluorescence intensity
values were used to create comparison graphs. Error bars, where
plotted, show standard deviations of samples in triplicates.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Inhibition of DDR and NF-κB pathways
in topo-inhibited cells accelerates cell death
Camptothecin (CPT) and its derivatives interfere
with topoisomerase, halting DNA replication and therefore cell
division. However, DNA damage repair (DDR) competent cells could
repair the damage and maintain cell viability. Prior studies have
also shown that colon cancer cells exposed to topoisomerase
inhibitors activate pathways that modulate cell survival and immune
cell functions, including DDR response and NF-κB activation. To
examine the effects of interference with these two pathways in
colon cancer cells exposed to CPT, we determined cell cycle and
cell death parameters for SW620 (MSS) or HCT116 (MSI) cells
co-treated with CPT and selective inhibitors of the two pathways
(SM-7368 as NF-κB pathway inhibitor; and AZD7762, KU60019 and VE822
as DDR pathway inhibitors targeting either Chk kinase, ATM kinase
or ATR kinase, respectively). To best delineate the effects of the
combinations, the cells were treated with individual drug
concentrations that did not induce massive death. Fig. 1A shows a representative histogram
from SW620 cells treated with control, single agents and
combinations of the drugs. As shown in Fig. 1A-C, while CPT, SM-7368 (SM7) or
AZD7762 (AZD), KU60019 (KU), or VE822 individually did not increase
the sub-G1 population in both cell types at the concentrations we
used, combination treatments markedly increased the sub-G1
population indicative of apoptotic cell death. Therefore,
inhibition of both the DDR pathways activated by checkpoint
kinases, and NF-κB activated in response to DNA-targeted
therapeutics may augment cell death induced by drugs that interfere
with topoisomerase enzyme. HCT116 (MSI) cells treated similarly
also showed increased sub-G1 although the relative amount of cell
death due to the combination treatment was lower (Fig. S1), and NF-κB inhibition or ATR
appeared to induce more cell death in these cells (Fig. 1C).
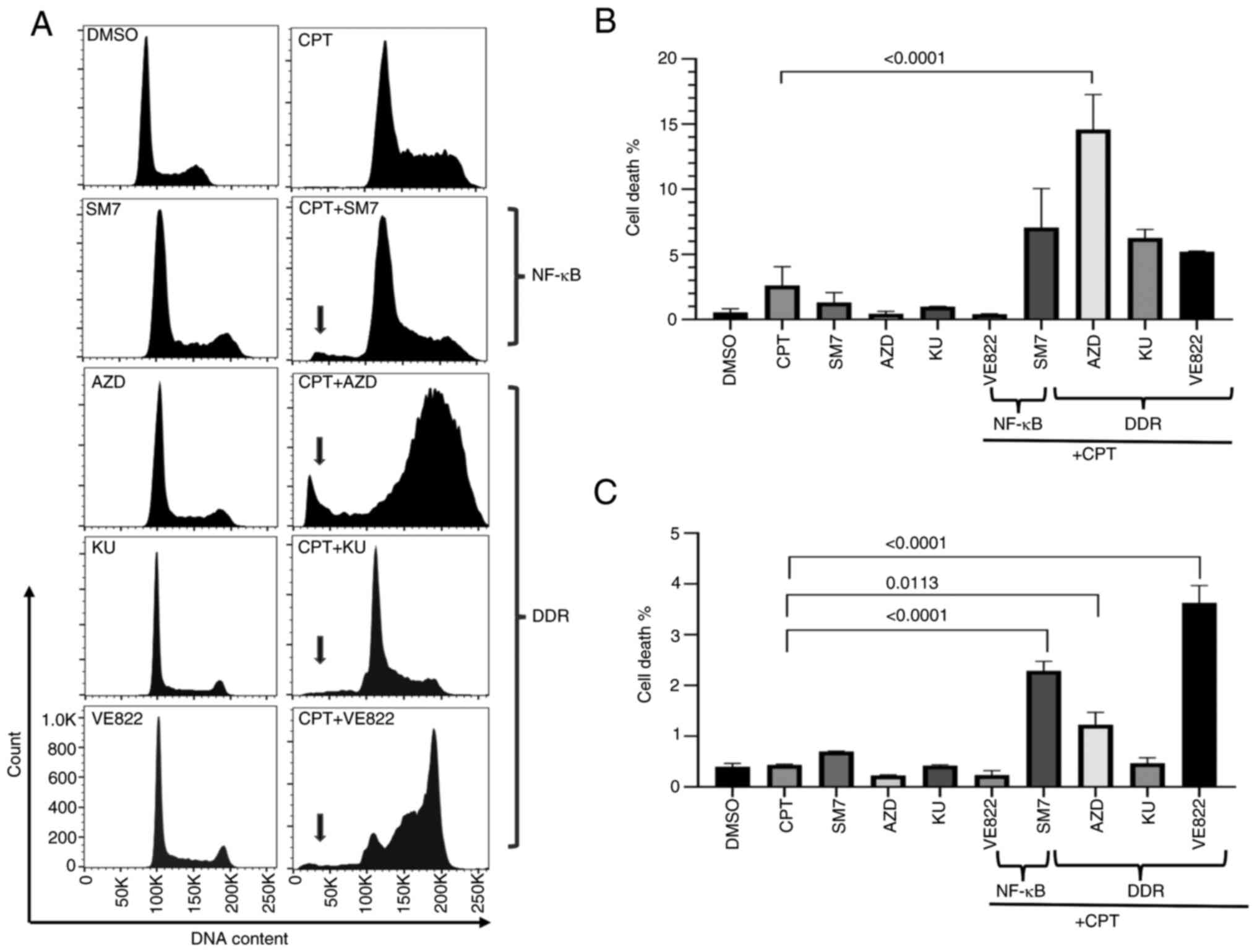 | Figure 1.Inhibition of DDR pathways in
CPT-treated cells enhances cell death. SW620 cells were treated
with vehicle (DMSO), single agents (CPT, SM7, AZD, KU or VE822 at
the concentrations indicated in materials and methods), or a
combination of CPT and the pathway-inhibitor drugs as shown. Cell
cycle profiles of treated cells were analyzed by propidium iodide
flow cytometry. Shown are (A) representative histograms from SW620
cells and graphs from technical replicates of the experiment from
(B) SW620 and (C) HCT116 cells. Arrows point to the to the
sub-G1 population indicative of cell death. AZD,
AZD7762; DMSO, dimethyl sulfoxide; CPT, camptothecin; DDR,
DNA-damage response; KU, KU60019; SM7, SM-7366. |
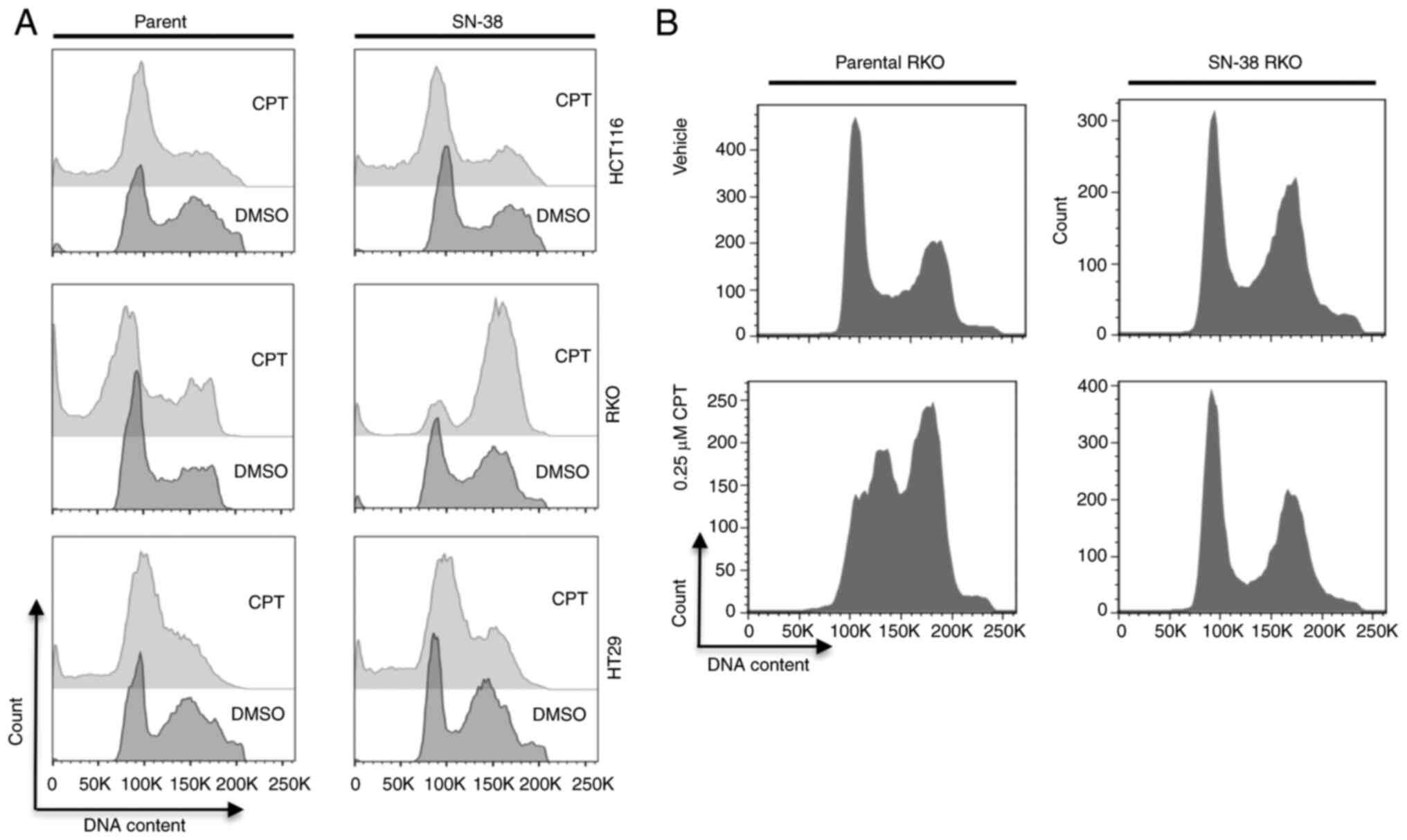
Adaptive tolerance through extended
exposure to topoisomerase inhibitor irinotecan (SN-38) may alter
cell cycle response of colon cancer cells
Adaptive resistance to cancer therapy agents is
known to impact drug efficacy and recurrence. To simulate these
characteristics, we generated SN-38 (irinotecan)-tolerant cells
from HCT116 (MSI), RKO (MSI) or HT-29 (MSS) cell lines by
continuous exposure of the cells to SN-38 over a period of 8 months
as described in materials and methods. We used these cells to
examine the cell cycle response after treatment for 24 h with 1 µM
CPT, a concentration at which peak NF-κB signaling activity was
detected in many treated colon cancer cells (29,30).
As a result (Fig. 2A), we did not
find marked differences in the cell cycle profiles and sub-G1
population profiles for parental or SN-38 tolerant HCT116 and HT-29
cells. However, while parental RKO cells displayed a high
percentage of sub-G1 population, the SN-38-tolerant cells arrested
in G2 with no increase in cell death (sub-G1 population). When we
reduced the concentration of CPT to 0.25 µM for these cells, the
parental RKO cells predominantly arrested in S-G2 phase of the cell
cycle, whereas the SN-38 tolerant cells showed no cell cycle
response, displaying profiles indistinguishable from the parental
cells (Fig. 2B). Therefore, at
least in RKO cells, the threshold for G2-shifted response was
elevated by acquired tolerance to topoisomerase inhibition. This
suggested that adaptive resistance or tolerance to topoisomerase
inhibition may alter, though not universally, the drug response of
cells upon exposure to a higher concentration of drugs of the same
mechanism of action. As both HCT116 and RKO cells are MSI, yet they
differ in their responses, it is not obvious if MS status is the
factor for the observed difference between the two.
Adaptive tolerance through extended
exposure to topoisomerase inhibitor irinotecan (SN-38) may alter
expression of PD-L1 in colon and colorectal cancer cells
We and others have previously shown that colon
cancer cells increase the surface expression of PD-L1 upon
treatment with DNA-damaging drugs (30–34).
Here, we undertook experiments to determine the impact of extended
exposure to SN-38 on the expression of this immunoregulatory
protein. First, we established the surface detection of PD-L1 by
immunofluorescence staining of RKO cells that abundantly express
PD-L1 (Fig. 3A). We then verified
that topoisomerase I or II inhibitors, i.e., CPT, IRI (both inhibit
topo I) or VP-16, (topo II) all induced the upregulation of PD-L1
in SW620 cells. In parallel, the cells were treated with 5-FU a
nucleoside analog that interferes with DNA replication and RNA
transcription. Gamma-interferon, a known inducer of immune
regulatory genes, was also included (Fig. 3A and B). Additionally,
immunoblotting of cell lysates prepared from SW620 cells treated
with varying concentrations of IRI or CPT was performed (Fig. 3C).
 | Figure 3.PD-L1 surface expression in response
to treatment is reduced in cells that acquire tolerance to
topoisomerase inhibition. (A) Detection of PD-L1 on the surface of
colon cancer cells; two RKO cells stained with specific antibody
(upper two rows) and one stained with isotype control (bottom row)
are shown. Phase contrast (left columns) and fluorescence (right
columns) microphotographs of the cells were taken at a
magnification of ×400. Topoisomerase inhibitors (VP-16, CPT and
IRI) are potent inducers of PD-L1 expression on cell surface. (B)
PD-L1 staining intensity histograms from SW620 cells treated with
the indicated drugs are shown. Histograms of the median
fluorescence intensities are presented. (C) Concentration-dependent
increase in PD-L1 expression following treatment with both CPT and
irinotecan. Immunoblots from SW620 cells treated with IRI or CPT at
the indicated concentrations are shown. Membranes were re-probed
with Topo I antibody to show drug activity. ns, non-specific bands
to show equivalent loading. (D-F) Flow cytometry comparison of cell
surface expression of PD-L1 in Par or SN-38 tolerant (SN38) (D)
HCT116, (E) HT-29 or (F) RKO cells treated with vehicle (DMSO) or
CPT (0.25 µM) and stained with isotype or PD-L1 antibody. Dot plots
for PD-L1 (x-axis) and FSC (y-axis) from one of three technical
duplicates are shown. Note that gates for each cell type were set
to capture comparable percentages of cells in the control (isotype)
plots, to allow measurements of relative PD-L1 expressions under
baseline (DMSO) treatment or drug (CPT) treatment conditions. CPT,
camptothecin; FSC, forward scatter; IRI, irinotecan; Par, parental;
PD-L1, programmed death-ligand 1; Topo I, topoisomerase I; γ-IFN,
gamma interferon; 5-FU, 5-fluorouracil; No Ab, no antibody; ns,
non-specific. |
These results showed that topoisomerase inhibitors
(IRI, CPT and VP-16 are strong inducers of PD-L1 and the induction
of PD-L1 is concentration-dependent, whereby low or high
concentrations did not induce expression. After establishing the
PD-L1 induction and surface expression, we tested if adaptive
tolerance to SN-38 would alter the PD-L1 surface expression in
colon or colorectal cancer cells. Parental and tolerant cells were
compared. As shown in Fig. 3D-F
all three tested parental cells (HCT116, HT-29, RKO), showed PD-L1
upregulation to variable degrees upon exposure to IRI, but the
SN-38 adapted cells showed no increase at the concentration and
time to which the parental cells responded. This suggested that the
mechanism of PD-L1 upregulation is linked to the mechanism of the
drug activity, and adaptation to the inhibitor may lead to
non-responsive state. Although this lack of increase in PD-L1
levels may seem to render the cancer cells readily targetable by
immune cells, other factors beside the expression of PD-L1 may
determine the outcomes of the interaction of these adapted cancer
cells with immune cells.
Interference with NF-κB and DDR
pathways modestly alters the upregulation of surface PD-L1 by
topoisomerase inhibition
Next we wanted to evaluate the effect of inhibitors
of NF-κB or DDR pathways on the upregulation of PD-L1 expression on
the surface of topoisomerase inhibited colon cancer cells derived
from MSS or MSI origin. For this, we used inhibitors of NF-κB
(SM-7366), Chk kinase (AZD), ATM kinase (KU60019), or ATR kinase
(VE822) either alone or in combination with IRI, and determined the
surface expression of PD-L1 on colon cancer cells. As shown on
Fig. 4, co-treatment of the MSS
SW620 (A-B) or MSI HCT116 (C-D) cells with the inhibitors only
modestly altered the surface expression of PD-L1 beyond that
induced by irinotecan. Nevertheless, a consistent decrease in the
median fluorescence intensity for PD-L1 was noticed in both cell
types when Chk kinase inhibitor was combined with topoisomerase
inhibitor (Fig. 4B and D).
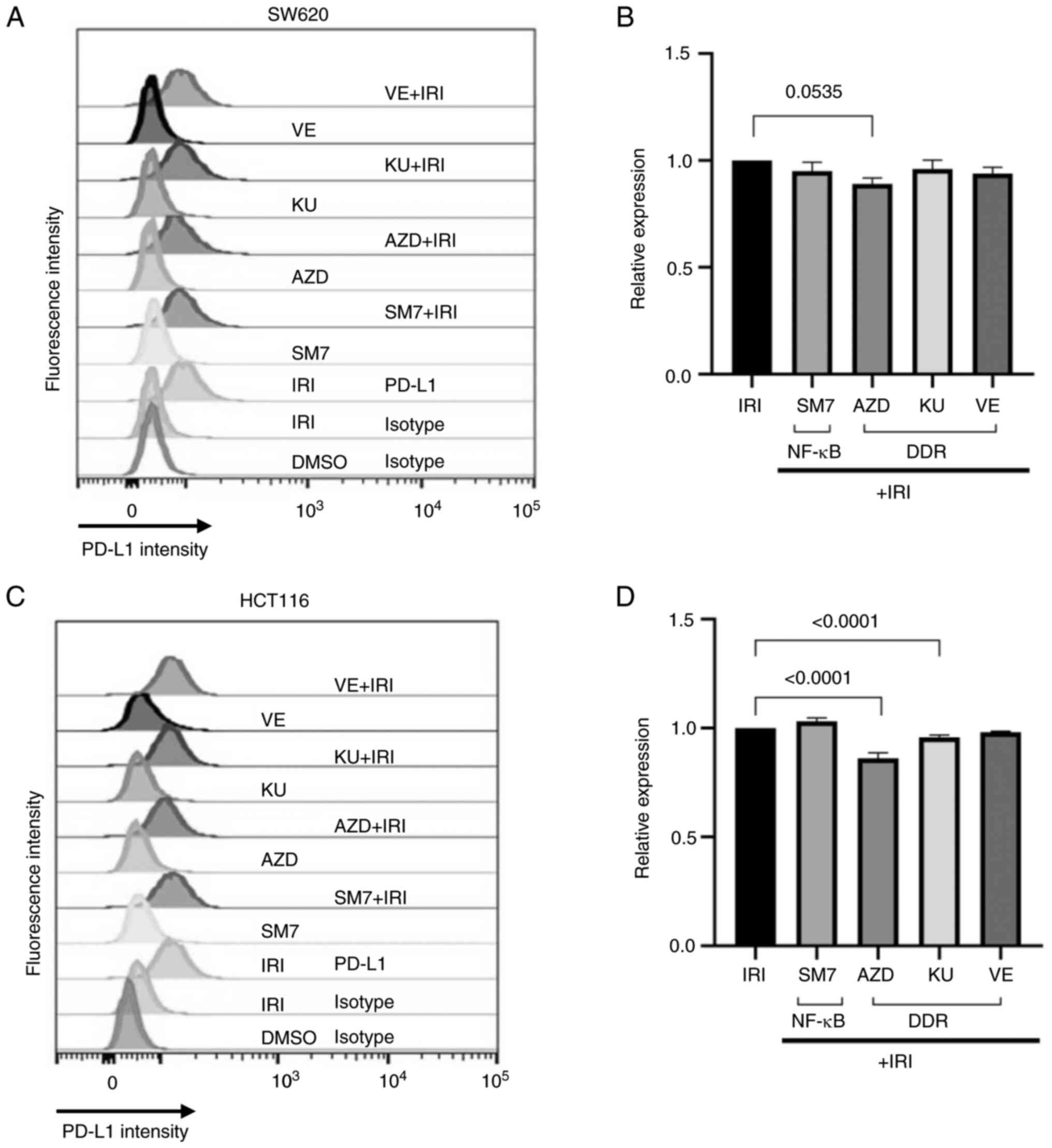 | Figure 4.NF-κB or DDR inhibitors only
moderately alter the induction of PD-L1 by IRI. (A and B) SW620 or
(C and D) HCT116 cells were treated with vehicle (DMSO), IRI, NF-κB
inhibitor (SM7), Chk kinase inhibitor (AZD), ATM inhibitor (KU) or
ATR inhibitor (VE) as single agents or in combination with IRI
(+IRI). (A and C) Surface expression of PD-L1 was measured by flow
cytometry using PD-L1-specific antibodies. DMSO or IRI-treated
cells were stained using a control (isotype) antibody to control
for background or non-specific reactions. (B and D) PD-L1
expression in combination-treated SW620 and HCT116 cells,
respectively, relative to the expression induced by IRI alone.
Graphs were generated from three technical duplicates. Combination
treatments did not increase PD-L1 expression beyond the levels
obtained by IRI treatment, but the combination of IRI with AZD
moderately reduced the relative expression in both cell types. DDR,
DNA-damage response; IRI, irinotecan; SM7, SM-7368; Chk,
checkpoint; AZD, AZD7762; KU, KU60019; VE, VE822; PD-L1, programmed
death-ligand 1; ATM, ataxia telangiectasia-mutated; ATR, ataxia
telangiectasia mutated and Rad3-related. |
Topoisomerase inhibitor treatment
enhances the expression of MHC I on colon cancer cells
Cancers that arise from hypermutated cells are
considered readily visible to immune cells, and therefore better
candidates for immunotherapy. Immunologically, the visibility of
such tumors is mostly dependent on the MHC I-enabled cell surface
presentation of neo-antigens from mutant proteins. Therefore, MHC I
expression on the targeted cells is critical for the cytotoxic
effects of T-cells. To assess the levels of MHC I on colon cancer
cells treated with topoisomerase inhibitors, we determined
expression of MHC I on SW620 (MSS) cell line by using an antibody
that recognizes pan-MHC I molecules expressed on human cells. Flow
cytometry was used for the detection of surface expression. An
isotype antibody of the same class and source species was used as
control. As shown on Fig. 5A and
B, the MHC I antibody not only distinctly detected surface MHC
I proteins, but the surface expression of MHC I (stained by HLA I
antibodies) was increased after treatment. Inhibitors of
Topoisomerase I (CPT, IRI, Topotecan), topoisomerase II (VP-16),
and the control cytokine IFN-gamma increased the surface abundance
of MHC I. To check if other chemotherapy agents in colon cancer
therapy also upregulated the expression of MHC I on cell surfaces,
we included 5-FU, oxaliplatin, CPT, erlotinib, and taxol in similar
assays. Fig. 5C and D show the
results of these experiments, where the isotype antibody did not
detect MHC I in treated cells (Fig.
5C) and the variable effects of the other drugs on MHC I
(Fig. 5D). The results also show
that while CPT was the most potent inducer of MHC I, 5-FU,
Oxaliplatin and Taxol induced MHC I by only 13–19%, while
erlotinib, an EGFR inhibitor did not induce an increase under the
same conditions. A similar increase was also evident in additional
colon cancer cell lines (shown below in Fig. 6). These findings suggest that
exposure to DNA-damage inducing drugs and taxol, which causes
chromosomal mis-alignment and -segregation, may lead to
upregulation of MHC I in colon cancer cells.
 | Figure 5.MHC I surface expression is increased
in colon cancer cells treated with chemotherapy drugs. (A and B)
Increased surface expression induced by topoisomerase I inhibitor
drugs CPT, irinotecan and topotecan as shown by (A) histogram or
(B) dot plots comparing isotype and HLA I-antibodies in
vehicle-treated (DMSO) or drug-treated SW620 cells. (C and D) SW620
cells were treated with vehicle (DMSO) or the drugs 5-FU,
oxaliplatin, CPT, erlotinib or taxol. Surface expression of MHC I
was analyzed by flow cytometry using (C) isotype control or (D)
pan-MHC I antibody. CPT, camptothecin; IRI, irinotecan; MHC, major
histocompatibility complex; HLA, human leukocyte antigen; 5-FU,
5-fluorouracil; FSC, forward scatter; γ-IFN, γ interferon; Topot,
topotecan. |
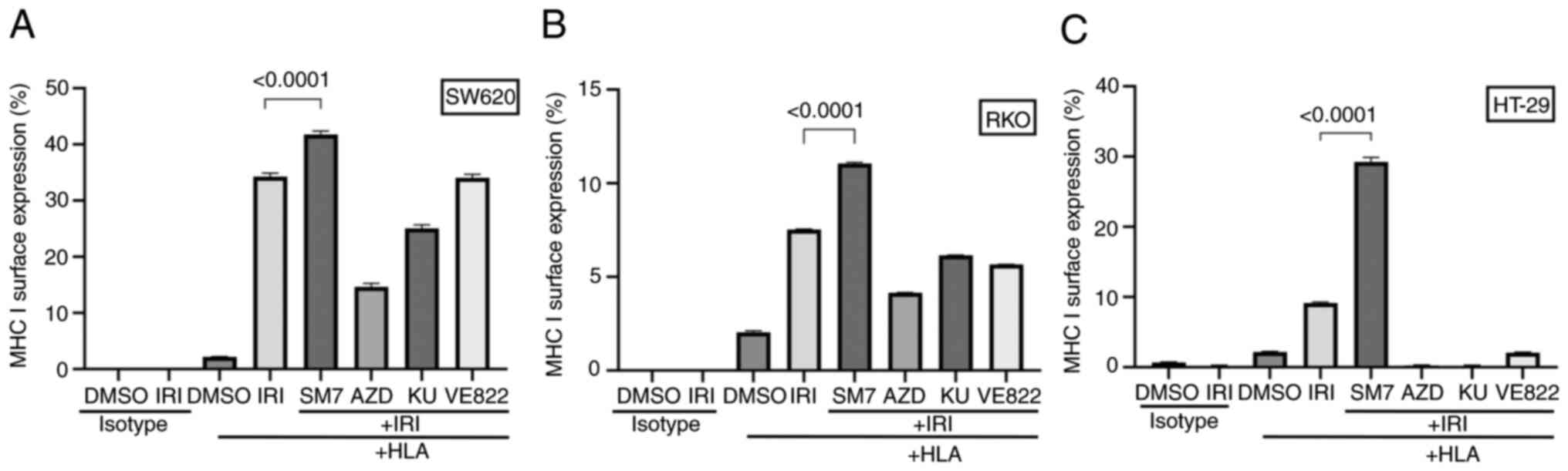 | Figure 6.Interference with the NF-κB pathway
enhances the surface expression of MHC I while interference with
DDR pathways suppresses the expression. RKO, SW620 or HT-29 cells
were treated with vehicle (DMSO) or IRI in combination with or
without the addition of SM7, AZD, KU or VE822 pathway inhibitors.
Shown are the MHC I surface staining median fluorescence graphs
from technical triplicates for (A) RKO, (B) SW620 or (C) HT-29
cells. While NF-κB inhibition consistently enhanced the expression
of MHC I in IRI-treated cells, DDR pathway inhibitors appeared to
interfere with the surface expression of MHC I. DDR, DNA-damage
response; IRI, irinotecan; MHC, major histocompatibility complex;
SM7, SM-7368; AZD, AZD7762; KU, KU60019; HLA, human leukocyte
antigen. |
Interference with NF-κB and DDR
pathways modulates the upregulation of MHC I expression induced by
topoisomerase inhibition
Once we established the upregulation of MHC I on
cell surface after topoisomerase inhibition, we wanted to examine
if such upregulation is modified by inhibitors of NF-κB (SM-7366),
Chk1 and Chk2 (AZD), ATM (KU60019), or ATR (VE822). Pathways
inhibited by these drugs are mediators of DDR and are targets of
clinical or investigational therapeutics. Inhibitors were tested
individually or in combination with IRI, and MHC I expression was
evaluated by flow cytometry.
As shown in Fig.
6A-C, NF-κB inhibitor (SM-7366) consistently enhanced the
surface expression of MHC I when combined with IRI, while it did
not have any enhancing effect by itself (not shown). On the other
hand, combination of AZD (chk1 and chk2 inhibitor) with IRI
consistently suppressed MHC I expression in the three cell lines we
tested. ATM inhibitor (KU60019) caused moderate to marked reduction
in the expression of MHC I in all three cell lines. On the other
hand, ATR inhibitor (VE822) showed variable effect depending on the
cell type, i.e. no change for SW620, moderate reduction in RKO, or
marked decrease in HT-29. These results showed that depending on
the pathway inhibited, the effect of topoisomerase inhibition on
MHC I expression may be enhanced, reduced, or remain unchanged.
Interestingly, in all three cell lines treated with the Chk-, or
ATM-inhibitors or in HT-29 cells treated with the ATR-inhibitor
VE822, we noticed not only lack of upregulation of MHC I but also
an active downregulation of the surface MHC I. In these
circumstances, the levels of MHC I on the surface was lower than
the baseline levels or found in vehicle treated control cells,
indicating a net reduction caused by the treatment. Taken together,
these findings suggest that the levels of MHC I on cancer cells can
be modulated by chemotherapeutic drugs and/or inhibitors, and this
outcome may provide an opportunity to render the cells visible to
immune cells in tumor microenvironment.
Adaptive tolerance through extended
exposure to topoisomerase inhibitor (SN-38) negatively impacts the
expression of MHC I on colon or colorectal cancer cells
Finally, to determine if acquired tolerance to
topoisomerase inhibition alters the surface expression of MHC I, we
compared parental HCT116 (MSI), RKO (MSI) or HT-29 (MSS) against
adaptively SN-38 tolerant cells derived from the three cell lines.
We treated the cells with IRI concentrations that enhanced MHC I
expression in parental cells and determined the surface expression
of MHC I by flow cytometry. As shown in Fig. 7A-C, in all the three cell lines we
tested, the percent increase in MHC I expression was much lower in
adaptively tolerant cells compared to the parental cells under the
same treatment conditions.
Discussion
In this study we show that clinically used
topoisomerase inhibitor drugs have the potential to alter the
expression of cell surface molecules that may make colon cancer
cells visible to immune cells. Firstly, we confirm that the protein
PD-L1 is upregulated most potently by both topoisomerase I and II
inhibitors and is detectable on cell surfaces. PD-L1 has been a
molecule of most interest in immunotherapy because its blockage has
enabled an unprecedented success in cancer therapy. Nevertheless,
some types of cancer, including the majority of CRC, remain
non-responsive for reasons yet to be defined. One of the approaches
to enhance the chances of immunotherapy success is to convert
‘cold’ or invisible tumors to ‘hot’ or immune-visible tumors by
manipulating cancer cells or their microenvironment. Studies are
ongoing to understand mechanism for invisibility of tumors such as
MSS colorectal cancer. Several combination therapies are also being
tested to enhance the potency of chemotherapy drugs by exploiting
the potential of DNA damaging drugs to generate neoantigens.
However, the results of such therapies are not yet available, and
the fundamental reason the cancer cells are invisible remains
incompletely understood.
Prior studies using various CRC, mammary, prostate,
and other cancer models (31–36)
have shown the upregulation of PD-L1, as well as MHC I and peptide
presentation by drugs that target critical signaling pathways,
including topoisomerase functions. Although MSS tumors constitute
the majority of CRC cases non-responsive to therapy, challenges
remain to study immunotherapy outcomes in MSS tumors, primarily due
to lack of suitable pre-clinical models. Here, we show that the
expression of both PD-L1 and MHC I is enhanced by treatment with
topoisomerase inhibitor drugs in both MSI and MSS cell types. Given
the critical function of MHC I to present cellular protein
breakdown peptides and neoantigens on the surface of cells, it is
reasonable to hypothesize that the newly expressed MHC I molecules
on cancer cells may contribute to the recognition by cytotoxic
T-cells in the microenvironment. In this regard, we also show for
the first time that drug-tolerant cells react differently compared
to naïve cells. Taken together, these observations make clinical
studies on the nature of immunomodulation by both MSS and relapsed
tumors very critical and impactful.
An important question about enhanced MHC I
expression is, what the peptides displayed on the newly expressed
MHC I proteins might be. Since an MHC molecule can bind different
varieties of peptides, it is not expected for a single peptide to
be displayed on the newly expressed MHC molecules. However, the
enhanced expression of these molecules correlates with an enhanced
turnover or generation of peptides, and therefore, the chances will
be greater for CD8+ T cells in the microenvironment to recognize
the neoantigens. However, the simultaneously increased expression
of PD-L1 on treated cancer cells may initiate a negative reaction
on T-cells, muting their activity on tumor cells. Nevertheless,
knowledge about the effect of a topoisomerase-targeted treatment on
the abundance of MHC I molecules on tumor cells may be helpful to
selectively identify individuals who may best benefit from
chemotherapy-immunotherapy combination.
Combination of chemotherapy with signaling pathway
inhibitors has been considered an alternative to enhance the
efficacy of cytotoxic drugs, while helping to reduce the dose
needed to achieve the same outcome. Therefore, we tested if the
combination of chemotherapy with specific pathway (NF-κB or DDR)
inhibitors would augment the effects of irinotecan, a clinically
used drug to treat colorectal cancer. Irinotecan enhances the
surface abundance of both PD-L1- and MHC I on colon cancer cells.
Our results from in vitro studies show that the simultaneous
inhibition of topoisomerase and NF-κB pathway consistently
upregulated the surface expression of MHC I, while inhibition of
neither NF-κB nor DDR pathways affected the PD-L1 expression
induced by topoisomerase inhibition. On the other hand, inhibition
of ATM or Chk 1/2 consistently interfered with the increase in MHC
I induced by topoisomerase inhibitor treatment. Although inhibition
of either of NF-κB or DDR inhibitors in topoisomerase inhibited
cells may induce apoptotic cell death, immune regulatory outcomes
of the combination-treatment may be different between NF-κB and DDR
inhibitors. The preliminary results shown here suggest that in
vivo studies need to be performed to identify if inhibition of
NF-κB pathway is more effective than inhibition of DDR pathways to
turn immunologically cold colon cancer microenvironment into a
‘hot’ one.
This suggests that immunotherapy that relies on MHC
I presentation of neoantigens may be challenged if cancer cells
acquire resistance to the chemotherapy. On the other hand, since
loss of MHC I renders cells visible to NK-cells, which also play
role in immunotherapy, alternative approaches will be needed to
treat such resistant cancers by activating resident NK-cells.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported through grants from the National
Institutes of Health (#U54CA118948, TU-CBR/RCMI #U54MD007585 shared
resources core facility) and Health and Human Services HHS/HRSA
(#D34HP00001). It was also supported by the VA Merit Review Award
(1I01BX 005143) and RCS Award (1IK6BX006029).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TS, PD and CY conceived and designed the study. MH,
VT, DB, SS, DG and TS were involved in experiments, and data
collection and analysis. MH, TS and PD wrote and revised the
manuscript. TS and MH confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ribas A and Wolchok JD: Cancer
immunotherapy using checkpoint blockade. Science. 359:1350–1355.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baybutt TR, Aka AA and Snook AE:
Immunotherapy in colorectal cancer: Where are we now? Curr
Colorectal Cancer Rep. 13:353–361. 2017. View Article : Google Scholar
|
|
3
|
Overman MJ, Ernstoff MS and Morse MA:
Where we stand with immunotherapy in colorectal cancer: Deficient
mismatch repair, proficient mismatch repair, and toxicity
management. Am Soc Clin Oncol Educ Book. 38:239–247. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Le DT, Durham JN, Smith KN, Wang H,
Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et
al: Mismatch repair deficiency predicts response of solid tumors to
PD-1 blockade. Science. 357:409–413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zumwalt TJ and Goel A: Immunotherapy of
metastatic colorectal cancer: Prevailing challenges and new
perspectives. Curr Colorectal Cancer Rep. 11:125–140. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Le DT, Hubbard-Lucey VM, Morse MA, Heery
CR, Dwyer A, Marsilje TH, Brodsky AN, Chan E, Deming DA, Diaz LA
Jr, et al: A blueprint to advance colorectal cancer
immunotherapies. Cancer Immunol Res. 5:942–949. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Becht E, de Reyniès A, Giraldo NA, Pilati
C, Buttard B, Lacroix L, Selves J, Sautès-Fridman C, Laurent-Puig P
and Fridman WH: Immune and stromal classification of colorectal
cancer is associated with molecular subtypes and relevant for
precision immunotherapy. Clin Cancer Res. 22:4057–4066. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Frey DM, Droeser RA, Viehl CT, Zlobec I,
Lugli A, Zingg U, Oertli D, Kettelhack C, Terracciano L and
Tornillo L: High frequency of tumor-infiltrating FOXP3(+)
regulatory T cells predicts improved survival in mismatch
repair-proficient colorectal cancer patients. Int J Cancer.
126:2635–2643. 2010.PubMed/NCBI
|
|
9
|
Salama P, Phillips M, Grieu F, Morris M,
Zeps N, Joseph D, Platell C and Iacopetta B: Tumor-infiltrating
FOXP3+ T regulatory cells show strong prognostic significance in
colorectal cancer. J Clin Oncol. 27:186–192. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
deLeeuw RJ, Kost SE, Kakal JA and Nelson
BH: The prognostic value of FoxP3+ tumor-infiltrating lymphocytes
in cancer: A critical review of the literature. Clin Cancer Res.
18:3022–3029. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang X, Liu M, Luo X, Zhu M, Huang S and
Pan X: The prognostic value of a tumor microenvironment-based
immune cell infiltration score model in colon cancer. Front Oncol.
11:7288422021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ghiringhelli F and Fumet JD: Is there a
place for immunotherapy for metastatic microsatellite stable
colorectal cancer? Front Immunol. 10:18162019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu Y, Zuo W, Shen X, Liu Y, Zhao Y, Xiong
Y, Cao H, Wang Y and Liang Z: NF-κB is involved in the regulation
of autophagy in mutant p53 cells in response to ionizing radiation.
Cell Death Discov. 7:1592021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu H, Mohan S and Natarajan M:
Radiation-triggered NF-κB activation is responsible for the
angiogenic signaling pathway and neovascularization for breast
cancer cell proliferation and growth. Breast Cancer (Auckl).
6:125–135. 2012.PubMed/NCBI
|
|
15
|
Huang TT, Wuerzberger-Davis SM, Seufzer
BJ, Shumway SD, Kurama T, Boothman DA and Miyamoto S: NF-kappaB
activation by camptothecin. A linkage between nuclear DNA damage
and cytoplasmic signaling events. J Biol Chem. 275:9501–9509. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brea-Calvo G, Siendones E, Sanchez-Alcázar
JA, de Cabo R and Navas P: Cell survival from chemotherapy depends
on NF-kappaB transcriptional up-regulation of coenzyme Q
biosynthesis. PLoS One. 4:e53012009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matsuoka S, Ballif BA, Smogorzewska A,
McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini
N, Lerenthal Y, et al: ATM and ATR substrate analysis reveals
extensive protein networks responsive to DNA damage. Science.
316:1160–1166. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Flynn RL and Zou L: ATR: A master
conductor of cellular responses to DNA replication stress. Trends
Biochem Sci. 36:133–140. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Patel SA and Minn AJ: Combination cancer
therapy with immune checkpoint blockade: Mechanisms and strategies.
Immunity. 48:417–433. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Twyman-Saint Victor C, Rech AJ, Maity A,
Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi
PM, et al: Radiation and dual checkpoint blockade activate
non-redundant immune mechanisms in cancer. Nature. 520:373–377.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gorzo A, Galos D, Volovat SR, Lungulescu
CV, Burz C and Sur D: Landscape of immunotherapy options for
colorectal cancer: Current knowledge and future perspectives beyond
immune checkpoint blockade. Life (Basel). 12:2292022.PubMed/NCBI
|
|
22
|
Zhou C, Jiang T, Xiao Y, Wang Q, Zeng Z,
Cai P, Zhao Y, Zhao Z, Wu D, Lin H, et al: Good tumor response to
chemoradioimmunotherapy in dMMR/MSI-H advanced colorectal cancer: A
case series. Front Immunol. 12:7843362021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Young KH, Baird JR, Savage T, Cottam B,
Friedman D, Bambina S, Messenheimer DJ, Fox B, Newell P, Bahjat KS,
et al: Optimizing timing of immunotherapy improves control of
tumors by hypofractionated radiation therapy. PLoS One.
11:e01571642016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang W, Wu L, Zhang J, Wu H, Han E and Guo
Q: Chemoimmunotherapy by combining oxaliplatin with immune
checkpoint blockades reduced tumor burden in colorectal cancer
animal model. Biochem Biophys Res Commun. 487:1–7. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thomas J, Leal A and Overman MJ: Clinical
development of immunotherapy for deficient mismatch repair
colorectal cancer. Clin Colorectal Cancer. 19:73–81. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shahda S, Noonan AM, Bekaii-Saab TS,
O'Neil BH, Sehdev A, Shaib WL, Helft PR, Loehrer PJ, Tong Y, Liu Z,
et al: A phase II study of pembrolizumab in combination with
mFOLFOX6 for patients with advanced colorectal cancer. J Clin
Oncol. 35 (15 Suppl):S35412017. View Article : Google Scholar
|
|
27
|
Sahin IH, Akce M, Alese O, Shaib W,
Lesinski GB, El-Rayes B and Wu C: Immune checkpoint inhibitors for
the treatment of MSI-H/MMR-D colorectal cancer and a perspective on
resistance mechanisms. Br J Cancer. 121:809–818. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dallas NA, Xia L, Fan F, Gray MJ, Gaur P,
van Buren G II, Samuel S, Kim MP, Lim SJ and Ellis LM:
Chemoresistant colorectal cancer cells, the cancer stem cell
phenotype, and increased sensitivity to insulin-like growth
factor-I receptor inhibition. Cancer Res. 69:1951–1957. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Samuel T, Fadlalla K, Gales DN, Putcha BD
and Manne U: Variable NF-κB pathway responses in colon cancer cells
treated with chemotherapeutic drugs. BMC Cancer. 14:5992014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bedi D, Henderson HJ, Manne U and Samuel
T: Camptothecin induces PD-L1 and immunomodulatory cytokines in
colon cancer cells. Medicines (Basel). 6:512019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang P, Su DM, Liang M and Fu J:
Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1)
surface expression in breast cancer cells and promote
PD-L1-mediated T cell apoptosi. Mol Immunol. 45:1470–1476. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Van Der Kraak L, Goel G, Ramanan K,
Kaltenmeier C, Zhang L, Normolle DP, Freeman GJ, Tang D, Nason KS,
Davison JM, et al: 5-Fluorouracil upregulates cell surface B7-H1
(PD-L1) expression in gastrointestinal cancers. J Immunother
Cancer. 4:652016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Iwai T, Sugimoto M, Wakita D, Yorozu K,
Kurasawa M and Yamamoto K: Topoisomerase I inhibitor, irinotecan,
depletes regulatory T cells and up-regulates MHC class I and PD-L1
expression, resulting in a supra-additive antitumor effect when
combined with anti-PD-L1 antibodies. Oncotarget. 9:31411–31421.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sato H, Niimi A, Yasuhara T, Permata TBM,
Hagiwara Y, Isono M, Nuryadi E, Sekine R, Oike T, Kakoti S, et al:
DNA double-strand break repair pathway regulates PD-L1 expression
in cancer cells. Nat Commun. 8:17512017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC
and Liu LF: Chemotherapeutics and radiation stimulate MHC class I
expression through elevated interferon-beta signaling in breast
cancer cells. PLoS One. 7:e325422012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou Y, Bastian IN, Long MD, Dow M, Li W,
Liu T, Ngu RK, Antonucci L, Huang JY, Phung QT, et al: Activation
of NF-κB and p300/CBP potentiates cancer chemoimmunotherapy through
induction of MHC-I antigen presentation. Proc Natl Acad Sci USA.
118:e20258401182021. View Article : Google Scholar : PubMed/NCBI
|