Introduction
Fenofibrate (FF), which is a peroxisome
proliferator-activated receptor (PPAR)-α agonist (1), has been widely used for the treatment
of hyperlipidemia since 1975 (2,3). In
vivo, FF is rapidly converted to fenofibric acid, which binds
to PPAR-α and forms a heterodimer complex with retinoid X receptor.
This complex then binds to PPAR response element (PPRE) to activate
transcription of target genes, including that of lipid metabolism
regulation (4). Beyond its
hypolipidemic effect, FF has been shown to have pleiotropic actions
in a PPAR-α-dependent or independent manner. Among them, the
anticancer property of FF has attracted much attention as a new
option for therapy (5). According
to previous reports, FF had cytotoxic effects on various tumor cell
lines derived from the brain, breast, liver, prostate, and lungs by
inducing apoptosis, cell cycle arrest, and motility inhibition
(5). However, in some of the
studies, FF concentrations that were higher than clinically
relevant blood concentrations (i.e., ≤50 µM) were used to exert
anticancer effects; interpretation of this result requires some
caution (6,7). Furthermore, no studies have evaluated
the anticancer effect of FF in combination with standard
chemotherapeutic drugs, such as cisplatin (CDDP) (8).
On the other hand, other in in vitro and
in vivo experiments, including animal models of diabetic
retinopathy and nephropathy and ischemia/ reperfusion-induced
cardiac injury, demonstrated that FF has a cytoprotective effect on
normal cells (4,9). Although the mechanism of FF-induced
cytoprotection is uncertain, it was reported to involve enhancement
of antioxidants. For example, FF has been shown to attenuate
oxidative damage to cardiomyocytes, retinal endothelial cells,
auditory hair cells, and skin keratinocytes (10–13).
Moreover, FF has been reported to reduce CDDP toxicity to renal
tubular cells and auditory hair cells, both of which can be
adversely affected by CDDP-containing chemotherapy (14,15).
Because oxidative stress comprises the major mechanism of CDDP
chemotherapy (16), we hypothesized
that concurrent treatment with FF may attenuate the anticancer
effect of CDDP.
Because both hyperlipidemia and cancer have become
increasingly prevalent, not a few patients with hyperlipidemia and
receiving FF need to commence chemotherapy for cancer. Therefore,
elucidating the impact of FF on CDDP chemotherapy is clinically
important. In this study, we determined the effect of FF at
clinically relevant blood concentrations on CDDP cytotoxicity to
lung cancer cells in vitro.
Materials and methods
Cell culture
Human non-small lung cancer cell lines A549
(CCL-185), H1299 (CRL-5803) and H441 (HTB-174) were obtained from
the American Type Culture Collection (Manassas, VA, USA); PC3
(JCRB0077) was obtained from the Japanese Collection of Research
Bioresources Cell Bank (Osaka, Japan). The cell lines were
authenticated by short tandem repeat profiling using the Promega
PowerPlex® 16 HS system (Promega Corporation, Madison,
WI). A549 cells were grown and maintained on type I collagen-coated
plates in Dulbecco's Modified Eagle medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine
serum (FBS, Biowest, Nuaillé, France). H1299 and PC3 cells were
grown and maintained in Roswell Park Memorial Institute (RPMI) 1640
medium (Gibco; Thermo Fisher Scientific, Inc.) containing 10% FBS.
The cells were incubated at 37°C in a humidified incubator
saturated with a gas mixture containing 5% CO2.
Confluent cells were treated with the following: i) FF (0–200 µM or
50 µM unless otherwise indicated; Sigma-Aldrich Japan, Tokyo,
Japan); ii) WY14643 (WY, 0–50 µM or 50 µM unless otherwise
indicated; Cayman Chemical, Ann Arbor, MI, USA); or iii) vehicle
alone (0.2% dimethyl sulfoxide) in RPMI1640 containing 0.5% FBS.
CDDP (0–40 µM or 40 µM unless otherwise indicated; Nippon Kayaku
Co., Ltd., Tokyo, Japan) was added to the culture medium after
12–48 h of treatment with FF or WY.
Cell survival assay
Cell survival was evaluated by Hoechst 33342 DNA
quantification assay, a colorimetric alamarBlue® assay,
and ATP quantification assay. For the Hoechst 33342 DNA
quantification assay, cells that were cultured in a 96-well plate
were lysed in 100 µl of distilled water, followed by a freeze-thaw
cycle. Thereafter, the cell lysates were solubilized in 100 µl of
TNE buffer (i.e., 10 mM Tris, 1 mM EDTA, and 2 M NaCl; pH 7.4)
containing 10 µg/ml of Hoechst 33342 (Sigma-Aldrich Japan, Tokyo,
Japan). The fluorescence intensities were read at an excitation
(Ex) of 350 nm and emission (Em) of 460 nm using a microplate
fluorometer (PerkinElmer Arvo X2, PerkinElmer Japan Co., Ltd.,
Tokyo, Japan). For the alamarBlue® assay, a one-tenth
volume of alamarBlue® reagent (Thermo Fisher Scientific)
was added to the culture medium in a 96-well plate for the last
four h of incubation. Thereafter, the absorbance was measured at
570 nm on a Benchmark Plus microplate immunoreader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) using 600 nm as a reference
wavelength. For the ATP quantification assay, the Cellno ATP assay
reagent (Toyo B-Net, Co, Ltd., Tokyo, Japan) was used according to
the manufacturer's instructions. Briefly, 100 µl of the lysis assay
solution provided by the manufacturer was added to confluent cell
cultures in a 96-well culture plate. After the plate was shaken for
1 min and incubated for 10 min at 23°C, luminescence was measured
in microplate luminometer (PerkinElmer Arvo X2, PerkinElmer Japan
Co., Ltd., Tokyo, Japan).
Measurement of cellular reactive
oxygen species (ROS) levels
A549 cells in 96-well plates were treated with or
without FF (50-µM) or WY (50-µM) for 12 h, followed by exposure to
40-µM CDDP for 48 h in the presence or absence of FF and WY. The
cells were then loaded with the cellular reactive oxygen species
(ROS) sensor CellROX® Green (5-µM) (Thermo Fisher
Scientific) for 30 min at 37°C in the presence of the nuclear dye
Hoechst33342. The medium was replaced with phosphate buffered
saline (PBS); the fluorescence intensities of the
CellROX® Green (Ex 485 nm, Em 530 nm) and Hoechst33342
(Ex 355 nm, Em 460 nm) dyes were recorded using microplate
fluorometer. The fluorescence intensities of CellROX®
Green were normalized to Hoechst33342 fluorescence in the
corresponding wells.
Superoxide dismutase assay
Cellular superoxide dismutase (SOD) activity was
determined using an SOD assay kit-WST (Dojindo Laboratories,
Kumamoto, Japan), according to the manufacturer's instructions.
Catalase activity assay
Cellular catalase activity was determined using the
EnzyChrom catalase assay kit (BioAssay Systems, Hayward, CA, USA),
according to the manufacturer's instructions.
Western blotting
Cell samples were lysed in a
radioimmunoprecipitation assay (RIPA) buffer [50 mM Tris
hydrochloride, 150 mM NaCl, 0.4 mM EDTA, 0.5% Nonidet P-40, and
0.1% sodium dodecyl sulfate (SDS); pH 7.4] containing a protease
inhibitor cocktail (Sigma-Aldrich Japan), a phosphatase inhibitor
cocktail (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and sodium
orthovanadate (1 mM). Nuclear proteins and cytoplasmic proteins
were extracted using a nuclear extraction kit (Active Motif,
Carlsbad, CA, USA), according to the manufacturer's instructions.
The samples were centrifuged at 10,000 × g for 30 min, and the
total protein concentration in the supernatants was assessed using
the DC protein assay kit (Bio-Rad Laboratories). After combining
with 5X SDS sample buffer (500 mM Tris, 5% 2-mercaptoethanol, 10%
glycerin, 2.5% SDS, 0.0125% bromophenol blue; pH 6.8), the samples
(20-µg protein/lane) were fractionated by SDS-polyacrylamide gel
electrophoresis and transferred to a polyvinylidene difluoride
membrane (EMD Millipore Immobilon®-P; Millipore, Co.,
Billerica, MA, USA). The membranes were blocked with 4% bovine
serum albumin (BSA, Biowest); probed with the primary antibodies
described below; diluted in an immunoreaction enhancer solution
(Can Get Signal® Solution 1; Toyobo Co., Ltd., Osaka,
Japan); and reacted with horseradish peroxidase (HRP)-conjugated
secondary antibodies, such as stabilized goat anti-rabbit IgG
(32460, Thermo Fisher Scientific) and stabilized goat anti-mouse
IgG (32430, Thermo Fisher Scientific). The immune complexes were
visualized using an enhanced chemiluminescence reagent (SuperSignal
West Pico; Thermo Fisher Scientific). The signal intensities were
quantified by densitometric scanning using ImageJ (version 1.49V;
National Institutes of Health, Bethesda, MD, USA).
The primary antibodies used in this study were mouse
monoclonal anti-β-actin (017-24573, Wako, Tokyo, Japan); rabbit
polyclonal anti-Lamin B1 (12987-1-AP, Proteintech Group, Inc.,
Tokyo, Japan); rabbit polyclonal anti-α-tubulin (PM054-7, Medical
& Biological Laboratories Co., Ltd., Tokyo, Japan); mouse
monoclonal anti-p53 (sc-126, Santa Cruz Biotechnology), rabbit
polyclonal anti-phosphorylated p53 (CSB-PA157242, Cusabio Biotech
Co., Ltd., Houston, TX, USA); mouse monoclonal anti-heat shock
protein 70 (HSP70) (SPA-810, Stressgen Biotechnologies Co., Ltd.,
Seoul, Korea); rabbit polyclonal anti-B-cell/CLL lymphoma 2 (Bcl-2)
(12789-1-AP, Proteintech Group); rabbit polyclonal anti-B-cell
lymphoma-extra large (Bcl-xL) (10783-1-AP, Proteintech Group);
rabbit polyclonal anti-Bcl-2-associated X protein (Bax)
(50599-2-Ig, Proteintech Group); mouse monoclonal anti-Bcl-2
antagonist of cell death (Bad) (B36420, BD Transduction
Laboratories, Lexington, KY, USA); rabbit polyclonal
anti-superoxide dismutase (SOD) 1 (GTX100554, GeneTex, Inc.,
Irvine, CA, USA); rabbit polyclonal anti-SOD2 (GTX116093, GeneTex);
rabbit polyclonal anti-heme oxygenase (HO)-1 (GTX101147, GeneTex);
rabbit polyclonal anti-catalase (GTX110704, GeneTex); rabbit
monoclonal anti-nuclear factor erythroid 2-related factor 2 (Nrf2)
(ab62352, Abcam, Cambridge, UK); mouse monoclonal anti-Kelch-like
ECH-associated protein 1 (Keap1) (M224-3, Medical & Biological
Laboratories Co., Ltd.); rabbit polyclonal anti-β-transduction
repeat containing protein (β-TrCP) (GTX102667, GeneTex); rabbit
polyclonal anti-aryl hydrocarbon receptor (AhR) (28727-1-AP,
Proteintech Group); and mouse monoclonal anti-ubiquitin (sc-8017,
Santa Cruz Biotechnology, Dallas, TX, USA).
Immunoprecipitation
Cells were lysed in Nonidet P-40 lysis buffer (50 mM
Tris hydrochloride, 140 mM NaCl, 1% NP-40, and 10% glycerol; pH
7.5) containing a protease inhibitor cocktail (Sigma-Aldrich,
Japan). Cell lysates containing an equal amount (820 µg) of protein
were incubated with 1 µg of rabbit monoclonal anti-Nrf2 (ab62352,
Abcam) and protein A/G plus agarose (sc-2003, Santa Cruz
Biotechnology) on a rotator shaker at 4°C overnight. The beads were
washed with RIPA buffer and boiled in 1X SDS sample buffer at 95°C
for 5 min. Proteins were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and immunoblotted as
described above.
Immunofluorescence staining
Cells in eight-chamber slides (Nunc®
Lab-Tek II® Chamber Slide System; Thermo Fisher
Scientific) were fixed with 3% paraformaldehyde and permeabilized
with 0.5% Triton® X-100 (Nacalai tesque, Inc., Kyoto,
Japan) in PBS for 10 min. After blocking the nonspecific binding
sites with 3% BSA, the slides were incubated with mouse monoclonal
anti-γH2A histone family member X (γH2AX) antibody (ab22551,
Abcam), followed by alpaca anti-mouse IgG1 (VHH) conjugated with
Alexa Fluor 488 (SA510328, Thermo Fisher Scientific) or rabbit
polyclonal anti-AhR (28727-1-AP) then by alpaca antirabbit IgG
(VHH) conjugated with Alexa Fluor 488 (SA510322, Thermo Fisher
Scientific). Thereafter, the cell nuclei were counterstained with
4′,6-diamidino-2-phenylindole (DAPI). Fluorescence images were
obtained using a microscope (Olympus IX71; Olympus Optical Co.,
Ltd., Tokyo, Japan) equipped with a digital camera. For γH2AX DNA
damage assay, the cells with ≥10 foci were determined to be
positive. For foci quantification, 100 cells were counted in each
sample, and percentages of the positive cells among the counted
cells were calculated.
Transcription factor activation
assay
Activation of the transcription factor Nrf2 was
assessed using a TranAM® Nrf2 Transcription Factor
Binding Assay kit (Active Motif Japan, Tokyo, Japan), according to
the manufacturer's instructions.
Cycloheximide chase assay
A549 cells were treated with 50-µM FF for 36 h,
followed by addition of 100 µg/ml of cycloheximide. After 0, 15,
30, and 60 min, the cells were lysed and processed for Western blot
analysis using rabbit monoclonal anti-Nrf2 antibody (ab62352) and
an HRP-conjugated secondary antibody. Signal intensities were
quantified by densitometric scanning using ImageJ (version 1.49V).
The half-lives (T1/2) of Nrf2 were calculated from
regression curve obtained from the plotted data of quantified
signal intensities normalized to β-actin at each time point.
Measurement of cytochrome P450 1A1
activity
Cytochrome P450 1A1 (CYP1A1) activity was determined
using P450-Glo CYP1A1 assay kit (Promega Corporation, Madison, WI,
USA), according to the manufacturer's instructions. The P450-Glo
assay value was normalized using CellTiter-Glo®
Luminescent Cell Viability Assay (Promega Corporation).
Reverse transcription-quantitative
polymerase chain reaction
This test was performed using TaqMan®
Fast Advanced Cells-to-CT Kit (Thermo Fisher Scientific) and
TaqMan® Gene Expression Assay (Thermo Fisher
Scientific). The predesigned human-specific primers with TaqMan
probes that were used in this study were ACTB TaqMan®
Gene Expression Assay (FAM) (assay ID: Hs99999903_m1, Thermo Fisher
Scientific); NFE2L2 TaqMan® Gene Expression Assay (FAM)
(assay ID: Hs00975961_g1, Thermo Fisher Scientific); AHR
TaqMan® Gene Expression Assay (FAM) (assay ID:
Hs00169233_m1, Thermo Fisher Scientific); and PPARA
TaqMan® Gene Expression Assay (FAM) (assay ID:
Hs00947536_m1, Thermo Fisher Scientific) (Table I). The ∆∆Cq method was used to
calculate the fold gene expression of NFE2L2 or AhR. ACTB was used
as a housekeeping gene to normalize the Ct values (17). The formulas used in this study were
as follows: ∆Cq=Cq (gene of interest)-Cq (housekeeping gene);
∆∆Cq=∆Cq (Sample)-∆Cq (Control average); Fold gene
expression=2−(∆∆Cq).
 | Table I.Reverse transcription-quantitative
polymerase chain reaction-based TaqMan® gene expression
assays. |
Table I.
Reverse transcription-quantitative
polymerase chain reaction-based TaqMan® gene expression
assays.
| Target | Exon
boundaries | TaqMan gene
expression assay ID | Accession no. |
|---|
| ACTB | 1-1 | Hs99999903_m1 | NM_001101 |
| NFE2L2 | 4-5 | Hs00975961_g1 | NM_006164 |
| AHR | 6-7 | Hs00169233_m1 | NM_001621 |
| PPARA | 4-5 | Hs00947536_m1 | NM_001001928 |
Small interfering RNA
transfection
Knockdown of AhR and PPARA was achieved by treating
A549 cells with small interfering RNA (siRNA) duplexes that
comprised four different predesigned sequences that target the
human AhR mRNA sequence (accession number P35869; cat. no.
L-004990-00-0005; Horizon Discovery Ltd., Cambridge, UK) and the
human PPARA mRNA sequence (accession number AY206718; cat. no.
L-003434-00-0005; Horizon Discovery Ltd.) (Table II). For the control experiment,
cells were treated with scrambled nontargeting siRNAs (catalogue
no. D-001810-10; Horizon Discovery). siRNAs were transfected using
a transfection reagent (DharmaFECT 1, Horizon Discovery), according
to the manufacturer's instructions.
| Table II.Sequences of the small interfering
RNAs. |
Table II.
Sequences of the small interfering
RNAs.
| Target | Sequence |
|---|
| AHR |
|
|
Sense-1 |
5′-GCAAGUUAAUGGCAUGUUUUU-3′ |
|
Antisense-1 |
3′-UUCGUUCAAUUACCGUACAAA-5′ |
|
Sense-2 |
5′-GAACUCAAGCUGUAUGGUAUU-3′ |
|
Antisense-2 |
3′-UUCUUGAGUUCGACAUACCAU-5′ |
|
Sense-3 |
5′-GCACGAGAGGCUCAGGUUAUU-3′ |
|
Antisense-3 |
3′-UUCGUGCUCUCCGAGUCCAAU-5′ |
|
Sense-4 |
5′-GCAACAAGAUGAGUCUAUUUU-3′ |
|
Antisense-4 |
3′-UUCGUUGUUCUACUCAGAUAA-5′ |
| PPARA |
|
|
Sense-1 |
5′-CCCGUUAUCUGAAGAGUUCUU-3′ |
|
Antisense-1 |
3′-UUGGGCAAUAGACUUCUCAAG-5′ |
|
Sense-2 |
5′-GCUUUGGCUUUACGGAAUAUU-3′ |
|
Antisense-2 |
3′-UUCGAAACCGAAAUGCCUUAU-5′ |
|
Sense-3 |
5′-GACUCAAGCUGGUGUAUGAUU-3′ |
|
Antisense-3 |
3′-UUCUGAGUUCGACCACAUACU-5′ |
|
Sense-4 |
5′-GGGAAACAUCCAAGAGAUUUU-3′ |
|
Antisense-4 |
3′-UUCCCUUUGUAGGUUCUCUAA-5′ |
| Nontargeting |
|
|
Sense-1 |
5′-UGGUUUACAUGUCGACUAAUU-3′ |
|
Antisense-1 |
3′-UUACCAAAUGUACAGCUGAUU-5′ |
|
Sense-2 |
5′-UGGUUUACAUGUUGUGUGAUU-3′ |
|
Antisense-2 |
3′-UUACCAAAUGUACAACACACU-5′ |
|
Sense-3 |
5′-UGGUUUACAUGUUUUCUGAUU-3′ |
|
Antisense-3 |
3′-UUACCAAAUGUACAAAAGACU-5′ |
|
Sense-4 |
5′-UGGUUUACAUGUUUUCCUAUU-3′ |
|
Antisense-4 |
3′-UUACCAAAUGUACAAAAGGAU-5′ |
Statistical analysis
The mean and standard error of the mean were used to
express the data. Statistical analyses were carried out using
Microsoft Excel X with the Statcel 3 (OMS, Tokyo, Japan) add-in
software. The Welch t-test, or one-way or two-way analysis of
variance (ANOVA) was used as appropriate to determine significant
differences. If the results of the ANOVA were significant, the
Tukey-Kramer test or Dunnett's test was used as a post hoc for
multiple comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
Dose-dependent effect of FF on lung
cancer cell survival in the presence or absence of CDDP
We first examined whether treatment with FF at
concentrations of 0–200 µM affected the survival of A549 cells. As
shown in Fig. 1A, the Hoechst 33342
DNA assay and alamarBlue® assay showed that FF at ≥100
µM significantly reduced A549 cell survival. This result
corroborated that of a previous research, which showed the
anticancer effect of FF (5).
However, at concentrations of ≤50 µM, which correspond to
clinically achievable blood concentrations (7,18), FF
had no significant effect on A549 cell survival. We next examined
whether FF at ≤50 µM affected A549 cell survival in the presence of
CDDP. Fig. 1B shows that exposure
to 5–40 µM of CDDP reduced A549 cell survival. However, the
presence of FF at 50 µM significantly promoted A549 cell survival
after CDDP exposure, as shown by the Hoechst 33342 DNA,
alamarBlue®, and cellular ATP assays. Within a FF
concentration range of 25–50 µM, FF had a dose-dependent
pro-survival effect against CDDP (Fig.
1C). In addition, treatment with WY, which is a selective
agonist of PPAR-α, promoted A549 cell survival after CDDP exposure
in a dose-dependent manner (Fig.
1C). The pro-survival effect of FF against CDDP was attenuated
in the presence of the PPAR-α antagonist GW6471 (Fig. 1D), indicating that the pro-survival
effect of FF against CDDP was, at least in part, secondary to its
PPAR-α agonistic activity. These findings implied that FF at 25 or
50 µM had a cytoprotective effect on A549 cells against CDDP,
whereas FF at ≥50 µM, albeit unachievable in clinical practice, had
a cytotoxic effect. We also examined the effect of FF on other
non-small cell lung cancer cell lines, including H1299, PC3, and
H441, to determine whether the FF attenuation of CDDP cytotoxicity
was peculiar to A549 cells. In each of these cell lines, treatment
with FF at 50 µM reduced the CDDP-induced cell death (Fig. 1E).
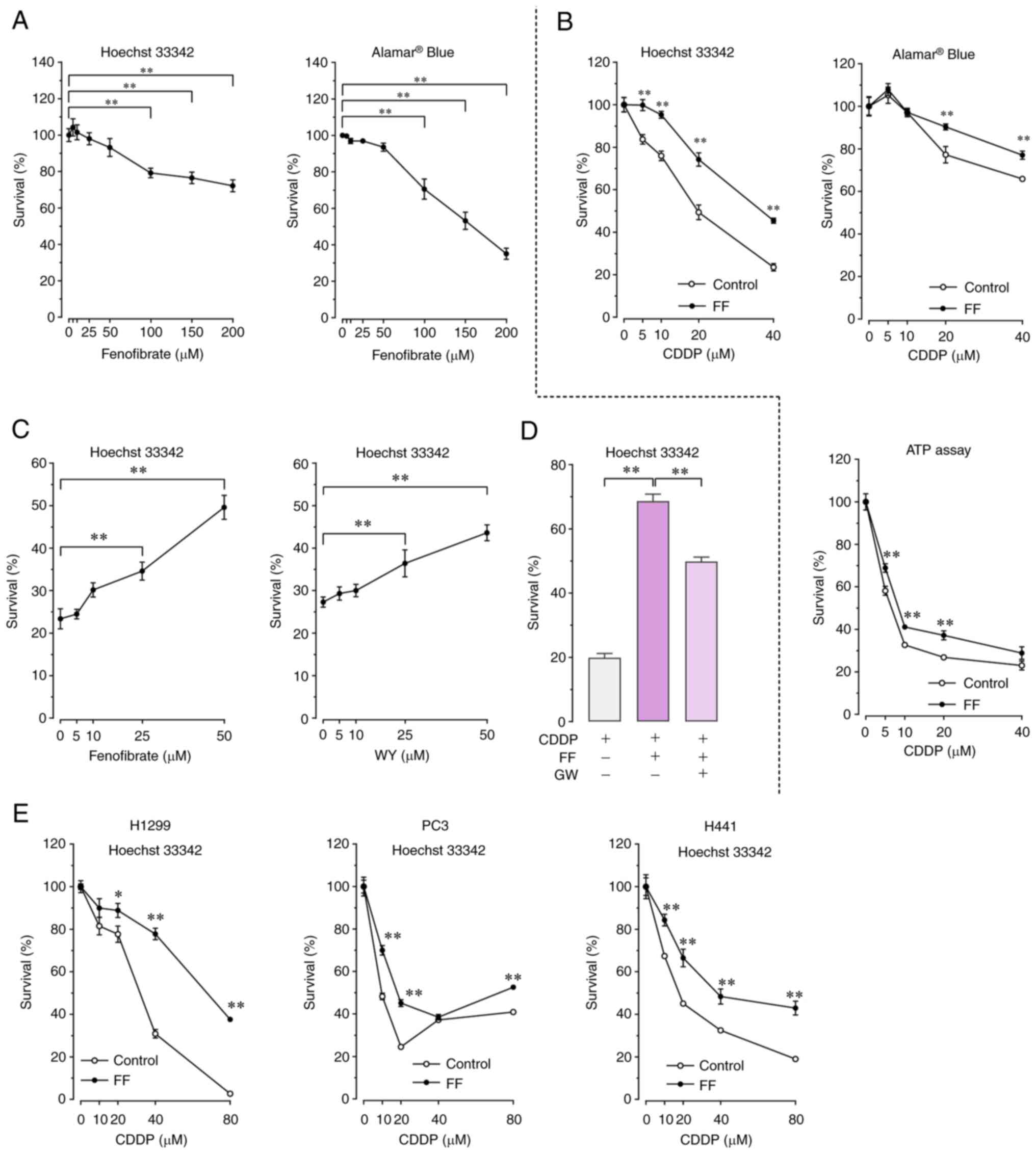 | Figure 1.Dose-dependent effect of FF on A549
cell survival in the presence or absence of CDDP. (A) To study the
effect of FF on cell survival in the absence of CDDP, A549 cells
were treated with 5–200 µM of FF or vehicle alone for 72 h. Cell
survival was determined by Hoechst 33342 DNA quantification assay
(n=8) and alamarBlue® assay (n=8). Data are expressed as
the mean and standard error of the mean. **P<0.01 using
the one-way ANOVA and Dunnett's test. (B) To study the effect of FF
on cell survival in the presence of CDDP, A549 cells were
pretreated with 50-µM FF or vehicle alone for 12 h, followed by
exposure to 40-µM CDDP for 72 h in the presence or absence of FF.
Cell survival was determined by Hoechst 33342 DNA quantification
assay (n=6), alamarBlue® assay (n=8), and ATP
quantification assay (n=6). Data are expressed as the mean and
standard error of the mean. **P<0.01 vs. control using
the Welch t-test. (C) To study the dose-dependent effects of FF and
WY on cell survival in the presence of CDDP, A549 cells were
pretreated with 5–50 µM of FF, 5–50 µM of WY, or vehicle alone for
12 h, followed by exposure to 40 µM CDDP for 72 h in the presence
or absence of FF or WY. Cell survival was determined using Hoechst
33342 DNA quantification assay (n=6). Data are expressed as the
mean and standard error of the mean. **P<0.01 using the
one-way ANOVA and Dunnett's test. (D) To study the inhibitory
effect of GW on FF-induced promotion of cell survival, A549 cells
were pretreated with 50 µM FF in the presence or absence of 5 µM GW
for 12 h, followed by exposure to 40 µM CDDP for 72 h. Cell
survival was determined by Hoechst 33342 DNA quantification assay
(n=6). Data are expressed as the mean and standard error of the
mean. **P<0.01 using the one-way ANOVA and Tukey-Kramer
test. (E) For survival analysis of other lung cancer cells, H1299,
PC3, and H441 cells were pretreated with 50 µM FF or vehicle alone
for 12 h, followed by exposure to 10–80 µM of CDDP for 72 h in the
presence or absence of FF. Cell survival was determined by Hoechst
33342 DNA quantification assay (n=8). *P<0.05 and
**P<0.01 vs. control using the Welch t-test. FF,
fenofibrate; CDDP, cisplatin; WY, WY14643; GW, GW6471. |
FF treatment did not modulate the DNA
damage response elicited by CDDP exposure
Next, we examined whether FF modulated CDDP-induced
DNA damage, which is thought to contribute to the mechanism of CDDP
cytotoxicity (16). CDDP exposure
evoked a DNA damage response in A549 cells, as demonstrated by
phosphorylation of H2AX (γH2AX) (19) (Fig. 2A
and B) and phosphorylation of the p53 protein (Fig. 2C). However, the presence of FF or WY
at 50 µM had no effect on the DNA damage response to CDDP. We also
evaluated the expression of the Bcl-2 family proteins, which are
thought to regulate apoptosis following CDDP-induced DNA damage
(20). The expression of the
proapoptotic proteins Bax and Bad, as well as the antiapoptotic
proteins Bcl-2 and Bcl-x, were comparable in A549 cells, regardless
of exposure to CDDP and of the presence of FF and WY (Fig. 2D). These findings indicated that FF
and WY did not modulate the DNA damage response elicited by CDDP.
In addition, treatment of A549 cells with FF and WY had no effect
on the expression of the HSP70 protein, which is thought to
contribute to CDDP resistance (21), regardless of exposure to CDDP
(Fig. 2C).
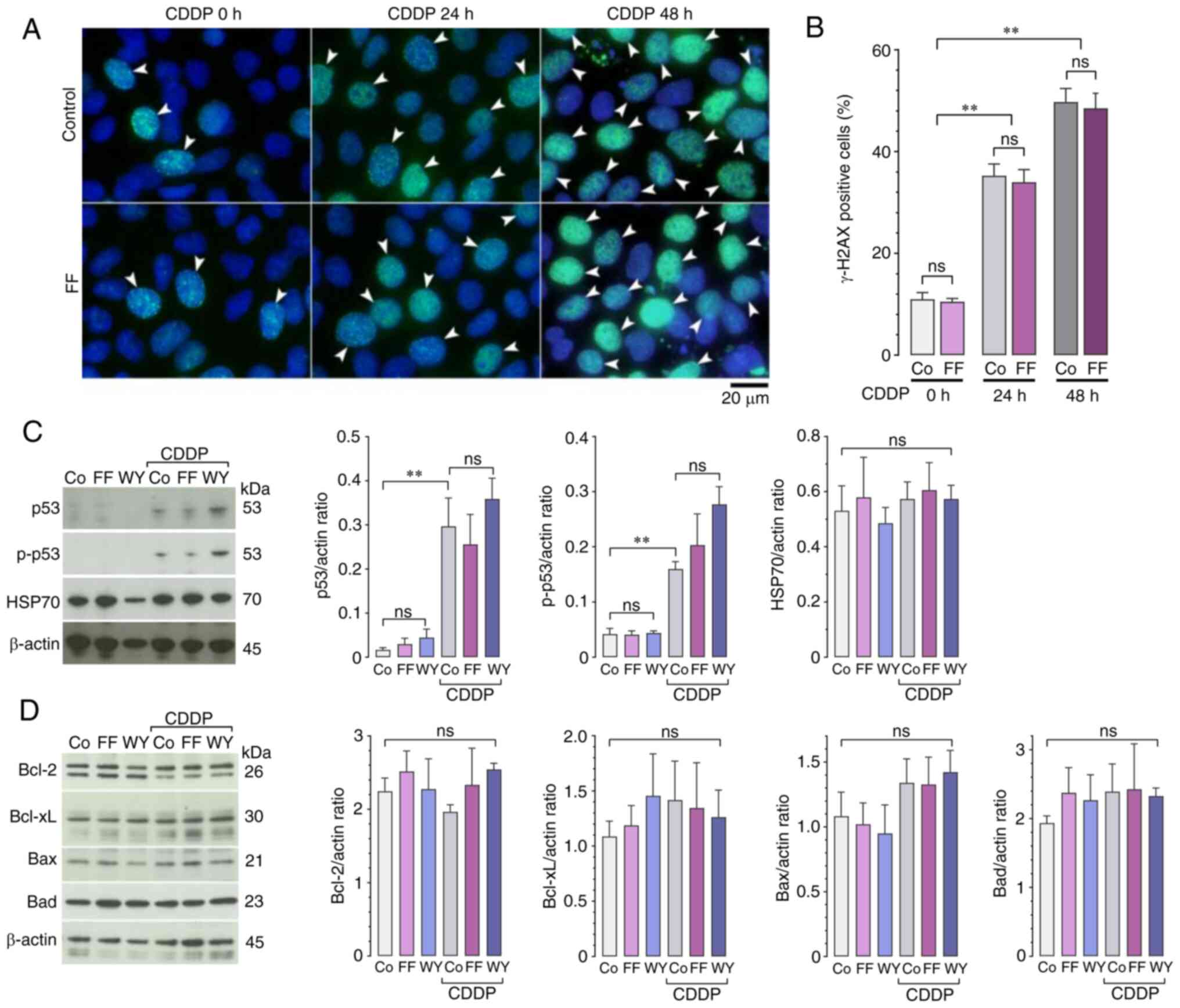 | Figure 2.Effects of FF and WY on CDDP-induced
DNA damage response. A549 cells were pretreated with 50 µM FF, 50
µM WY, or vehicle alone for 24 h, followed by exposure to 40 µM
CDDP for 24 or 48 h (i.e., 24 h unless indicated) in the presence
or absence of FF or WY. (A and B) Immunofluorescence of γH2AX
expression. (A) Representative images of γH2AX expression (green);
cell nuclei were counterstained with DAPI (blue); the arrowheads
indicate γH2AX-positive cells. (B) The percentages of
γH2AX-positive cells are shown. Data are expressed as the mean and
standard error of the mean (n=4). **P<0.01 using the
two-way ANOVA and Tukey-Kramer test t; ns, not significant; Co,
control. (C and D) Western blot analyses for the expression of p53,
p-p53, and HSP70 (C); and Bcl-2, Bcl-xL, Bax, and Bad (D) are
shown. The relative protein levels were estimated using
densitometry and normalized to the level of β-actin as a loading
control. Data are expressed as the mean and standard error of the
mean (n=4). **P<0.01 using the one-way ANOVA and
Tukey-Kramer test. FF, fenofibrate; WY, WY14643; CDDP, cisplatin;
γH2AX, γH2A histone family member X; DAPI,
4′,6-diamidino-2-phenylindole; p-p53, phosphorylated p53; HSP70,
heat shock protein 70; Bcl-2, B-cell/CLL lymphoma 2; Bcl-xL, B-cell
lymphoma-extra large; Bax, Bcl-2-associated X protein; Bad, Bcl-2
antagonist of cell death. |
FF treatment reduced CDDP-induced ROS
accumulation by enhancing antioxidant activity
CDDP cytotoxicity results not only from a DNA damage
response but also from ROS generation (16). In this study, CDDP exposure of A549
cells increased the cellular ROS levels (Fig. 3A); this corroborated the findings of
previous researches (22,23). However, the presence of FF or WY at
50 µM significantly reduced the cellular ROS levels (Fig. 3A) and promoted A549 cell survival
after exposure to exogenous hydrogen peroxide (Fig. 3B). Based on these findings, we
hypothesized that rather than reducing oxidant production, FF and
WY enhanced the activity of antioxidants. Consistent with our
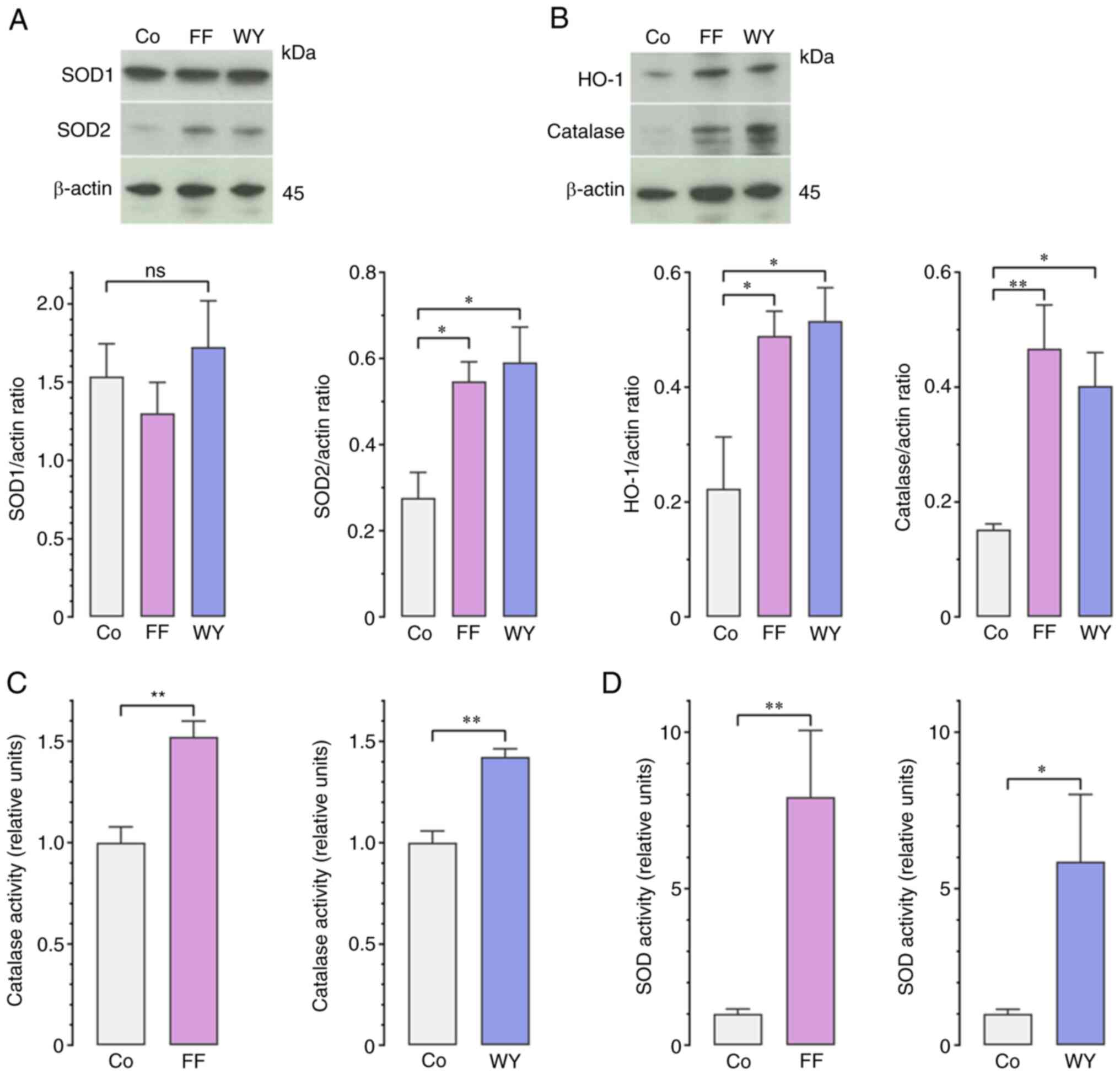
expectations, FF or WY at 50 µM enhanced the protein expressions of
mitochondrial Mn SOD (SOD2), HO-1, and catalase (Fig. 4A and B), as well as the enzyme
activities of SOD and catalase (Fig. 4C
and D). Both FF and WY did not affect the protein expression of
cytosolic Cu/Zn SOD (SOD1), which was expressed at a high level in
A549 cells, regardless of exposure to FF and WY (Fig. 4A) (24). These findings indicated that FF and
WY reduced cellular ROS by enhancing the antioxidant activity of
A549 cells.
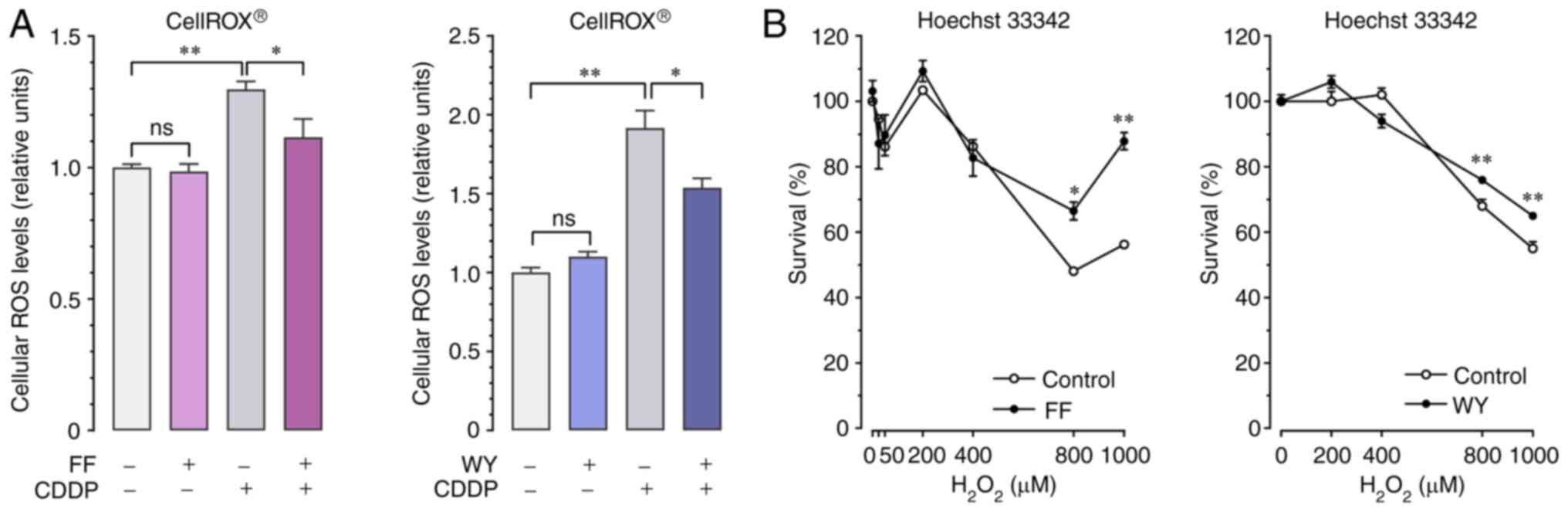 | Figure 3.Effects of fenofibrate FF and WY on
CDDP-induced cellular ROS accumulation and
H2O2-induced cell death. (A) For the
quantitative analysis of cellular ROS levels, A549 cells were
pretreated with 50-µM FF, 50 µM WY, or vehicle alone for 12 h,
followed by exposure to 40 µM CDDP for 48 h in the presence or
absence of FF or WY. Cellular ROS levels were estimated using the
CellROX® Green assay. Data are expressed as the mean and
standard error of the mean (n=12). *P<0.05 and
**P<0.01 using the one-way ANOVA and Tukey-Kramer test;
ns, not significant. (B) To analyze cell survival after
H2O2 exposure, A549 cells were pretreated
with 50 µM FF, 50 µM WY, or vehicle alone for 36 h, followed by
exposure to 25–1,000 µM of H2O2 for 48 h in
the presence or absence of FF or WY. Cell survival was determined
by Hoechst 33342 DNA quantification assay. Data are expressed as
the mean and standard error of the mean (n=8). *P<0.05
and **P<0.01 vs. control using the Welch t-test. FF,
fenofibrate; WY, WY14643; CDDP, cisplatin; ROS, reactive oxygen
species. |
FF treatment enhanced the expression
and activation of Nrf2 transcription factor
Next, we examined whether the enhancement of
antioxidants by FF was mediated by the activation of Nrf2, which is
a transcription factor that binds to the antioxidant response
element (ARE) to stimulate the transcription of antioxidant genes,
such as those of SOD, HO-1 and catalase (25,26).
Treatment with FF or WY at 50 µM increased the transcription,
translation, nuclear translocation, and sequence-specific
DNA-binding activity of Nrf2 in A549 cells (Fig. 5A-D). The Nrf2 protein undergoes
rapid ubiquitination and proteasomal degradation upon its binding
to Keap1-Cullin3 and β-TrCP-Cullin1 (27,28).
Therefore, we examined whether FF affected the ubiquitination and
degradation of Nrf2. Western blot analyses of
Nrf2-immunoprecipitated proteins by anti-ubiquitin antibody
demonstrated no significant difference in the ubiquitination level
of Nrf2 in the presence and absence of FF (Fig. 5E). A cycloheximide chase assay
showed similar half-lives (T1/2) of the Nrf2 protein in
the presence (17.8 min) or absence (16.1 min) of FF at 50 µM
(Fig. 5F). This result indicated
that FF had no effect on Nrf2 protein degradation. Furthermore, the
presence of FF and WY had no effect on the protein expression of
Keap1 and β-TrCP (Fig. 5G). The
extremely low basal Keap1 level in A549 cells was presumably
secondary to hypermethylation of the Keap1 promoter in these cells
(29,30). These findings implied that treatment
of A549 cells with FF enhanced the transcription, translation, and
activation of Nrf2 without affecting its degradation. Consistent
with those found with A549 cells, treatment with FF at 50 µM
enhanced the transcription of the Nrf2 gene in H1299, PC3,
and H441 cells (Fig. 5H).
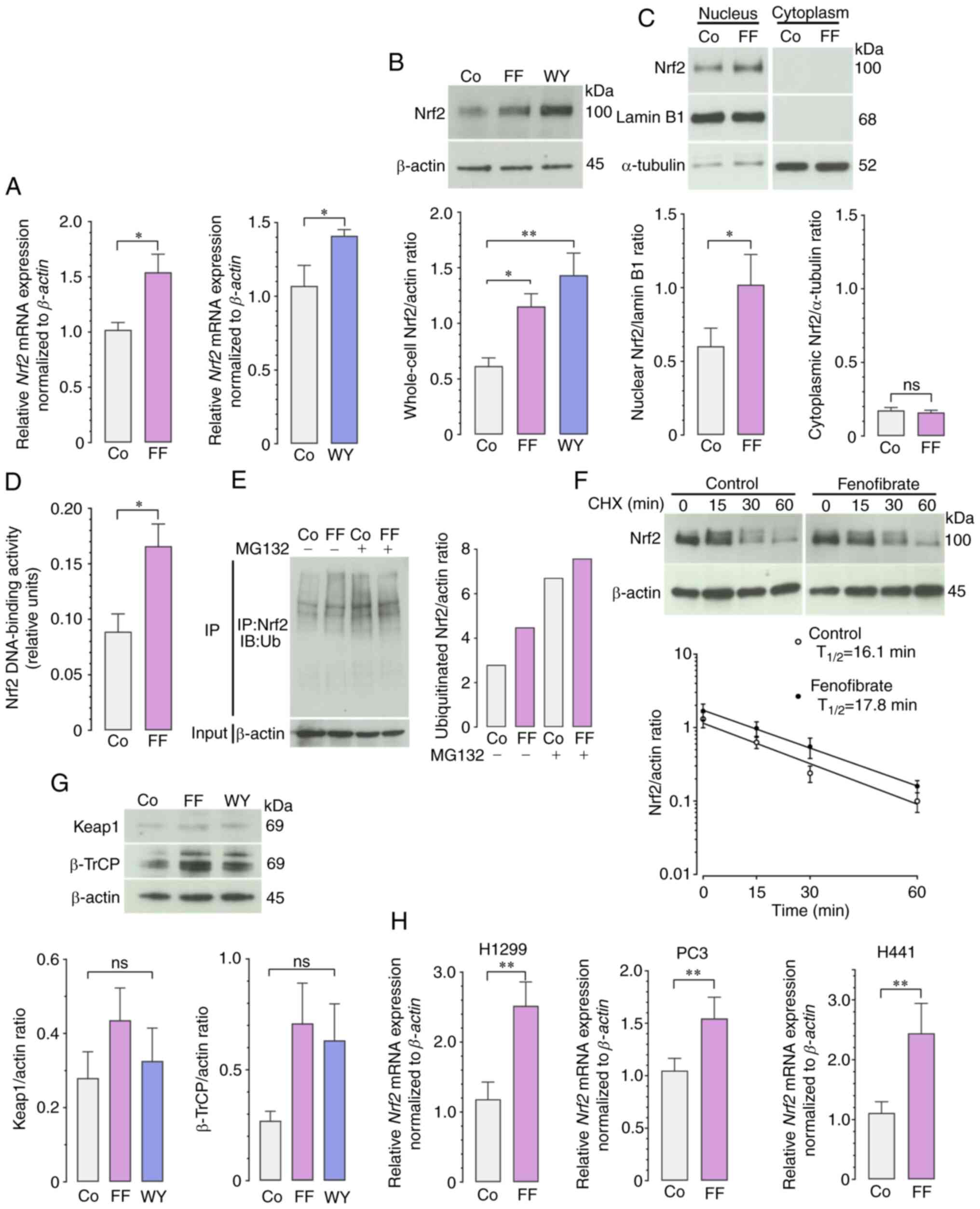 | Figure 5.Effects of FF and WY on the
transcription, translation, activation, and degradation of Nrf2.
(A-F) A549 cells were treated with 50 µM FF, 50 µM WY, or vehicle
alone for 36 h. (A) Reverse transcription-quantitative polymerase
chain reaction was used to analyze Nrf2 gene expression. The
expression levels of Nrf2 mRNA were normalized to the level
of β-actin mRNA as a housekeeping gene. Data are expressed
as the mean and standard error of the mean (n=10). *P<0.05 using
the Welch t-test (n=10); Co, control. (B) Western blot analysis for
whole-cell Nrf2 protein expression was done. The relative protein
levels were estimated using densitometry and normalized to the
level of β-actin as a loading control. Data are expressed as the
mean and standard error of the mean (n=4). *P<0.05 and
**P<0.01 using the one-way ANOVA and Dunnett's test. (C) Western
blot analysis for nuclear and cytoplasmic Nrf2 protein was done;
the relative protein levels were normalized to lamin B1 and
α-tubulin, respectively. Data are expressed as the mean and
standard error of the mean (n=4). *P<0.05 using the Welch t-test
(n=4). (D) The sequence-specific DNA-binding activity of Nrf2 is
shown. Data are expressed as the mean and standard error of the
mean (n=8). *P<0.05 using the Welch t-test. (E) Ubiquitination
level of Nrf2. A549 cells were treated with or without 10 µM of
MG132, a protease inhibitor, 16 h before the preparation of cell
lysates, which were used for the immunoprecipitation of Nrf2 using
antiNrf2 antibody. The ubiquitination levels of Nrf2 were examined
by immunoblotting using antiUb antibody. The levels were reduced in
the absence of MG132, which was a negative control of experiments.
(F) Nrf2 protein degradation was assessed by cycloheximide chase
assay. A549 cells were treated with 50-µM FF for 36 h, followed by
addition of 100-µg/ml cycloheximide. After 0, 15, 30, and 60 min,
the cells were lysed and processed for Western blot analysis for
determination of whole-cell Nrf2 protein content. The relative
protein levels were estimated using densitometry, normalized to the
protein levels of β-actin as a loading control, and plotted against
time. Data are expressed as the standard error of the mean (n=4).
(G) Western blot analysis for Keap1 and β-TrCP; the relative
protein levels were normalized to the level of β-actin as loading
control (n=4). (H) Reverse transcription-quantitative polymerase
chain reaction was done to analyze the gene expression of
Nrf2 in H1299, PC3, and H441 cells after 12 h of treatment
of with 50 µM FF. The expression levels of Nrf2 mRNA were
normalized to the level of β-actin mRNA as a housekeeping
gene. Data are expressed as the mean and standard error of the mean
(n=7-9). **P<0.01 using the Welch t-test; FF, fenofibrate; WY,
WY14643; Nrf2, nuclear factor-erythroid 2-related factor 2; Ub,
ubiquitinin; Keap1, Kelch-like ECH-associated protein 1; β-TrCP,
β-transduction repeat containing protein; Co, control; ns, not
significant. |
FF treatment enhanced Nrf2 expression
by stimulating AhR expression
The promoter region of Nrf2 was reported to possess
a xenobiotic response element (XRE) (31), which implies that the AhR can bind
to the XRE after heterodimerizing with its partner AhR nuclear
translocator and activate Nrf2 gene transcription (32). Furthermore, the promoter region of
AhR was reported to possess PPRE (33), which implies that upon activation by
FF, PPAR-α can bind to the PPRE after heterodimerizing with the
retinoid X receptor and activate AhR gene transcription.
Based on these previous researches, we examined whether the
FF-induced enhancement of Nrf2 expression was secondary to a
preceding enhancement of AhR expression by FF. As shown in Fig. 6A-D, treatment with FF or WY at 50 µM
increased the transcription, translation, and nuclear translocation
of AhR, as well as the enzyme activity of CYP1A1, which is
increasingly expressed when AhR binds to the XRE (34,35).
Consistent with those found with A549 cells, treatment with FF at
50 µM enhanced the transcription of the AhR gene in H1299,
PC3, and H441 cells (Fig. 6E).
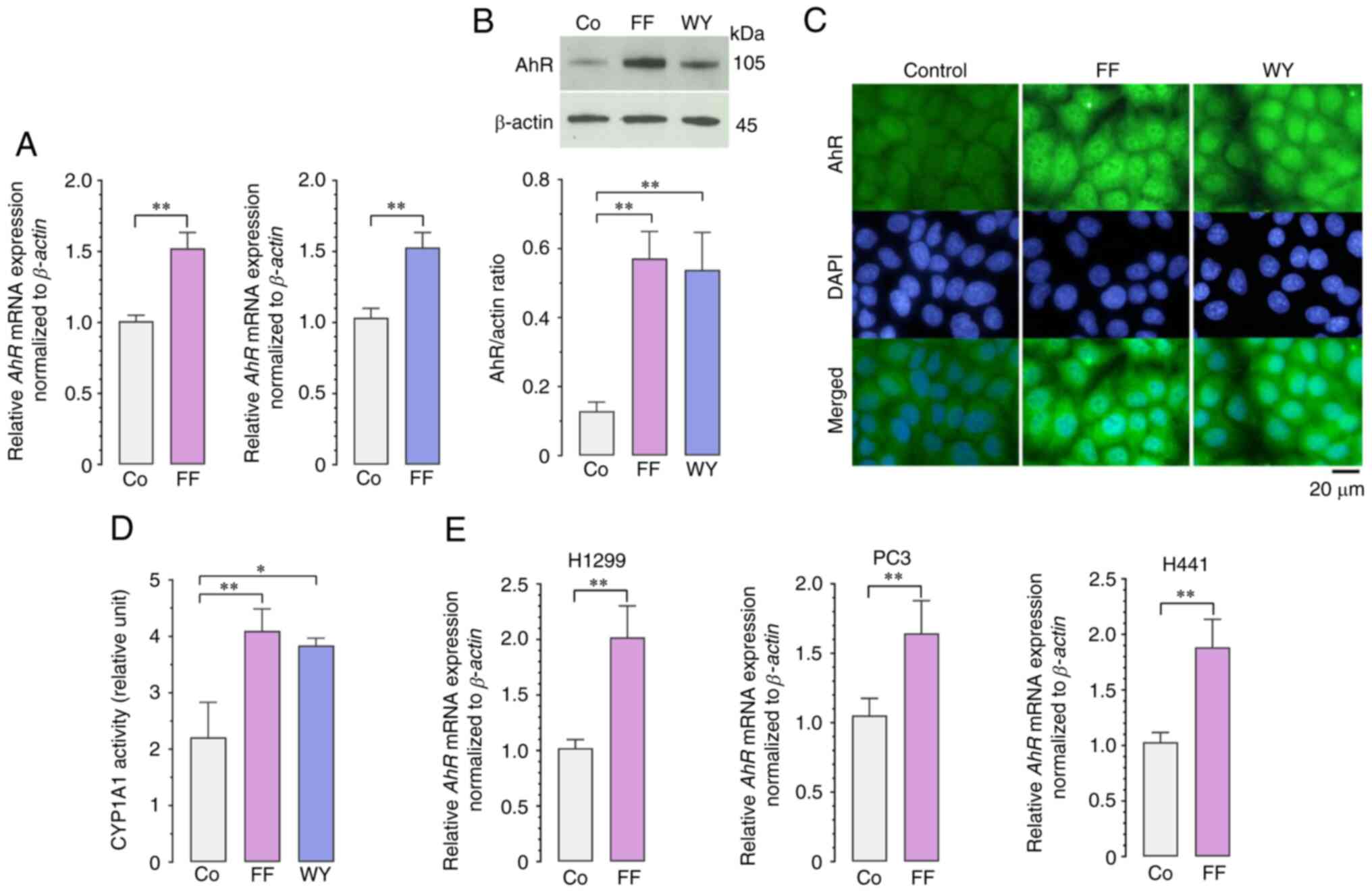 | Figure 6.Effects of FF and WY on the
transcription, translation, and activation of the AhR. (A-D) A549
cells were treated with 50 µM FF, 50 µM WY, or vehicle alone for 36
h. (A) Reverse transcription-quantitative polymerase chain reaction
was performed to analyze AhR expression. The expression
levels of AhR mRNA were normalized to the level of
β-actin mRNA as a housekeeping gene. Data are expressed as
the mean and standard error of the mean (n=10). **P<0.01 using
the Welch t-test. (B) Western blot analysis for AhR protein
expression was done. The relative protein levels were estimated
using densitometry and normalized to the level of β-actin as a
loading control. **P<0.01 using the one-way ANOVA and Dunnett's
test. (C) Representative images of immunofluorescence show
increased cellular expression and nuclear translocation of AhR
(green) after treatment with FF or WY. Cell nuclei were
counterstained with DAPI (blue). (D) Measurement of CYP1A1 activity
is shown. Data are expressed as the mean and standard error of the
mean (n=4). *P<0.05 and **P<0.01 using the one-way ANOVA and
Dunnett's test. (E) Reverse transcription-quantitative polymerase
chain reaction was performed to analyze the gene expression of
AhR in H1299, PC3, and H441 cells after 12 h of treatment of
with 50 µM FF. The expression levels of AhR mRNA were
normalized to the level of β-actin mRNA as a housekeeping
gene. Data are expressed as the mean and standard error of the mean
(n=7-9). **P<0.01 using the Welch t-test. FF, fenofibrate; WY,
WY14643; AhR, aryl hydrocarbon receptor; DAPI,
4′,6-diamidino-2-phenylindole; CYP1A1, cytochrome P450 1A1; Co,
control. |
Knockdown of the AhR gene with siRNA
transfection reduced the effects of FF and WY, which would have
otherwise increased Nrf2 gene transcription and promoted the
survival of CDDP-exposed A549 cells (Fig. 7A-C). Accumulating evidence indicated
that PPAR-α-independent mechanisms are involved in the pleiotropic
effects of FF on various pathophysiological processes (36,37).
However, the knockdown of the PPARA gene with siRNA transfection
reduced the stimulatory effect of FF on AhR gene transcription,
although the AhR gene inhibition by the PPARA siRNA transfection
was not statistically significant (P=0.08) (Fig. 7D and E). These findings indicated
that the PPAR-α agonists FF and WY stimulated the expression of AhR
that binds to the XRE, which in turn stimulated the expression of
Nrf2 that binds to ARE to activate antioxidant expression, thereby,
resulting in A549 cell protection from CDDP cytotoxicity.
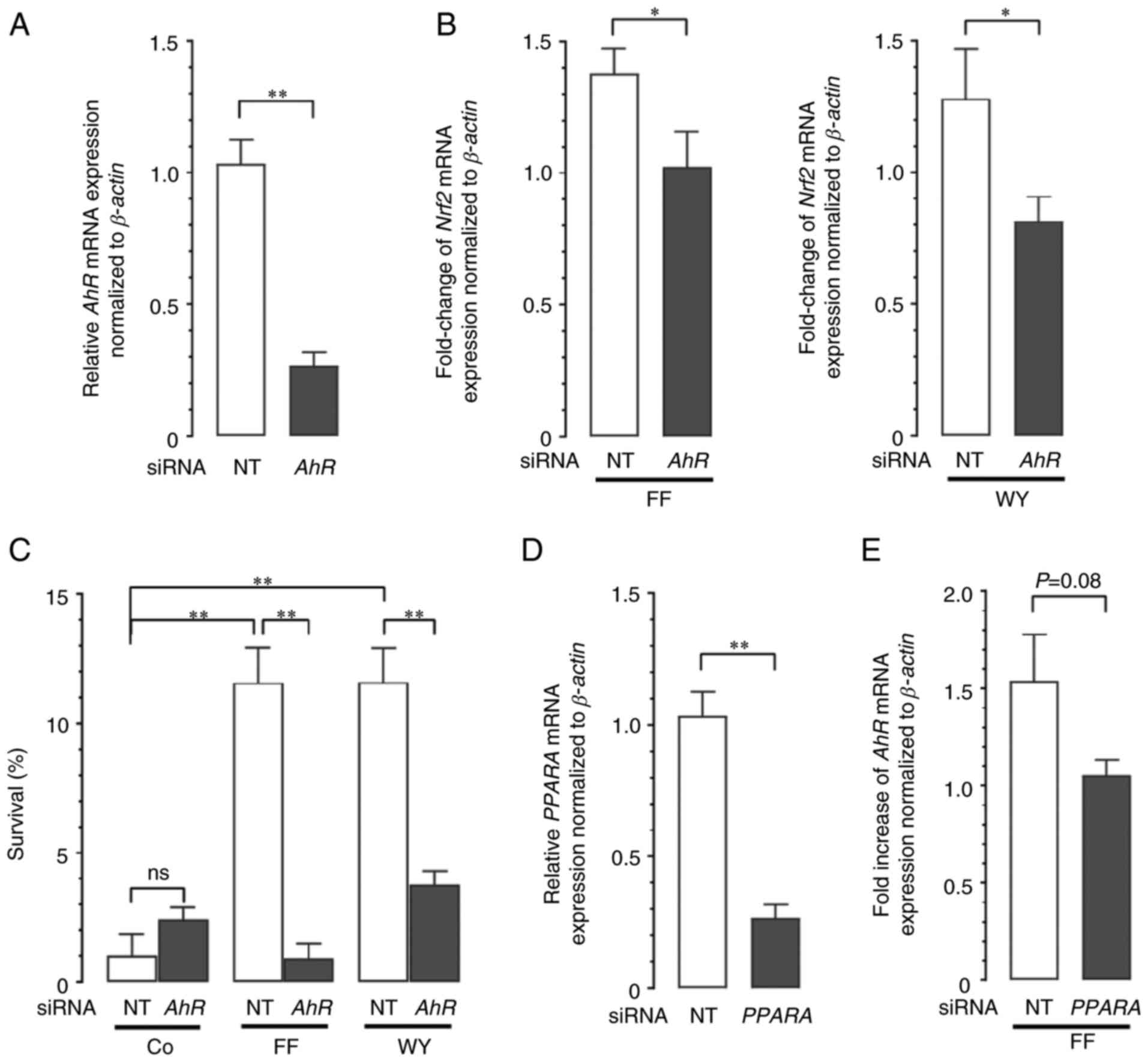 | Figure 7.Effect of AhR gene and
PPARA gene knockdown with siRNA. (A) Knockdown efficiency of
AhR siRNA. A549 cells were transfected with AhR-targeting or
NT siRNA. The expression levels of AhR mRNA at 48 h were
determined by reverse transcription-quantitative polymerase chain
reaction and normalized to the level of β-actin mRNA as a
housekeeping gene. Data are expressed as the mean and standard
error of the mean (n=10). **P<0.01 using the Welch t-test. (B)
Effect of AhR gene knockdown on Nrf2 gene expression.
A549 cells were transfected with AhR-targeting or NT siRNA,
followed by treatment with 50 µM FF, 50 µM WY, or vehicle alone for
36 h. The expression levels of Nrf2 mRNA were normalized to
the level of β-actin mRNA as a housekeeping gene. Data are
expressed as the mean and standard error of the mean (n=8 for FF
treatment, n=10 for WY treatment). *P<0.05 using the Welch
t-test. (C) For cell survival analysis, A549 cells transfected with
AhR-targeting or NT siRNA were pretreated with 50 µM FF, 50 µM WY,
or vehicle alone for 12 h followed by exposure to 40 µM CDDP for 72
h in the presence or absence of FF or WY. Cell survival was
determined using Hoechst 33342 DNA quantification assay. Data are
expressed as the mean and standard error of the mean (n=6).
**P<0.01 using the one-way ANOVA and Tukey-Kramer test. (D)
Knockdown efficiency of PPARA siRNA. A549 cells were transfected
with PPARA-targeting or NT siRNA. The expression levels of
PPARA mRNA at 24 h were determined by reverse
transcription-quantitative polymerase chain reaction and normalized
to the level of β-actin mRNA as a housekeeping gene. Data
are expressed as the mean and standard error of the mean (n=10).
**P<0.01 using the Welch t-test. (E) Effect of PPARA gene
knockdown on AhR gene expression. A549 cells were
transfected with PPARA-targeting or NT siRNA, followed by treatment
with 50 µM FF or vehicle alone for 12 h. The expression levels of
AhR mRNA were normalized to the level of β-actin mRNA
as a housekeeping gene. Data are expressed as the mean and standard
error of the mean (n=8). AhR, aryl hydrocarbon receptor; PPARA,
peroxisome proliferator-activated receptor-α; siRNA, small
interfering RNA; NT, nontargeting; Nrf2, nuclear factor erythroid
2-related factor 2; FF, fenofibrate; WY, WY14643; CDDP, cisplatin;
ns, not significant; Co, control. |
Discussion
According to previous reports, FF may have an
anticancer effect (5). However, in
the current study, we demonstrated that the effect of FF on lung
cancer cells depended on its concentration. We discovered that FF
at ≤50 µM, which is a clinically relevant blood concentration
(7,18), attenuated CDDP cytotoxicity to lung
cancer cells, whereas FF at ≥100 µM, albeit clinically
unachievable, had an anticancer effect. The mechanism of FF
attenuation of CDDP cytotoxicity involved PPAR-α-dependent AhR
expression, which in turn stimulated Nrf2 expression and
antioxidant production, resulting in lung cancer cell protection
from CDDP-evoked oxidative damage (Fig.
8). Our findings suggested that the concomitant use of FF with
CDDP may compromise the efficacy of chemotherapy.
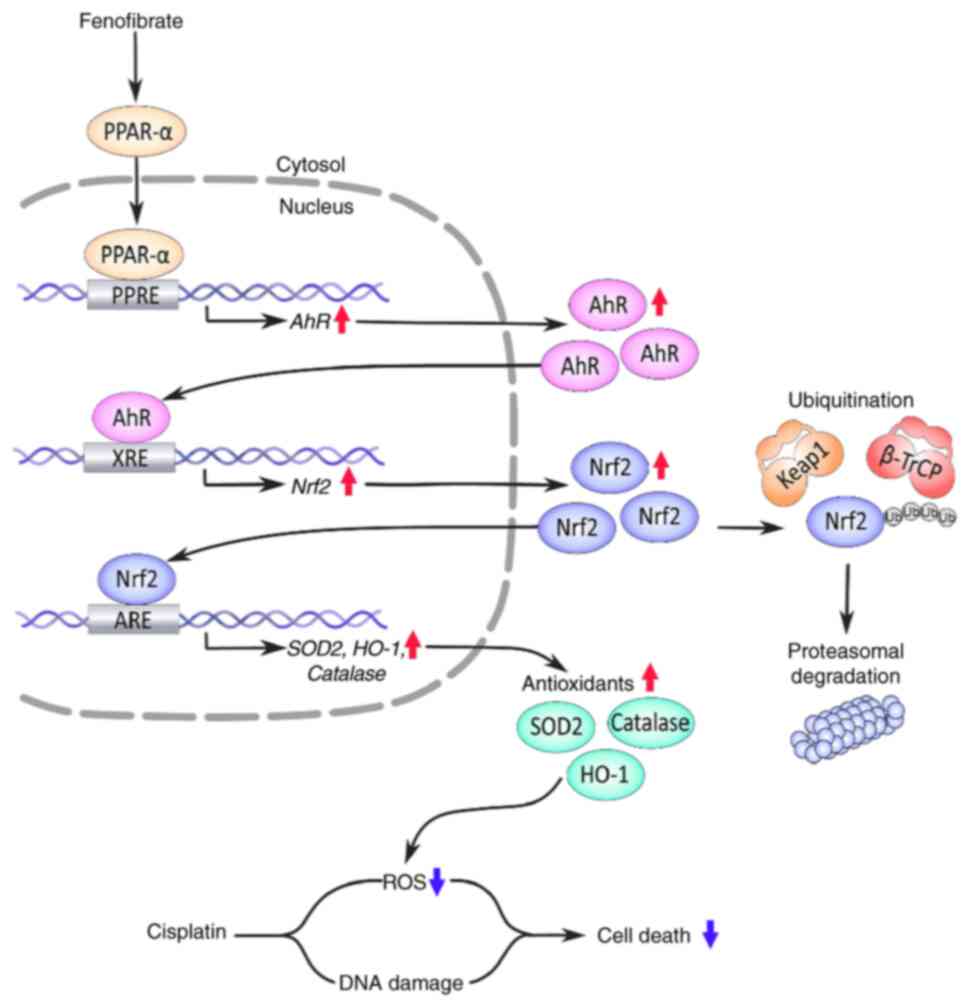 | Figure 8.Graphical summary of the signaling
pathway that mediates fenofibrate (FF)-induced cytoprotection
against cisplatin toxicity. ↑, increase; ↓, decrease; PPAR-α,
peroxisome proliferator-activated receptor-α; PPRE, peroxisome
proliferator response element; AhR, aryl hydrocarbon receptor; XRE,
xenobiotic response element; Nrf2, Nuclear factor erythroid
2-related factor 2; ARE, antioxidant response element; SOD2,
superoxide dismutase 2; CAT, catalase; HO-1, Heme oxygenase-1; ROS,
reactive oxygen species; Keap1, Kelch-like ECH-associated protein
1; β-TrCP, β-transduction repeat containing protein; Ub,
ubiquitin. |
It is understood that ROS generation contributes to
CDDP cytotoxicity, whereas antioxidants generated by the Nrf2-ARE
pathway cause CDDP resistance (25,38).
Our findings indicate that FF attenuation of CDDP cytotoxicity was
secondary to Nrf2-dependent antioxidant generation but not to the
modulation of p53-dependent DNA damage response. This conclusion
was supported by our observation that FF was capable of reducing
CDDP cytotoxicity in p53-deficient H1299 cells and p53-proficient
A549 cells.
Nrf2 activation is tightly regulated both
transcriptionally and post-translationally (27,28).
The posttranslational regulation of Nrf2 utilizes the Nrf2
degradation system, which is driven by E3 ubiquitin ligase
complexes that involve cytosolic Keap1-Cullin3 and nuclear
β-TrCP-Cullin1, which ubiquitinate and direct Nrf2 for rapid
proteasomal degradation (27,28).
Our findings on A549 cells showed that FF increased the Nrf2
protein by stimulating its transcription but not by affecting its
degradation. These findings were contrast to those of a previous
study on hepatoma cells, which showed that FF increased the Nrf2
protein post-translationally by triggering p62-dependent Keap1
degradation (39). Therefore, FF
may activate Nrf2 either during transcription or after
transnationally in a cell type-specific manner. In addition to Nrf2
activation, other mechanisms may underlie the FF enhancement of
antioxidant activity. For example, FF may directly activate the
transcription of antioxidant genes (40), because the promoter regions for the
SOD1 (41), SOD2
(42), and catalase
(43) genes were reported to
possess the PPRE, to which PPAR-α can bind when activated by FF. In
addition, FF may inhibit cellular ROS generation by interfering
with the ROS-producing pathway that involves nuclear factor kappa-B
and phosphoinositol 3-kinase/Akt (13,40).
Our findings indicated that FF activated the
PPAR-α-PPRE-AhR-XRE-Nrf2-ARE cascade pathway to stimulate
antioxidant generation. The presence of this cascade was verified
by two previous gene sequence studies, which individually
identified the PPAR-α-binding site PPRE in the promoter region of
AhR (33) and the AhR binding site
XRE in the promoter region of Nrf2 (31). Interestingly, another gene sequence
study has identified the Nrf2 binding site ARE in the promoter
region of AhR (44). These studies
implied that the binding of AhR to XRE activates the expression of
Nrf2, which binds to the ARE, thereby, allowing AhR expression in
turn. This suggested the existence of a mutually synergistic
interaction between Nrf2 and AhR, which may amplify the
antioxidative response that is initially triggered by PPAR-α
activation upon FF treatment.
In this study FF enhanced the protein expressions of
mitochondrial Mn SOD (SOD2), HO-1, and catalase. However, other
antioxidants may also be involved in the FF-mediated attenuation of
CDDP cytotoxicity because Nrf2 was found to stimulate the
transcription of many other antioxidant genes, including SOD3,
glutathione peroxidase, glutathione reductase, thioredoxin,
thioredoxin reductase, and peroxiredoxin (45).
Consistent with the findings of a previous study
(5), the present study found that
FF had an anticancer effect. However, in our study on A549 cells,
this effect required high FF concentrations of ≥100 µM, which
exceed the clinically relevant blood concentrations of 50 µM
(7,18). To the best of our knowledge, no
previous studies administered higher doses of similar drugs in
short cycles for cancer therapy; however, the present study
hypothesized that higher doses of FF could be administered in short
cycles with long intervals in combination with the intermittent
administration of standard chemotherapeutic agents. In previous
studies on different cancer cell lines, FF was found to exert an
anticancer effect by inducing apoptosis, cell cycle arrest, and
motility inhibition (5). Different
molecular mechanisms for the anticancer effect of FF had been
reported; these included the activation of AMP-activated protein
kinase, fork-head box O1, and fork-head box O3A; the inhibition of
Akt and extracellular-signal-regulated kinase; and the accumulation
of cellular ROS (5).
Apart from the impact of FF on cancer chemotherapy,
its therapeutic role as a promising antioxidant enhancer has
attracted much attention (4,40).
According to previous in vitro studies FF protected several
normal cells from oxidative damage (10–13,46).
Moreover, other studies using animal models of diabetic retinopathy
and nephropathy, and ischemia/reperfusion-induced cardiac injury
showed that FF protected these organs from damage secondary to
oxidative stress in (5,47,48).
However, the molecular mechanism of its antioxidative action
remains unknown. The current study provided mechanistic insights
into understanding the antioxidative property of FF.
In conclusion, we discovered that FF at clinically
relevant concentrations attenuated CDDP cytotoxicity to lung cancer
cells by enhancing the antioxidant defense system through
activation of a pathway that involves the
PPAR-α-PPRE-AhR-XRE-Nrf2-ARE. Although further exploration will be
needed, our study suggested that in patients receiving CDDP-based
chemotherapy for lung cancer, caution is required when FF is
concomitantly used. Although the anticancer property of FF has
recently attracted much attention, it required high FF
concentrations that exceeded clinically relevant
concentrations.
Acknowledgements
The authors would like to thank Mrs. Eriko Kurosawa
(Tokyo Medical University Ibaraki Medical Center) for providing
technical assistance.
Funding
This work was supported by a Grant-in-Aid for Scientific
Research from the Ministry of Education and Science, Japan (grant
no. 21K08189); Nippon Boelinger Ingelheim Co., Ltd., MSD K.K.;
Astellas Pharma Inc.; and Novartis Pharma K.K.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MK and KA conceived and designed the project;
acquired, analyzed and interpreted the data; and wrote the original
draft. SA and HN analyzed and interpreted the data. MK and KA
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AhR
|
aryl hydrocarbon receptor
|
|
ANOVA
|
analysis of variance
|
|
ARE
|
antioxidant response element
|
|
ARNT
|
AhR nuclear translocator
|
|
BSA
|
bovine serum albumin
|
|
FBS
|
fetal bovine serum
|
|
PBS
|
phosphate buffered saline
|
|
PPAR
|
peroxisome proliferator-activated
receptor
|
|
PPRE
|
PPAR response element
|
|
ROS
|
reactive oxygen species
|
|
XRE
|
xenobiotic response element
|
References
|
1
|
Patsouris D, Mandard S, Voshol PJ, Escher
P, Tan NS, Havekes LM, Koenig W, März W, Tafuri S, Wahli W, et al:
PPARalpha governs glycerol metabolism. J Clin Invest. 114:94–103.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Koltai T: Fenofibrate in cancer:
Mechanisms involved in anticancer activity. F1000Res. 4:552015.
View Article : Google Scholar
|
|
3
|
Abdel Magid AM, Abbassi MM, Iskander EEM,
Mohamady O and Farid SF: Randomized comparative efficacy and safety
study of intermittent simvastatin versus fenofibrate in
hemodialysis. J Comp Eff Res. 6:413–424. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Noonan JE, Jenkins AJ, Ma JX, Keech AC,
Wang JJ and Lamoureux EL: An update on the molecular actions of
fenofibrate and its clinical effects on diabetic retinopathy and
other microvascular end points in patients with diabetes. Diabetes.
62:3968–3975. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lian X, Wang G, Zhou H, Zheng Z, Fu Y and
Cai L: Anticancer roperties of fenofibrate: A repurposing use. J
Cancer. 9:1527–1537. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vlase L, Popa A, Muntean D and Leucuta SE:
Pharmacokinetics and comparative bioavailability of two fenofibrate
capsule formulations in healthy volunteers. Arzneimittelforschung.
60:560–563. 2010.PubMed/NCBI
|
|
7
|
Davies SP, Mycroft-West CJ, Pagani I, Hill
HJ, Chen YH, Karlsson R, Bagdonaite I, Guimond SE, Stamataki Z, De
Lima MA, et al: The hyperlipidaemic drug fenofibrate significantly
reduces infection by SARS-CoV-2 in cell culture models. Front
Pharmacol. 12:6604902021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luty M, Piwowarczyk K, Łabędź-Masłowska A,
Wróbel T, Szczygieł M, Catapano J, Drabik G, Ryszawy D,
Kędracka-Krok S, Madeja Z, et al: Fenofibrate augments the
sensitivity of drug-resistant prostate cancer cells to docetaxel.
Cancers (Basel). 11:772019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Balakumar P, Sambathkumar R, Mahadevan N,
Muhsinah AB, Alsayari A, Venkateswaramurthy N and Dhanaraj SA:
Molecular targets of fenofibrate in the cardiovascular-renal axis:
A unifying perspective of its pleiotropic benefits. Pharmacol Res.
144:132–141. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li J, Wang P, Chen Z, Yu S and Xu H:
Fenofibrate ameliorates oxidative stress-induced retinal
microvascular dysfunction in diabetic rats. Curr Eye Res.
43:1395–1403. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sekulic-Jablanovic M, Petkovic V, Wright
MB, Kucharava K, Huerzeler N, Levano S, Brand Y, Leitmeyer K, Glutz
A, Bausch A and Bodmer D: Effects of peroxisome proliferator
activated receptors (PPAR)-γ and -α agonists on cochlear protection
from oxidative stress. PLOS One. 12:e01885962017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hsu YJ, Lin CW, Cho SL, Yang WS, Yang CM
and Yang CH: Protective effect of fenofibrate on oxidative
stress-induced apoptosis in retinal-choroidal vascular endothelial
cells: Implication for diabetic retinopathy treatment. Antioxidants
(Basel). 9:7122020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cortes-Lopez F, Sanchez-Mendoza A,
Centurion D, Cervantes-Perez LG, Castrejon-Tellez V, Del
Valle-Mondragon L, Soria-Castro E, Ramirez V, Sanchez-Lopez A,
Pastelin-Hernandez G, et al: Fenofibrate protects cardiomyocytes
from hypoxia/reperfusion- and high glucose-induced detrimental
effects. PPAR Res. 2021:88953762021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thongnuanjan P and Soodvilai S,
Chatsudthipong V and Soodvilai S: Fenofibrate reduces
cisplatin-induced apoptosis of renal proximal tubular cells via
inhibition of JNK and p38 pathways. J Toxicol Sci. 41:339–349.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim SJ, Park C, Lee JN and Park R:
Protective roles of fenofibrate against cisplatin-induced
ototoxicity by the rescue of peroxisomal and mitochondrial
dysfunction. Toxicol Appl Pharmacol. 353:43–54. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Desager JP, Horsmans Y, Vandenplas C and
Harvengt C: Pharmacodynamic activity of lipoprotein lipase and
hepatic lipase, and pharmacokinetic parameters measured in
normolipidaemic subjects receiving ciprofibrate (100 or 200 mg/day)
or micronised fenofibrate (200 mg/day) therapy for 23 days.
Atherosclerosis. 124 (Suppl):S65–S73. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ivashkevich A, Redon CE, Nakamura AJ,
Martin RF and Martin OA: Use of the γ-H2AX assay to monitor DNA
damage and repair in translational cancer research. Cancer Lett.
327:123–133. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yip KW and Reed JC: Bcl-2 family proteins
and cancer. Oncogene. 27:6398–6406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Endo H, Yano M, Okumura Y and Kido H:
Ibuprofen enhances the anticancer activity of cisplatin in lung
cancer cells by inhibiting the heat shock protein 70. Cell Death
Dis. 5:e10272014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Berndtsson M, Hägg M, Panaretakis T,
Havelka AM, Shoshan MC and Linder S: Acute apoptosis by cisplatin
requires induction of reactive oxygen species but is not associated
with damage to nuclear DNA. Int J Cancer. 120:175–180. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kikuchi R, Iwai Y, Tsuji T, Watanabe Y,
Koyama N, Yamaguchi K, Nakamura H and Aoshiba K: Hypercapnic tumor
microenvironment confers chemoresistance to lung cancer cells by
reprogramming mitochondrial metabolism in vitro. Free Radic Biol
Med. 134:200–214. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fujii J, Homma T and Osaki T: Superoxide
radicals in the execution of cell death. Antioxidants(Basel).
11:5012022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shaw P and Chattopadhyay A: Nrf2-ARE
signaling in cellular protection: Mechanism of action and the
regulatory mechanisms. J Cell Physiol. 235:3119–3130. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee C: Collaborative power of Nrf2 and
PPARγ activators against metabolic and drug-Induced oxidative
injury. Oxid Med Cell Longev. 2017:13781752017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kobayashi A, Kang MI, Okawa H, Ohtsuji M,
Zenke Y, Chiba T, Igarashi K and Yamamoto M: Oxidative stress
sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to
regulate proteasomal degradation of Nrf2. Mol Cell Biol.
24:7130–7139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Taguchi K and Yamamoto M: The KEAP1-NRF2
system in cancer. Front Oncol. 7:852017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Probst BL, McCauley L, Trevino I, Wigley
WC and Ferguson DA: Cancer cell growth is differentially affected
by constitutive activation of NRF2 by KEAP1 deletion and
pharmacological activation of NRF2 by the synthetic triterpenoid,
RTA 405. PLoS One. 10:e01352572015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang R, An J, Ji F, Jiao H, Sun H and Zhou
D: Hypermethylation of the Keap1 gene in human lung cancer cell
lines and lung cancer tissues. Biochem Biophys Res Commun.
373:151–154. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Miao W, Hu L, Scrivens PJ and Batist G:
Transcriptional regulation of NF-E2 p45-related factor (NRF2)
expression by the aryl hydrocarbon receptor-xenobiotic response
element signaling pathway: Direct cross-talk between phase I and II
drug-metabolizing enzymes. J Biol Chem. 280:20340–20348. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Larigot L, Juricek L, Dairou J and Coumoul
X: AhR signaling pathways and regulatory functions. Biochim Open.
7:1–9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Villard PH, Caverni S, Baanannou A, Khalil
A, Martin PG, Penel C, Pineau T, Seree E and Barra Y: PPARalpha
transcriptionally induces AhR expression in Caco-2, but represses
AhR pro-inflammatory effects. Biochem Biophys Res Commun.
364:896–901. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Coelho NR, Pimpão AB, Correia MJ,
Rodrigues TC, Monteiro EC, Morello J and Pereira SA:
Pharmacological blockage of the AHR-CYP1A1 axis: A call for in vivo
evidence. J Mol Med (Berl). 100:215–243. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Terashima J, Habano W, Gamou T and Ozawa
S: Induction of CYP1 family members under low-glucose conditions
requires AhR expression and occurs through the nuclear
translocation of AhR. Drug Metab Pharmacokinet. 26:577–583. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Majeed Y, Upadhyay R, Alhousseiny S, Taha
T, Musthak A, Shaheen Y, Jameel M, Triggle CR and Ding H: Potent
and PPARα-independent anti-proliferative action of the
hypolipidemic drug fenofibrate in VEGF-dependent angiosarcomas in
vitro. Sci Rep. 9:63162013. View Article : Google Scholar
|
|
37
|
Kikuchi R, Maeda Y, Tsuji T, Yamaguchi K,
Abe S, Nakamura H and Aoshiba K: Fenofibrate inhibits TGF-β-induced
myofibroblast differentiation and activation in human lung
fibroblasts in vitro. FEBS Open Bio. 11:2340–2349. 2021.(Epub ahead
of print). View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Raghunath A, Sundarraj K, Nagarajan R,
Arfuso F, Bian J, Kumar AP, Sethi G and Perumal E: Antioxidant
response elements: Discovery, classes, regulation and potential
applications. Redox Biol. 17:297–314. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Park JS, Kang DH, Lee DH and Bae SH:
Fenofibrate activates Nrf2 through p62-dependent Keap1 degradation.
Biochem Biophys Res Commun. 465:542–547. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim T and Yang Q:
Peroxisome-proliferator-activated receptors regulate redox
signaling in the cardiovascular system. World J Cardiol. 5:164–174.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim YH, Yoo HY, Chang MS, Jung G and Rho
HM: C/EBP alpha is a major activator for the transcription of rat
Cu/Zn superoxide dismutase gene in liver cell. FEBS Lett.
401:267–270. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ding G, Fu M, Qin Q, Lewis W, Kim HW,
Fukai T, Bacanamwo M, Chen YE, Schneider MD, Mangelsdorf DJ, et al:
Cardiac peroxisome proliferator-activated receptor gamma is
essential in protecting cardiomyocytes from oxidative damage.
Cardiovasc Res. 76:269–279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Girnun GD, Domann FE, Moore SA and Robbins
ME: Identification of a functional peroxisome
proliferator-activated receptor response element in the rat
catalase promoter. Mol Endocrinol. 16:2793–2801. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shin S, Wakabayashi N, Misra V, Biswal S,
Lee GH, Agoston ES, Yamamoto M and Kensler TW: NRF2 modulates aryl
hydrocarbon receptor signaling: Influence on adipogenesis. Mol Cell
Biol. 27:7188–7197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ma Q: Role of nrf2 in oxidative stress and
toxicity. Annu Rev Pharmacol Toxicol. 53:401–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sun C, Song B, Sheng W, Yu D, Yang T, Geng
F, Fang K, Jiao Y, Zhang J and Zhang S: Fenofibrate attenuates
radiation-induced oxidative damage to the skin through fatty acid
binding protein 4 (FABP4). Front Biosci (Landmark Ed). 27:2142022.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kadian S, Mahadevan N and Balakumar P:
Differential effects of low-dose fenofibrate treatment in diabetic
rats with early onset nephropathy and established nephropathy. Eur
J Pharmacol. 698:388–396. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ibarra-Lara L, Sánchez-Aguilar M,
Sánchez-Mendoza A, Del Valle-Mondragón L, Soria-Castro E,
Carreón-Torres E, Díaz-Díaz E, Vázquez-Meza H, Guarner-Lans V and
Rubio-Ruiz ME: Fenofibrate therapy restores antioxidant protection
and improves myocardial insulin resistance in a rat model of
metabolic syndrome and myocardial ischemia: The role of angiotensin
II. Molecules. 22:312016. View Article : Google Scholar : PubMed/NCBI
|