Introduction
Osteosarcoma is the most common primary bone tumor
in childhood and adolescence. Recent progresses in treatment
consisting of surgery and adjuvant chemotherapy have improved
prognosis. However, still ~30% of patients do not have long-term
survival, mainly due to uncontrollable metastasis (1–4). Thus,
novel and safe solutions for overcoming therapeutic resistance must
urgently be developed.
Previously, we established a mouse osteosarcoma
model using bone marrow stromal cells derived from Ink4a/Arf
null mice by overexpression of c-MYC (5). When these osteosarcoma cells,
designated AXT cells, were inoculated into C57BL/6 syngeneic mice,
lethal tumors with metastatic lesions rapidly develop that mimic
the pathophysiology of human osteoblastic osteosarcoma (6–9). Since
osteosarcoma develops from mesenchymal origin, agents that suppress
the function of mesenchymal cells such as fibroblasts might also
exert anti-osteosarcoma in vivo.
Nintedanib (formerly known as BIBF 1120) is an oral
small-molecule anti-fibrotic drug that is a triple angiokinase
inhibitor; the drug inhibits fibroblast growth factor (FGF),
vascular endothelial growth factor (VEGF), and platelet-derived
growth factor (PDGF) signaling pathways (10–12).
Nintedanib is FDA-approved for treating idiopathic pulmonary
fibrosis (IPF) and advanced lung adenocarcinoma (12–15).
Preclinical in vitro and in vivo studies have shown
that nintedanib exerts an anti-tumor effect in various types of
cancers including non-small cell lung cancer, renal cell carcinoma,
cholangiocarcinoma, colorectal cancer, ovarian cancer, prostate
cancer, and pancreatic ductal adenocarcinoma (10,16–18).
Importantly, in addition to directly targeting malignant tumor
cells, nintedanib exerts an anti-tumor effect by increasing the
sensitivity of the tumor to other anti-tumor drugs and affecting
the tumor microenvironment (16,18–20).
The influence of nintedanib on the microenvironment suppresses the
function of cancer-associated fibroblasts (CAFs) that critically
support cancer progression (21–23).
Therefore, in vitro studies alone are not sufficient to
evaluate the anti-tumor effect of nintedanib and in vivo
studies are needed. Clinically, in addition to benefits in
non-small cell lung cancer, nintedanib has efficacy in ovarian
cancer and mesothelioma either as adjuvant therapy after
chemotherapy or in combination with chemotherapy (24,25).
A previous study showed that nintedanib suppresses
lung metastasis of osteosarcoma cells by inhibiting fibrotic
remodeling of osteosarcoma stem cells, suggesting that nintedanib
is useful for treating osteosarcoma metastasis (26). However, the anti-tumor effect of
nintedanib in osteosarcoma has not been fully elucidated. In this
study, the clinical potential of nintedanib for treating
osteosarcoma was examined in vitro and in a mouse model that
mimics human disease, and the molecular mechanisms of nintedanib
activity were examined.
Materials and methods
Cell culture
Mouse osteosarcoma AXT cells, which were previously
established from Ink4a/Arf-null mice by overexpression of
human c-MYC (5), or human
osteosarcoma cells (U2OS (#HTB-96), MG63 (#CRL-1427), Saos2
(#HTB-85) purchased from ATCC (Manassas, VA, USA) were cultured in
IMDM (Nacalai Tesque, Kyoto, Japan) or McCoy's 5A medium
(Thermo-Fischer Scientific, Carlsbad, CA, USA), respectively,
containing 10% FBS under 5% CO2 at 37°C (27,28).
Mouse bone marrow stromal cells (wild-type or Ink4a/Arf null BMSCs)
were established as previously described (5). Briefly, bone marrow cells from
C57BL/6J or Ink4a/Arf null mice were collected from femurs and
tibias and hematopoietic cells were depleted with antibodies to
CD45 and lineage-specific antigens. Adherent cells were cultured in
IMDM supplemented with 20% FBS and used as BMSCs within ten
passages.
Reagents
Nintedanib (#HY-50904 MedChemExpress, Monmouth
Junction, NJ, USA) was dissolved in dimethyl sulfoxide (DMSO) at a
stock concentration of 20 mM.
Cell viability evaluation
AXT cells or human osteosarcoma MG63, Saos2, and
U2OS cells (5×102 in 50 µl of IMDM for AXT cells or
McCoy's 5A medium for MG63, Saos2, and U2OS cells, containing 10%
FBS) were cultured in a 96-well cell culture plate. Then, 50 µl of
the corresponding medium containing test reagents at twice the
desired concentrations was added to each well. Cell viability was
evaluated using a Cell Titer Glo assay kit (Promega, Madison, WI,
USA). Assays were performed at least in triplicate and data are
shown as the means ± SD relative (fold) against the corresponding
control value for cells incubated in the absence of test
reagents.
Endothelial cell tube formation
assay
A 96-well cell culture plate was coated with 30 µl
of Matrigel (Corning, Corning, NY, USA) and Human Umbilical Vein
Endothelial Cells (HUVEC (#KE-4109) purchased from CRABO, Osaka,
Japan) cultured with Endothelial Cell Growth Medium (Takara, Shiga,
Japan) were seeded after passing through the 40 mm-cell strainer
(2×104 in 45 µl of the culture medium per each well).
Then, 25 µl of the culture medium with or without 400 nM or 4 µM of
nintedanib was added and the final total volume was 100 µl. Tube
formation was evaluated after 14 h.
Mouse care
All mouse experiments were performed in accordance
with the guidelines of Hoshi University, and the present study was
approved by the Committee on Animal Research of Hoshi University
(approval number: 20-071). Mice were housed in ventilated cages
(five mice/cage; floor area 501 cm2) under specific
pathogen-free conditions. Mice were fed a standard chow diet and
water ad libitum. Rooms were maintained at 22°C and kept on
a 12 h light and dark cycle with inspection every weekday to ensure
that they were not under distress throughout the experiments.
Tumor xenograft model
The detailed duration of the experiments and the
number of animals used was shown in main figures. Briefly,
1–2×106 AXT cells were suspended in 100 µl of IMDM and
injected subcutaneously into the bilateral flanks (two injections
in total) of 7-week-old female C57BL/6J mice (SLC, Shizuoka, Japan)
or 20-week-old female C57BL/6 SCID mice (The Jackson Laboratory,
Bar Harbor, ME, USA) under inhalation anesthesia with 4% isoflurane
(Wako, Tokyo, Japan) for the induction and 2% for maintenance.
Nintedanib at 50 mg/kg was orally administered once
a day. The endpoint criteria were as follows: i) The mean tumor
diameter exceeds 20 mm. ii) The combined tumor burden is more than
15% of body weight (~20 g of 7-week-old mice). iii) Occurrence of
ulceration, infection, or necrosis of tumor. iv) Body weight loss
is more than 20% of weight. The results did not reach the endpoint
criteria in this study. Bilateral two tumors developed in each
mouse in this study. The major and minor axes of the bilateral
tumors were measured, and the estimated tumor volume was calculated
according to the guideline of Washington State University
(https://iacuc.wsu.edu/documents/2017/12/tumor-burden-guidelines.pdf/).
Estimated tumor volume
(mm3)=d2 × D/2. D and d were the major and
minor diameter in mm, respectively. The maximum diameter and the
volume of bilateral tumors observed in each mouse was listed in
Table SI. When the mice were
euthanized, a lethal dose (200 mg/kg) of pentobarbital sodium
(Tokyo Kasei Kogyo, Tokyo, Japan) was intraperitoneally injected.
Death was confirmed by cardiac arrest. None of the mice was
unexpectedly dead or found dead during the study.
Immunoblot analysis
2X Laemmli sample buffer (Bio-Rad, Hercules, CA,
USA) supplemented with β-mercaptoethanol was used for the
preparation of cell lysate. Immunoblot analyses were performed
according to standard semi-dry transfer methods using 5–20%
gradient precast polyacrylamide gels (ePAGEL, ATTO, Tokyo, Japan).
The primary antibodies were listed in Table SII. Signal intensities were
quantitated using the ImageJ software (29), and the relative (fold) values
against the corresponding control value normalized against the
intensities of the α-Tubulin bands are shown.
Phospho-kinase array
Activated tyrosine kinase in vivo was
screened using the mouse RTK phosphorylation antibody array C1 kit
(RayBiotech, Norcross, GA, USA). The array map is shown in Table SIII. AXT cells were inoculated into
C57BL/6J mice and nintedanib was administrated. The tumor lysate
from two tumors of two mice was extracted by disruption using a
BioMasher homogenizer (Nippi, Tokyo, Japan). The total cell lysate
used was 500 µg.
Cell-cycle analysis
Trypsinized cells were washed with PBS, fixed with
70% ethanol for ≥48 h at −20°C, washed twice with ice-cold PBS, and
stained with PBS supplemented with 10 µg/ml propidium iodide and 20
µg/ml RNase. The DNA content of 20,000 singlet cells was measured
by FACSVerse (BD Biosciences, San Jose, CA, USA).
Evaluation of apoptosis by flow
cytometry
Trypsinized cells were washed with ice-cold PBS, and
stained with allophycocyanin-conjugated annexin V (eBioscience,
Carlsbad, CA, USA) and propidium iodide. Stained cells were
analyzed by FACSVerse. FlowJo software (Tree Star, Ashland, OR,
USA) was used for the data management.
Reverse transcription (RT) and
quantitative (q)PCR analysis
Total RNA was extracted and RT and qPCR analyses
were performed using the NucleoSpin RNA kit and PrimeScript
(Takara). To evaluate circulating tumor cells (CTCs), whole blood
was collected from the right ventricle of euthanized mice and total
RNA was extracted from 200 µl blood with a NucleoSpin RNA blood kit
(Takara, Shiga, Japan). Since GFP is constitutively expressed in
AXT cells, CTCs were quantitated based on the expression level of
Gfp relative to Actb mRNA. The sequences of PCR
primers are as follows: GFP forward:
5′-GACGTAAACGGCCACAAGTT-3′, reverse: 5′-TTGCCGGTGGTGCAGATGAA-3′,
Ccnd1 forward: 5′-CAACAACTTCCTCTCCTGCT-3′, reverse:
5′-ACTCCAGAAGGGCTTCAATC-3′, Actb forward:
5′-CAACCGTGAAAAGATGACCC-3′, reverse: 5′-TACGACCAGAGGCATACAG-3′.
qPCR analysis was performed with StepOne thermal cycler (Thermo
Fisher Scientific) with the 2-step protocol; 60 sec at 95°C, then
40 cycles of 15 sec at 95°C and 60 sec at 60°C followed by the
melting and dissociation curve analysis.
Immunohistochemistry (IHC)
Immunohistochemical analysis was performed according
to standard procedures (5,6,8).
Deparaffinized sections were stained with primary antibodies listed
in Table SII. Hematoxylin was used
for nuclear staining.
Immunofluorescence
Deparaffinized tumor sections were stained with
primary antibodies to aSMA (Abcam) derived from rabbit and to CD31
(eBiosience) derived from rat. Alexa Fluor 555-conjugated
anti-rabbit and Alexa Fluor 647-conjugated anti-rat secondary
antibody (both from Abcam) were used. DAPI solution (Dojindo,
Kumamoto, Japan) was used for nuclear staining. Samples were
observed with FV3000 confocal microscopy (Olympus, Tokyo,
Japan).
Statistical analysis
Unless indicated otherwise, quantitative data are
expressed as the means ± SD relative to the control value. All
assays were performed at least in triplicate. Data were analyzed
with a Student's t-test or one-way analysis of variance (ANOVA)
with the Dunnet post hoc test, using Graph Pad Prism 9 (GraphPad
Software, San Diego, CA, USA). P<0.05 was considered to indicate
a statistically significant difference.
Results
Nintedanib suppresses osteosarcoma
cell growth
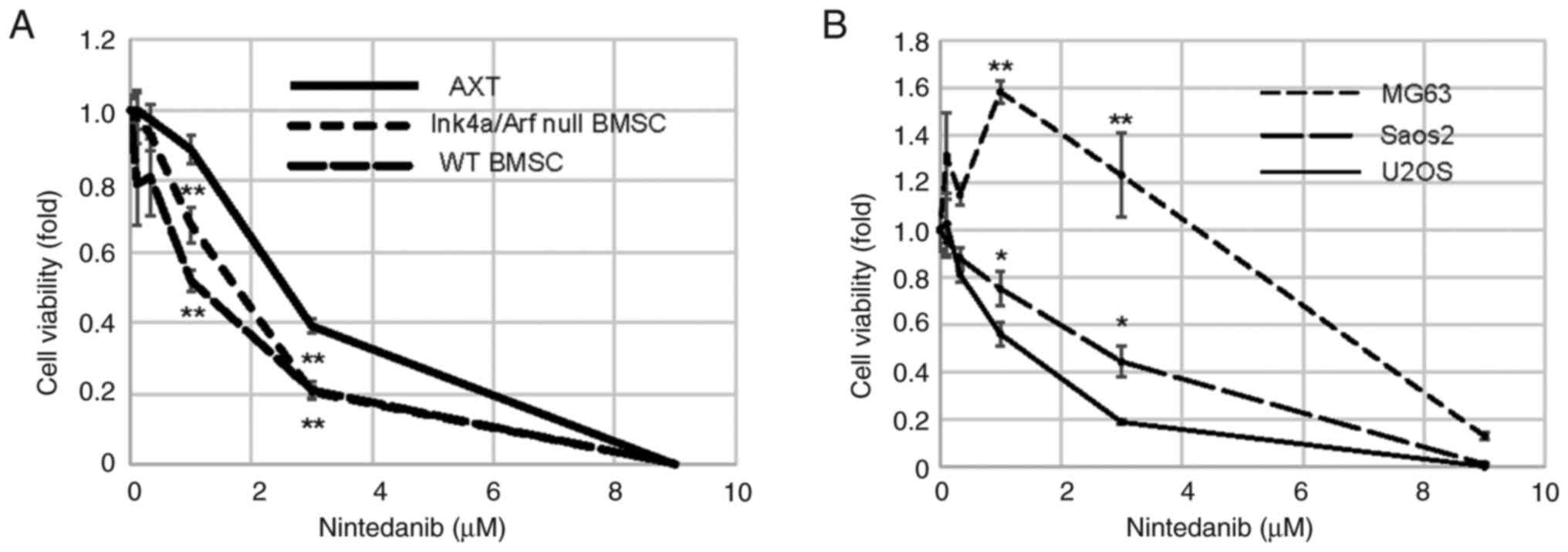
First, the anti-proliferative effect of nintedanib
on osteosarcoma cells in vitro was evaluated. Nintedanib
decreased the viability of mouse osteosarcoma AXT cells in a
concentration-dependent manner (Fig.
1A). To examine the effect of nintedanib on the non-tumor
counterparts of AXT cells, Ink4a/Arf null BMSC and wild-type BMSC
derived from the C57BL/6 background were used (5). The growth of both cell lines was also
inhibited by nintedanib in a concentration-dependent manner and
these cell lines were more sensitive to nintedanib than AXT cells
(Fig. 1A). Next, the
anti-proliferative effect of nintedanib on human osteosarcoma cells
was evaluated. Similar to findings with AXT cells, nintedanib had a
direct anti-proliferative effect on human U2OS and Saos2
osteosarcoma cells, but the effect of the drug on MG63 cells was
less pronounced (Fig. 1B) and
concentrations of nintedanib ≤3 µM did not suppress growth. These
results indicate that the effect of nintedanib in osteosarcoma is
dependent on cell type in vitro.
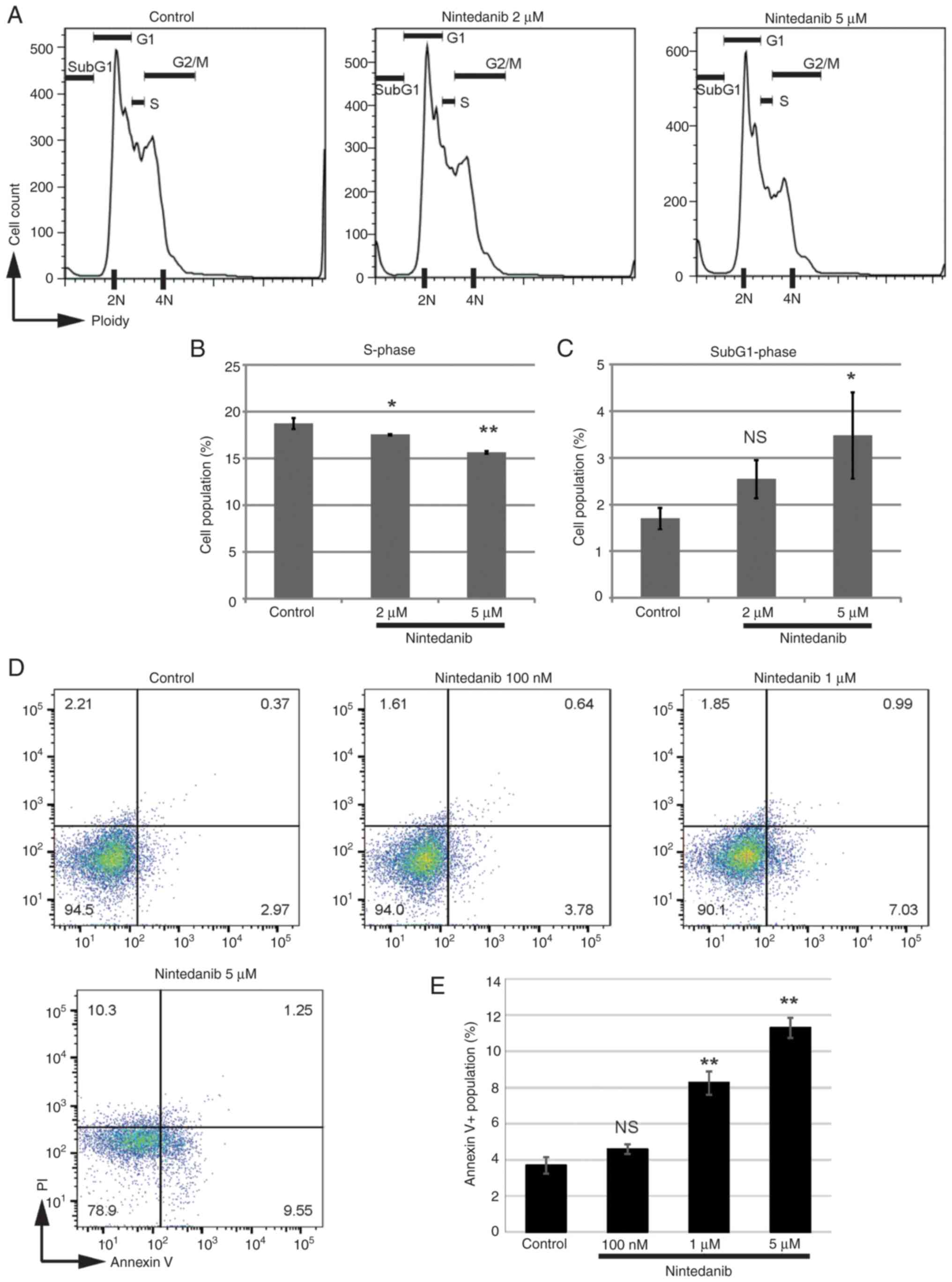
Nintedanib suppresses cell-cycle
progression and survival signals in osteosarcoma cells
To elucidate the mechanisms underlying the
suppression of osteosarcoma cell growth by nintedanib, the
cell-cycle status was examined. A 20 h exposure of AXT cells to
nintedanib decreased the fraction in S-phase (Fig. 2A and B). Treatment with nintedanib
increased the sub-G1 fraction (Fig.
2C), indicating the emergence of apoptotic cells. To further
evaluate the induction of apoptosis, AXT cells were treated with
nintedanib for 23 h. Although the level of annexin V-positive
apoptotic cells was not high, nintedanib concentration-dependently
increased the percentage of this cell type (Fig. 2D and E). Together, these findings
suggest that nintedanib inhibited cell-cycle progression and
increased apoptotic cells in AXT cells.
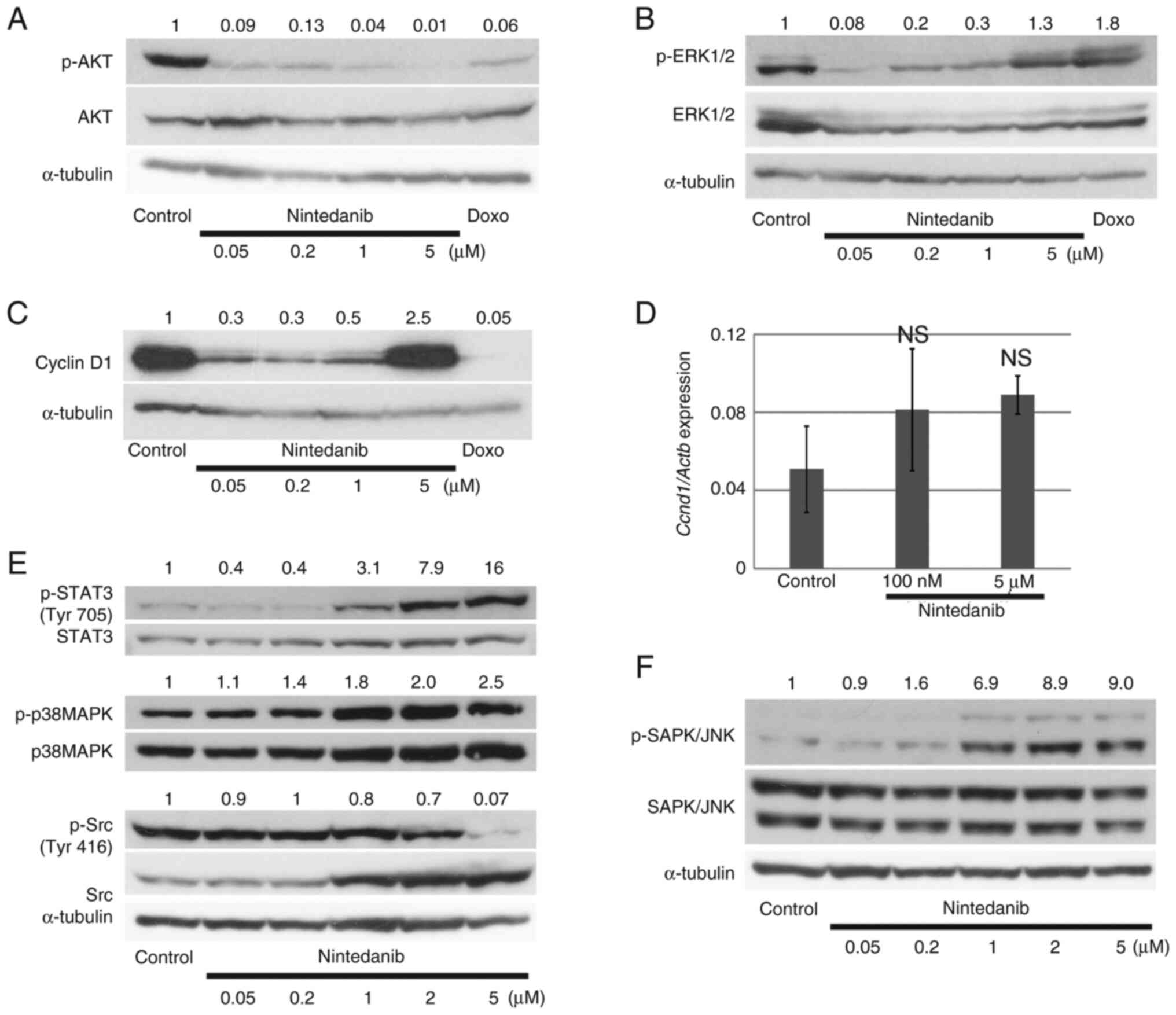
To clarify the molecular events induced by
nintedanib treatment, we evaluated activated kinases by
immunoblotting. Phosphorylation of AKT was decreased by 21.5 h
treatment with nintedanib or doxorubicin as a positive control
(Fig. 3A). The phosphorylation
level of ERK1/2 was also attenuated by nintedanib treatment.
However, this suppression was not concentration-dependent and
ERK1/2 activation was restored by 5 µM nintedanib treatment
(Fig. 3B). The expression level of
cyclin D1 was downregulated by nintedanib treatment. Notably,
similar to the ERK1/2 phosphorylation result, 5 µM nintedanib
restored cyclin D1 protein levels to control values (Fig. 3C). Cells treated with 5 µM
nintedanib had higher gene expression levels of cyclin D1 than
controls, but the level was not statistically significant (Fig. 3D). Thus, the high expression level
of cyclin D1 induced by 5 µM nintedanib could be mainly
attributable to the accumulation of cyclin D1 protein.
We further examined the activation of signaling
molecules downstream of growth factor or cytokine receptors such as
IL-6 and CXCL8 which are suggested to be important to promote
osteosarcoma metastasis (30–32).
Phosphorylation level of STAT3 was not high under the culture with
10% FBS containing medium and tended to be suppressed only at low
concentration of nintedanib like p-ERK (Fig. 3E). p38MAPK and SAPK/JNK were
activated by the supplement of nintedanib (Fig. 3E and F). These molecules might not
be activated as downstream of cytokine or growth factor receptors.
On the other hand, Src was inactivated by the supplement of high
concentration of nintedanib, consistent with a previous report that
nintedanib suppresses many kinases related to Src activation at
high concentrations (10).
Collectively, these results showed that nintedanib
altered the expression levels of intracellular signaling molecules,
but the effect of nintedanib on these molecules was not necessarily
concentration-dependent.
Anti-osteosarcoma effect of nintedanib
in vivo
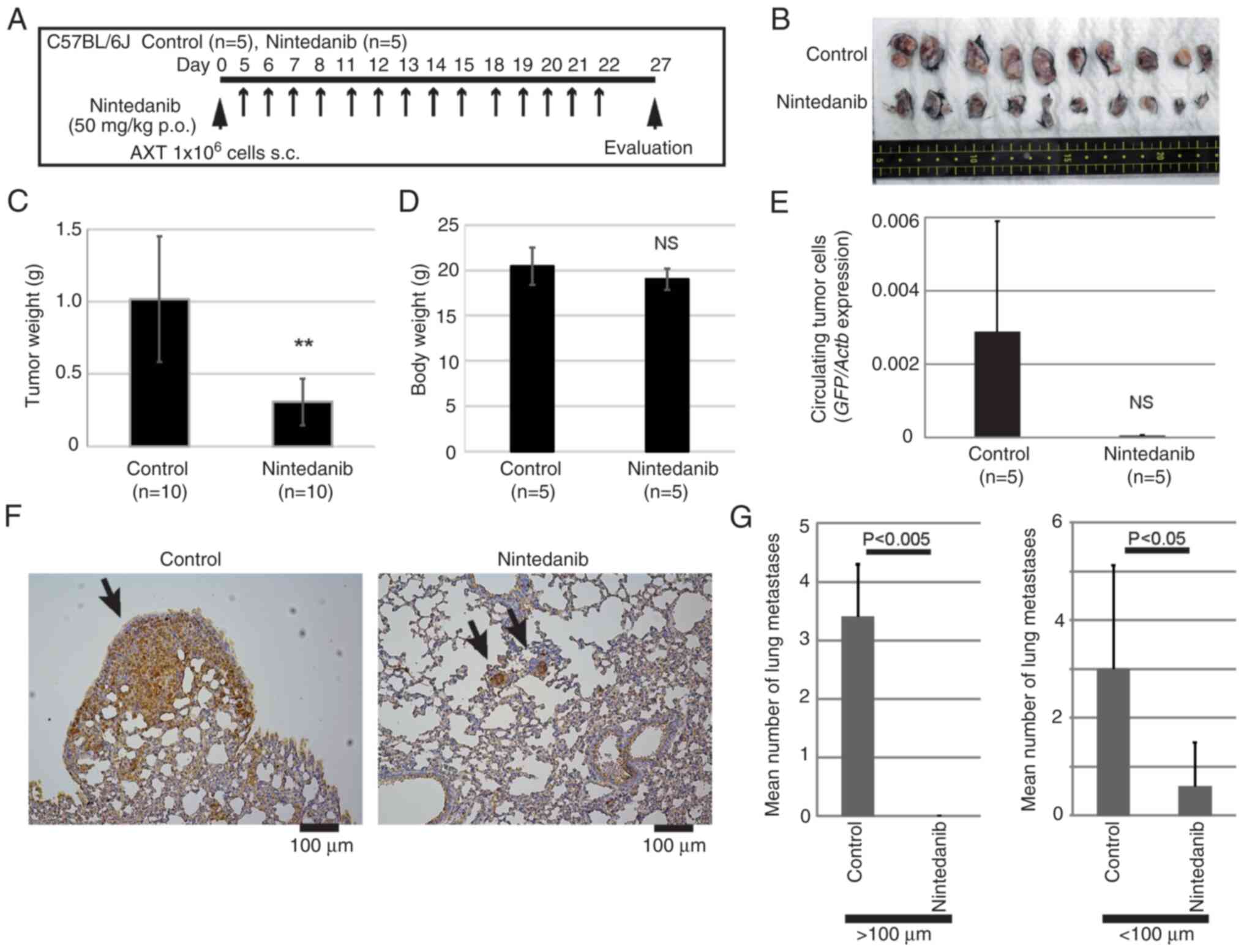
We examined whether nintedanib might exert
anti-tumor activity against AXT cells in vivo. AXT cells
were inoculated into syngeneic C57BL/6 mice and then the mice were
treated with a single daily dose of nintedanib at 50 mg/kg
(Fig. 4A). Previous toxicity tests
using mice reported that no major adverse events occurred when
daily 100 mg/kg nintedanib was orally administered for 14 days, and
the approximate lethal single dose is over 2,000 mg/kg. The in
vivo studies using mouse IPF models demonstrated that a daily
dose of 24.9–83 mg/kg nintedanib in mice was efficacious and
tolerated when administered for 10 to 30 days (URL: [Ofev,
INN-nintedanib (europa.eu)],
[https://www.ema.europa.eu/en/documents/assessment-report/ofev-epar-public-assessment-report_en.pdf]).
Therefore, we set 50 mg/kg of nintedanib to evaluate the
anti-tumorigenic effect.
Administration of nintedanib significantly decreased
the primary tumor size and weight (Fig.
4B and C). Treatment with nintedanib did not affect the mouse
body weight (Fig. 4D) and all mice
were alive, well, and not exhausted during the treatment. Since AXT
cells were labeled with GFP, circulating tumor cells could be
quantitated (27,28). Treatment with nintedanib decreased
the number of circulating tumor cells, although the effect was not
statistically significant (Fig.
4E). Consistent with those findings, nintedanib also
significantly reduced the levels of lung metastatic lesions
(Fig. 4F and G). Notably, large
metastatic lesions over 100 µm diameter were not detected in
nintedanib-treated mice.
Therefore, these findings show that nintedanib as a
single agent exerted an anti-tumor effect on primary lesions and
metastatic growth of osteosarcoma.
Nintedanib attenuates PDGFR activation
in vivo
To elucidate the molecular mechanisms underlying the
anti-osteosarcoma effect in vivo, tumor lysate was prepared
from tumors developed in mice inoculated with AXT cells as shown in
Fig. 5A, and then the
phosphorylation levels of receptor tyrosine kinases per unit of
protein were compared. Phosphorylation was still detected in 31 of
71 kinases in the lysate from nintedanib-treated mice (Fig. 5B). FGFR, PDGFR, and VEGFR are the
molecular targets for nintedanib (10–12),
but phosphorylation levels of FGFR1 and FGFR2 were very weak and
not affected by nintedanib treatment. Activation of PDGFRα/β,
VEGFR2, and VEGFR3 could not be detected using this phosphorylation
array system.
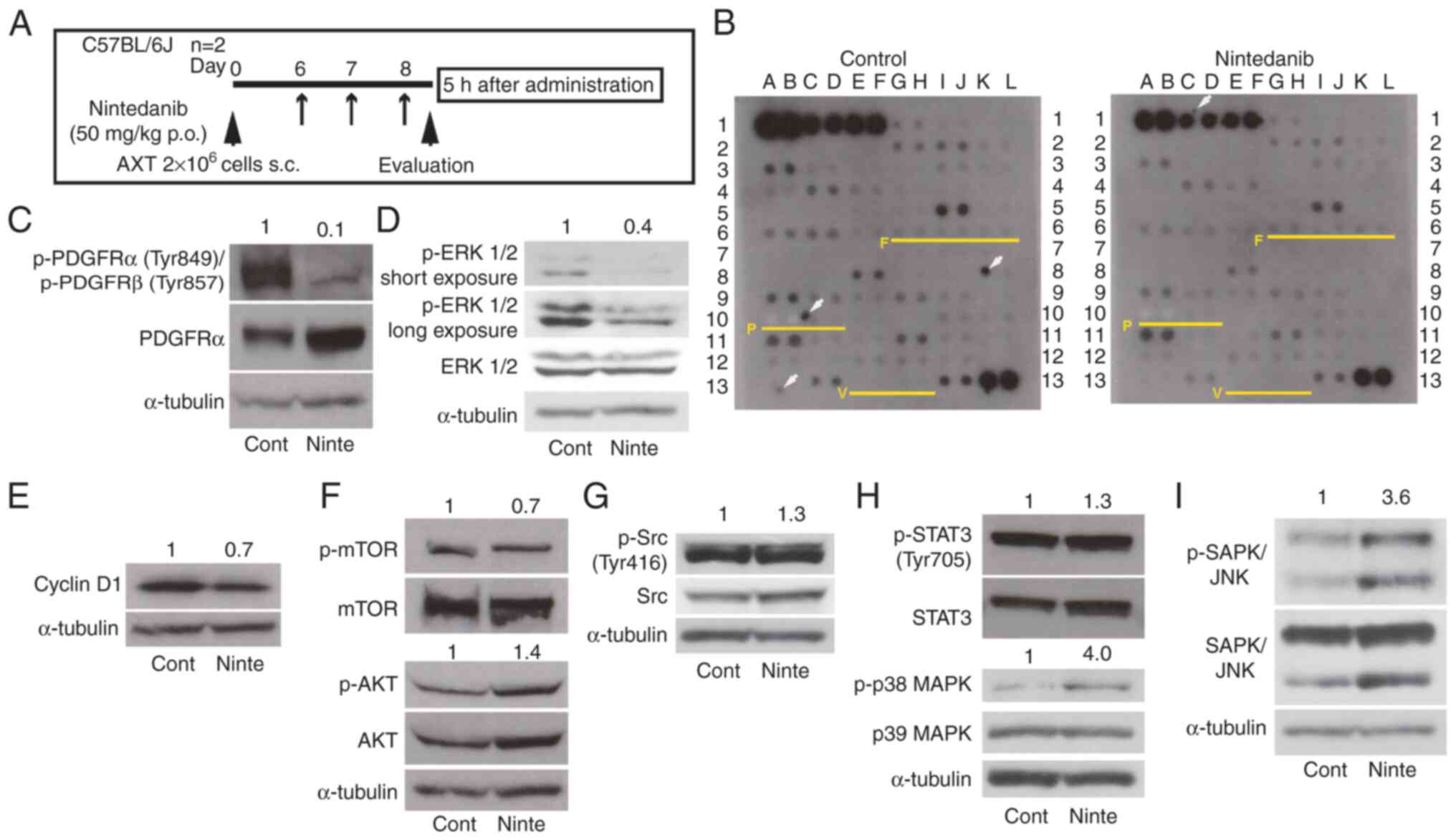 | Figure 5.Kinase activation in AXT cells with
treatment of nintedanib in vivo. (A) A schedule of cell
transplantation and nintedanib administration. (B) Tumor lysates
were prepared from mice treated with or without nintedanib. Protein
(500 µg per sample) was analyzed on a mouse phospho-receptor
tyrosine kinase array. Each antibody was spotted in duplicate. The
array map is shown in Table SIII.
F: FGFRs, P: PDGFRs, V: VEGFR2/3. White arrows: nonspecific spots.
Immunoblot analysis of (C) p-PDGFRs and PDGFRa, (D) p-ERK1/2 and
ERK1/2, (E) CyclinD1, (F) p-mTOR, mTOR, p-AKT and AKT, (G) p-Src
and Src, (H) p-STAT3, STAT3, p-p38 MAPK and p38 MAPK, and (I) p-JNK
and JNK using the same tumor lysate analyzed in (B). The relative
(fold) values of the phosphorylated forms of indicated molecules
and Cyclin D1 against the corresponding control value normalized
against the intensities of the α-Tubulin bands are shown. p-,
phosphorylated. |
To further evaluate the activation levels of PDGFRs
and VEGFR2, the same lysate was subjected to immunoblotting. The
activation of PDGFRα/β was attenuated by nintedanib administration
(Fig. 5C), suggesting that
nintedanib inhibited its molecular targets in vivo.
Phosphorylation of Tyr951 or Tyr996 of VEGFR2 was not detected
(data not shown). We examined the activation status of downstream
molecules. Consistent with our in vitro results at low
concentrations, nintedanib decreased ERK1/2 phosphorylation levels
(Figs. 3B and 5D). Cyclin D1 was slightly downregulated
(Fig. 5E). However, phosphorylation
levels of mTOR and AKT were not affected by nintedanib (Fig. 5F).
We evaluated the activation of PDGFR in AXT cells
in vitro and compared to the result from the in vivo
samples (Fig. S1A). Our previous
studies using immunoprecipitation western blotting showed that
PDGFR activation was difficult to detect under the culture with FBS
containing medium without supplement of PDGF ligands (7). In addition, under the presence of
serum in vitro, survival of AXT cells was not dependent on
the PDGFR signaling. Consistent with our previous findings, the
activation of PDGFR in vitro was considerably weaker than
that in vivo. Interestingly, the expression level of PDGFRα
increased with nintedanib treatment, but the associated receptor
activation was not observed (Fig.
S1B). In in vivo tumor samples, phosphorylation status
of Src and STAT3 was not attenuated by nintedanib administration
(Fig. 5G and H). On the other hand,
like in vitro results, p38MAPK and SAPK/JNK were activated
by nintedanib administration (Figs. 3E,
F, 5H and I). These results
suggest that nintedanib does not block STAT3 signaling under 50
mg/kg administration, and p38MAPK and SAPK/JNK might not be
activated as downstream of cytokine or growth factor receptors.
Nintedanib suppresses tumor
vasculature formation but does not inhibit the enrichment of
αSMA-positive cells to osteosarcoma
To clarify nintedanib-mediated histological changes
and the mechanisms underlying anti-osteosarcoma effect in
vivo, immunohistochemical analyses were performed.
Previous reports suggest that nintedanib exerts
anti-tumor activity by inhibiting the functions of CAFs or
fibrogenic reprogramming that confers to metastatic ability to
osteosarcoma cells (21,22,26).
α-smooth muscle actin (αSMA) is a useful marker for smooth muscle
cells and a part of fibroblasts including myofibroblasts or CAFs
(33,34). In osteosarcoma developed according
to Fig. 4A, αSMA positive cells
consisted of a variety of cell types; a part of osteosarcoma cells,
fibroblastic cells surrounding osteosarcoma (possibly including
CAFs), CD31-negative cells localized around blood vessels, and
CD31/αSMA double-positive cells (Figs.
6A and S2). However, these
cells were also abundant in the tumors of nintedanib-treated mice,
indicating that nintedanib treatment did not result in quantitative
changes in αSMA-positive cells.
A previous report suggested that nintedanib promoted
anti-tumor immunity and potentiated the effects of immune
checkpoint inhibitors (23).
However, tumor-infiltrating CD8-positive T cells were not numerous
in either control or nintedanib-treated mice (Fig. 6B). In addition, nintedanib exhibited
anti-osteosarcoma activity in C57BL/6 SCID mice, in which T- and
B-cell function is obsolete (Fig. 6C
and D).
Next, we evaluated the effect of nintedanib on
vasculature formation in osteosarcoma. Immunohistochemical staining
revealed that nintedanib reduced the formation of CD31-positive
ducts, despite the variable abundance of tumor vasculatures
(Fig. 7A and B). To evaluate the
effect of nintedanib on vascular formations more directly,
endothelial cell tube formation assay was performed. HUVEC formed
tubes in 14 h in Matrigel and they became thinner and interrupted
by the supplement of nintedanib compared to the control (Fig. 7C and D). Nintedanib treatment also
attenuated the viability of HUVEC (Fig.
7E).
These findings indicated that nintedanib exerted
anti-tumor activity in our osteosarcoma model mainly by inhibiting
tumor vascular formation.
Discussion
In this study, we examined the anti-osteosarcoma
effect of nintedanib in vitro and in vivo. Notably,
BMSCs were more sensitive to nintedanib treatment than osteosarcoma
AXT cells (Fig. 1A). Previous
studies indicate that nintedanib exhibits therapeutic effects in
idiopathic pulmonary fibrosis, which is an approved clinical
indication of nintedanib, by inhibiting growth factor-induced cell
proliferation (35,36). BMSCs might show higher sensitivity
to nintedanib via the same growth factor-dependent mechanisms. On
the other hand, AXT cells, whose viability is driven by oncogenes,
might be less dependent on growth factors than BMSCs. Human
osteosarcoma cell lines had different susceptibilities to
nintedanib, and MG63 cells were less sensitive than the other cell
types (Fig. 1B). Several studies
have suggested that cell lines of the same cancer type have
different susceptibilities to nintedanib (16–18).
In AXT cells, nintedanib treatment decreased the
S-phase population and induced apoptosis in a
concentration-dependent manner (Fig.
2). However, the concentrations of nintedanib needed to produce
this effect were much higher than the IC50 value of nintedanib for
inhibiting FGFR, PDGFR, and VEGFR, which is less than 150 nM
(10). The suppression of signal
activation other than its initial targets might be involved in
decreasing the S-phase population and inducing apoptosis in
vitro.
The phosphorylation of AKT was suppressed by
nintedanib, while a high concentration re-activated ERK1/2
(Fig. 3B). This paradoxical
phenomenon was also observed in human bladder cancer cell lines;
the authors of that study suggested that high-concentration
nintedanib activates signals that are independent of those that
control cancer cell sensitivity and viability (19). High-concentration nintedanib in
vitro might activate a complex cross-talk of PI3K, MAPK, and
other signaling pathways.
Importantly, single-agent nintedanib suppressed the
formation of both primary and metastatic lesions of osteosarcoma
(Fig. 4). Since PDGFR activation
was inhibited in vivo and p-ERK was also attenuated, these
findings show that nintedanib inhibited its target molecules in
vivo. In in vitro experiments, nintedanib only inhibited
cell growth by 10%, even when AXT cells were continuously exposed
to 1 µM of nintedanib for 3 days (Fig.
1A). On the other hand, in vivo nintedanib
administration reduced tumor size by more than half compared with
the control (Figs. 4C and 6D). These findings suggest that nintedanib
exerts a stronger anti-tumor effect in vivo. Notably, the
nintedanib-mediated inhibition of p-AKT seen in vitro was
not observed in vivo (Fig.
5F). Unlike the in vitro environment, the tumor
microenvironment consists of complex structures and various cells
and factors related to tumor cells (37,38).
The continuous activation of AKT that was observed even in the
presence of nintedanib is likely due to activation signals from
upstream molecules that are not the target molecules of nintedanib
(Fig. 5B).
Histological analysis showed that nintedanib
significantly suppressed osteosarcoma angiogenesis (Fig. 7A and B). Immunoblot analysis using
tumor lysate could not detect the activation of VEGFR, although
VEGF is a critical factor for angiogenesis. It is plausible that
nintedanib also might block VEGFR activation, which contributes to
angiogenesis at levels that were undetectable in this study.
Nintedanib administration did not reduce
αSMA-positive cells in osteosarcoma (Figs. 6A and S2). Notably, many tyrosine kinases were
still phosphorylated in tumors of nintedanib-treated mice (Fig. 5B). The factors involved in the
activation of these kinases might function to maintain the
enrichment of CAFs. For instance, EphA3 was shown to be important
for the recruitment of CAFs in a mouse lung cancer model (39). The constitutive activation of
JAK1was suggested to confer sustained proinvasive ability to CAFs
(40).
A previous study showed that nintedanib blocked
FGFR-mediated fibrogenic reprogramming of osteosarcoma cells that
conferred metastatic ability to primary tumor cells (26). This study clearly demonstrated that
originally Fibronectin (FN)/αSMA-negative Well5 cells in the
primary bone lesion were changed to FN/αSMA-positive cells during
metastatic process through the activation of FGFR signaling. Our
previous study also suggests that FGF2 released from the tumor
environment suppresses osteogenesis and promotes proliferation and
migration of osteosarcoma cells (9). However, regarding the fibrogenic
properties, unlike Well5 cells, the primary lesions already
contained a lot of αSMA-positive osteosarcoma cells (Fig. 6A), suggesting that AXT osteosarcoma
cells in primary lesions originally possess highly malignant
properties without the need for FGFR signaling.
Collectively, the results of this study showed that
nintedanib has a direct anti-tumor effect on osteosarcoma at
concentrations higher than those to block its initial targets in
vitro, and more importantly, suppresses osteosarcoma
progression in vivo. Nintedanib possibly exerts
anti-osteosarcoma effect by modulating the tumor environment rather
than directly targeting tumor cells. Therefore, nintedanib could be
an effective option for refractory osteosarcoma.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Dr Ikuyo Ishimatsu
(Keio University) for their technical assistance; and Ms. Honami
Ichikawa, Ms. Sae Ichikawa and Ms. Riko Karakama (undergraduate
students; Hoshi University) for their experimental assistance.
Funding
This work was supported by KAKENHI grant from the Japan Society
for the Promotion of Science (JSPS) (grant no. 21K07134). This work
was technically supported by the KAKENHI project JP16H06276
(AdAMS).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
TS, AS and AM confirm the authenticity of all the
raw data. TS contributed to conceptualization, design, acquisition
of data, data curation, formal analysis, funding acquisition,
investigation, methodology, project administration, validation,
visualization and original draft preparation. AS contributed to
data curation, statistical analysis, analysis and interpretation of
data, and reviewing and editing. YF contributed to acquisition of
data. AM contributed to analysis and interpretation of data,
supervision and administrative support. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal care and procedures were performed in
accordance with the guidelines of Hoshi University, and the present
study was approved by the Committee on Animal Research of Hoshi
University (approval number: 20-071).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ritter J and Bielack SS: Osteosarcoma. Ann
Oncol. 21 (Suppl 7):vii320–vii325. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fletcher CDM, Unni KK and Mertens F:
Osteogenic tumours: WHO Classification Tumours of Soft Tissue and
Bone. IARC Press; Lyon: 2002
|
|
3
|
Jaffe N: Osteosarcoma: Review of the past,
impact on the future. The American experience. Cancer Treatment and
Res. 152:239–262. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moore DD and Luu HH: Osteosarcoma. Cancer
Treat Res. 162:65–92. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shimizu T, Ishikawa T, Sugihara E,
Kuninaka S, Miyamoto T, Mabuchi Y, Matsuzaki Y, Tsunoda T, Miya F,
Morioka H, et al: c-MYC overexpression with loss of Ink4a/Arf
transforms bone marrow stromal cells into osteosarcoma accompanied
by loss of adipogenesis. Oncogene. 29:5687–5699. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shimizu T, Sugihara E, Yamaguchi-Iwai S,
Tamaki S, Koyama Y, Kamel W, Ueki A, Ishikawa T, Chiyoda T, Osuka
S, et al: IGF2 preserves osteosarcoma cell survival by creating an
autophagic state of dormancy that protects cells against
chemotherapeutic stress. Cancer Res. 74:6531–6541. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yamaguchi SI, Ueki A, Sugihara E, Onishi
N, Yaguchi T, Kawakami Y, Horiuchi K, Morioka H, Matsumoto M,
Nakamura M, et al: Synergistic antiproliferative effect of imatinib
and adriamycin in platelet-derived growth factor
receptor-expressing osteosarcoma cells. Cancer Sci. 106:875–882.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kamel WA, Sugihara E, Nobusue H,
Yamaguchi-Iwai S, Onishi N, Maki K, Fukuchi Y, Matsuo K, Muto A,
Saya H and Shimizu T: Simvastatin-induced apoptosis in osteosarcoma
cells: A key role of RhoA-AMPK/p38 MAPK signaling in antitumor
activity. Mol Cancer Ther. 16:182–192. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shimizu T, Ishikawa T, Iwai S, Ueki A,
Sugihara E, Onishi N, Kuninaka S, Miyamoto T, Toyama Y, Ijiri H, et
al: Fibroblast growth factor-2 is an important factor that
maintains cellular immaturity and contributes to aggressiveness of
osteosarcoma. Mol Cancer Res. 10:454–468. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hilberg F, Roth GJ, Krssak M, Kautschitsch
S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel
A, Quant J, et al: BIBF 1120: Triple angiokinase inhibitor with
sustained receptor blockade and good antitumor efficacy. Cancer
Res. 68:4774–4782. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roth GJ, Heckel A, Colbatzky F, Handschuh
S, Kley J, Lehmann-Lintz T, Lotz R, Tontsch-Grunt U, Walter R and
Hilberg F: Design, synthesis, and evaluation of indolinones as
triple angiokinase inhibitors and the discovery of a highly
specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120). J
Med Chem. 52:4466–4480. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wind S, Schmid U, Freiwald M, Marzin K,
Lotz R, Ebner T, Stopfer P and Dallinger C: Clinical
pharmacokinetics and pharmacodynamics of nintedanib. Clin
Pharmacokinet. 58:1131–1147. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Richeldi L, Costabel U, Selman M, Kim DS,
Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G,
et al: Efficacy of a tyrosine kinase inhibitor in idiopathic
pulmonary fibrosis. New Engl J Med. 365:1079–1087. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Richeldi L, du Bois RM, Raghu G, Azuma A,
Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y,
et al: Efficacy and safety of nintedanib in idiopathic pulmonary
fibrosis. New Engl J Med. 370:2071–2082. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Reck M, Kaiser R, Mellemgaard A, Douillard
JY, Orlov S, Krzakowski M, von Pawel J, Gottfried M, Bondarenko I,
Liao M, et al: Docetaxel plus nintedanib versus docetaxel plus
placebo in patients with previously treated non-small-cell lung
cancer (LUME-Lung 1): A phase 3, double-blind, randomised
controlled trial. Lancet Oncol. 15:143–155. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kutluk Cenik B, Ostapoff K T, Gerber DE
and Brekken RA: BIBF 1120 (nintedanib), a triple angiokinase
inhibitor, induces hypoxia but not EMT and blocks progression of
preclinical models of lung and pancreatic cancer. Mol Cancer Ther.
12:992–1001. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Awasthi N and Schwartz RE: Profile of
nintedanib in the treatment of solid tumors: The evidence to date.
Onco Targets Ther. 8:3691–3701. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Awasthi N, Hinz S, Brekken RA, Schwarz MA
and Schwarz RE: Nintedanib, a triple angiokinase inhibitor,
enhances cytotoxic therapy response in pancreatic cancer. Cancer
Lett. 358:59–66. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marqués M, Corral S, Sánchez-Díaz M, Del
Pozo N, Martínez de Villarreal J, Schweifer N, Zagorac I, Hilberg F
and Real FX: Tumor and stromal cell targeting with nintedanib and
alpelisib overcomes intrinsic bladder cancer resistance. Mol Cancer
Ther. 22:616–629. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu J, Gao J, Wang A, Jiang Z, Qi S, Qi Z,
Liu F, Yu K, Cao J, Chen C, et al: Nintedanib overcomes drug
resistance from upregulation of FGFR signalling and
imatinib-induced KIT mutations in gastrointestinal stromal tumours.
Mol Oncol. 16:1761–1774. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gabasa M, Ikemori R, Hilberg F, Reguart N
and Alcaraz J: Nintedanib selectively inhibits the activation and
tumour-promoting effects of fibroblasts from lung adenocarcinoma
patients. Br J Cancer. 117:1128–1138. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yamanaka T, Harimoto N, Yokobori T,
Muranushi R, Hoshino K, Hagiwara K, Gantumur D, Handa T, Ishii N,
Tsukagoshi M, et al: Nintedanib inhibits intrahepatic
cholangiocarcinoma aggressiveness via suppression of cytokines
extracted from activated cancer-associated fibroblasts. Br J
Cancer. 122:986–994. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kato R, Haratani K, Hayashi H, Sakai K,
Sakai H, Kawakami H, Tanaka K, Takeda M, Yonesaka K, Nishio K and
Nakagawa K: Nintedanib promotes antitumour immunity and shows
antitumour activity in combination with PD-1 blockade in mice:
Potential role of cancer-associated fibroblasts. Br J Cancer.
124:914–924. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ledermann JA, Hackshaw A, Kaye S, Jayson
G, Gabra H, McNeish I, Earl H, Perren T, Gore M, Persic M, et al:
Randomized phase II placebo-controlled trial of maintenance therapy
using the oral triple angiokinase inhibitor BIBF 1120 after
chemotherapy for relapsed ovarian cancer. J Clin Oncol.
29:3798–3804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grosso F, Steele N, Novello S, Nowak AK,
Popat S, Greillier L, John T, Leighl NB, Reck M, Taylor P, et al:
Nintedanib plus pemetrexed/cisplatin in patients with malignant
pleural mesothelioma: Phase II results from the randomized,
placebo-controlled LUME-meso trial. J Clin Oncol. 35:3591–3600.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang W, Zhao JM, Lin J, Hu CZ, Zhang WB,
Yang WL, Zhang J, Zhang JW and Zhu J: Adaptive fibrogenic
reprogramming of osteosarcoma stem cells promotes metastatic
growth. Cell Rep. 24:1266–1277.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shimizu T, Kimura K, Sugihara E,
Yamaguchi-Iwai S, Nobusue H, Sampetrean O, Otsuki Y, Fukuchi Y,
Saitoh K, Kato K, et al: MEK inhibition preferentially suppresses
anchorage-independent growth in osteosarcoma cells and decreases
tumors in vivo. J Orthop Res. 39:2732–2743. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shimizu T, Sugihara E, Takeshima H,
Nobusue H, Yamaguchi R, Yamaguchi-Iwai S, Fukuchi Y, Ushijima T,
Muto A and Saya H: Depletion of R270C mutant p53 in osteosarcoma
attenuates cell growth but does not prevent invasion and metastasis
in vivo. Cells. 11:36142022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH image to ImageJ: 25 Years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Johnson DE, O'Keefe RA and Grandis JR:
Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev
Clin Oncol. 15:234–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ha H, Debnath B and Neamati N: Role of the
CXCL8-CXCR1/2 axis in cancer and inflammatory diseases.
Theranostics. 7:1543–1588. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gross AC, Cam H, Phelps DA, Saraf AJ, Bid
HK, Cam M, London CA, Winget SA, Arnold MA, Brandolini L, et al:
IL-6 and CXCL8 mediate osteosarcoma-lung interactions critical to
metastasis. JCI Insight. 3:e997912018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sappino AP, Skalli O, Jackson B, Schürch W
and Gabbiani G: Smooth-muscle differentiation in stromal cells of
malignant and non-malignant breast tissues. Int J Cancer.
41:707–712. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Orimo A, Gupta PB, Sgroi DC,
Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL
and Weinberg RA: Stromal fibroblasts present in invasive human
breast carcinomas promote tumor growth and angiogenesis through
elevated SDF-1/CXCL12 secretion. Cell. 121:335–348. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wollin L, Maillet I, Quesniaux V, Holweg A
and Ryffel B: Antifibrotic and anti-inflammatory activity of the
tyrosine kinase inhibitor nintedanib in experimental models of lung
fibrosis. J Pharmacol Exp Ther. 349:209–220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hostettler KE, Zhong J, Papakonstantinou
E, Karakiulakis G, Tamm M, Seidel P, Sun Q, Mandal J, Lardinois D,
Lambers C and Roth M: Anti-fibrotic effects of nintedanib in lung
fibroblasts derived from patients with idiopathic pulmonary
fibrosis. Respir Res. 15:1572014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Labrie M, Brugge JS, Mills GB and
Zervantonakis IK: Therapy resistance: Opportunities created by
adaptive responses to targeted therapies in cancer. Nat Rev Cancer.
22:323–339. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bejarano L, Jordāo MJC and Joyce JA:
Therapeutic targeting of the tumor microenvironment. Cancer Discov.
11:933–959. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vail ME, Farnsworth RH, Hii L, Allen S,
Arora S, Anderson RL, Dickins RA, Orimo A, Wu SZ, Swarbrick A, et
al: Inhibition of EphA3 expression in tumour stromal cells
suppresses tumour growth and progression. Cancers (Basel).
15:46462023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Albrengues J, Bertero T, Grasset E, Bonan
S, Maiel M, Bourget I, Philippe C, Herraiz Serrano C, Benamar S,
Croce O, et al: Epigenetic switch drives the conversion of
fibroblasts into proinvasive cancer-associated fibroblasts. Nat
Commun. 6:102042015. View Article : Google Scholar : PubMed/NCBI
|