Introduction
Blastic plasmacytoid dendritic cell neoplasm (BPDCN)
is a rare and aggressive hematological malignancy with a mostly
poor prognosis. In the United States, approximately 0.04 cases of
BPDCN per 100,000 individuals have been reported between 2008 and
2014, with a median overall survival (OS) of 8–20 months (1). The condition tends to occur in older
adults with a median age of 60–70 years and most individuals
diagnosed with the disease are male (2). BPDCN originates from plasmacytoid
dendritic cells, and its most common clinical manifestations are
cutaneous lesions, bone marrow involvement and leukemic
dissemination (3). CD4+
and CD56+ tumor cells are typical of BPDCN, and CD123,
transcription factor 4 (TCF4), T cell leukemia/lymphoma protein 1
(TCL1), CD304 and CD303 (also referred to as blood dendritic cell
antigen 2) are its main markers (4,5). BPDCN
does not express typical markers of a lymphoid or myeloid lineage
(6). There are no consensus
treatment guidelines for BPDCN (7).
Retrospective studies have demonstrated that regimens based on
lymphoid leukemia are associated with improved remission rates
compared with those based on myeloid leukemia or lymphoma (2,8).
Regardless of the regimen, however, disease relapses are frequent
and rapid (9,10). In addition, allogeneic stem cell
transplantation (alloSCT) is the only known potential cure for
BPDCN (3,11). The 5-year OS, disease-free survival,
recurrence and non-recurrence mortality rates for patients with
BPDCN who receive alloSCT are 51.2, 44.4, 32.2 and 23.3%,
respectively (11).
Myelofibrosis (MF) is a type of myeloproliferative
neoplasm (MPN), which is clinically characterized by progressive
anemia, splenomegaly and systemic symptoms (including fatigue,
night sweats and fever) (12). The
pooled annual incidence rate of primary myelofibrosis (PMF) in
Europe, North America and Oceania between 1946 and 2012 was
0.47/100,000 (13). The five
independent risk factors in the International Prognostic Scoring
System for PMF include age >65 years, hemoglobin <10 g/dl,
white blood cell count >25×109/l, circulating blasts
≥1% and presence of constitutional symptoms (14). Based on these variables, PMF is
categorized into four groups: Low-risk, intermediate-risk-1,
intermediate-risk-2 and high-risk, corresponding to a median OS of
11.3, 7.9, 4 and 2.3 years, respectively (14). Treatment of PMF requires
individualized assessment based on the spleen size, symptom burden
and risk stratification (15). Most
current pharmacologic treatments for PMF are palliative in scope,
such as Janus kinase 2 (JAK2) inhibitors (ruxolitinib) (12). The overall mortality rates before
and after ruxolitinib approval were 79.8 and 47.3%, respectively,
and the overall risk of death was reduced by 53% with ruxolitinib
approval (16). MF can be secondary
to several hematological malignancies, including chronic myeloid
leukemia, myelodysplastic syndrome and hairy cell leukemia
(17–19).
The present study reports the case of an elderly
male patient with BPDCN in combination with MF. Among previous
studies, only one case report was found on the conversion of PMF to
BPDCN (20); however, to the best
of our knowledge, there is no relevant literature on MF secondary
to BPDCN. The case is unique as the patient was CD4−,
with combined MF and mutations in the Tet methylcytosine
dioxygenase 2 (TET2) and NRAS proto-oncogene GTPase (NRAS) genes,
which is rare. Future research is required to further elucidate the
potential relationship between these two different diseases.
Case report
Case
In May 2021, a 70-year-old male patient gradually
developed a large, dark, purplish-red rash on the chest, abdomen
and upper back without concomitant symptoms, such as itching, pain
or fever, following a vaccination against severe acute respiratory
syndrome coronavirus 2. The patient said that they went to the
Dermatology Department of the Fourth Hospital of Hebei Medical
University (Shijiazhuang, China), and were treated with oral
antiallergic medication for 2 months (details unknown), after which
their skin returned to normal. In September 2021, the patient went
to Shijiazhuang People's Hospital (Shijiazhuang, China), for
incomplete intestinal obstruction, and on the third day of
hospitalization, a rash reappeared without any apparent trigger and
was more severe than the previous one. The patient was given
dexamethasone (5 mg intramuscular QD), ebastine (20 mg oral QD),
benadryl (20 mg intramuscular QD) and topical glycerite lotion, and
the rash symptomatically improved after 4 consecutive days of
medication. During hospitalization, whole body CT was examined, and
there were multiple enlarged lymph nodes in the abdominal cavity
and retroperitoneum (details unknown). In April 2022, the patient
developed symptoms of generalized malaise, and went to Shijiazhuang
Hospital of Traditional Chinese Medicine (Shijiazhuang, China), and
the repeat whole body CT suggested that the number of enlarged
lymph nodes in the abdominal cavity had increased compared with the
previous one, and the patient did not receive any treatment.
However, the condition was recurrent and gradually worsened. In May
2022, the patient was admitted to Hebei General Hospital
(Shijiazhuang, China) (Fig. 1A). A
physical assessment revealed the following: Temperature, 36.3°C
(normal range, 36.1–37.0°C); pulse, 88 beats/min (normal range,
60–100 beats/min); respiration, 22 breaths/min (normal range, 16–20
breaths/min); blood pressure, 112/69 mmHg (normal range,
90–130/60–80 mmHg); conscious; no jaundice. Skin and mucosal
examinations revealed a large, dark, purplish-red rash on the
chest, back (Fig. 1B and C) and
left inner thigh. A lymph node assessment showed palpable enlarged
lymph nodes on the left supraclavicular region, which were tough
and could be pushed without pressure pain, while the remaining
superficial lymph nodes were not enlarged. An abdominal examination
revealed no evidence of hepatosplenomegaly. Other findings were
unremarkable. The patient had a previous history of hypertension
for 10 years, regular administration of amlodipine besylate,
well-controlled blood pressure, and no other medical history,
including hematological neoplasms.
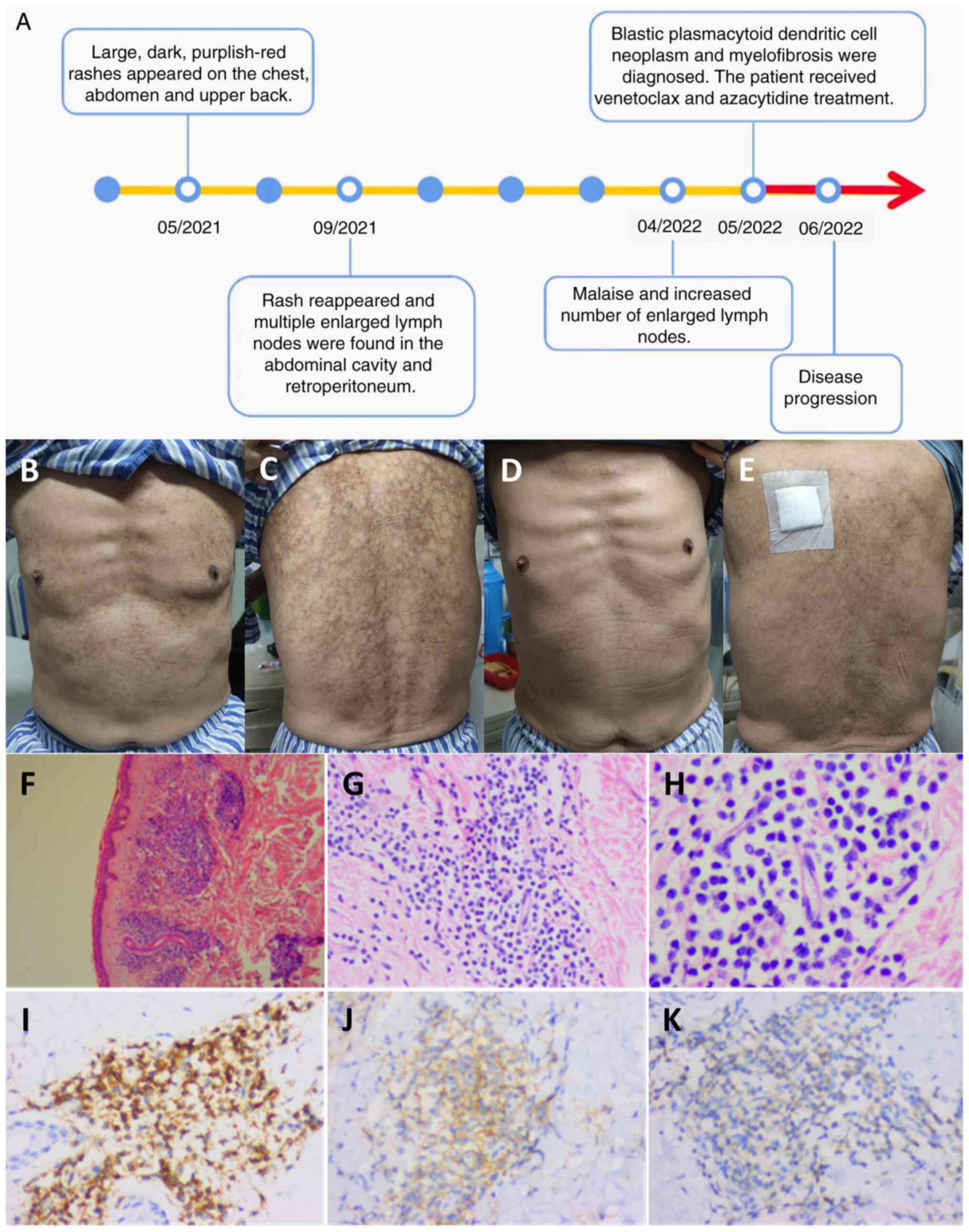 | Figure 1.Timeline, images and pathology of the
skin. (A) Timeline of disease progression. (B-E) Skin rashes on the
(B) chest and (C) back before treatment and on the (D) chest and
(E) back after 6 days of chemotherapy. Pathological findings of the
skin rashes, visualized using hematoxylin and eosin staining at (F)
×40, (G) ×200 and (H) ×400 magnification. (I) CD43+
(magnification, ×200), (J) CD56+ (magnification, ×200)
and (K) CD123+ (magnification, ×200)
immunohistochemistry staining results. No obvious lesions were seen
in the epidermis, but a large number of blastoid cells had
infiltrated around small blood vessels in the dermis and skin
appendages, which were single in morphology and medium in size,
with round or oval nuclei, fine-grained chromatin and a small
nucleolus. |
Laboratory assessment results at admission and after
one course of VA regimen (venetoclax 100 mg, D1; 200 mg, D2-28;
azacytidine 0.1 g subcutaneously, D1-7) are presented in Table I. White blood cells, lymphocytes,
cytokines interleukin (IL)-6 and IL-10 markedly decreased after
treatment compared with before, but C-reactive protein levels were
increased. Next-generation sequencing of peripheral blood, which
assessed the protein-coding regions of 267 genes strongly
associated with hematological disorders, revealed multiple
mutations, including exosome complex exonuclease RRP44 (DIS3;
13q22.1), NRAS (1p13.2), TET2 (4q24), CUB and Sushi multiple
domains 1 (CSMD1; 8p23.2), B-cell lymphoma (BCL)6 corepressor-like
protein 1 (Xq26.1) and platelet-derived growth factor receptor
(PDGFR)A (4q12) mutations (Table
II). However, no mutations were found in the FMS-related
receptor tyrosine kinase 3 (FLT3), JAK2, STAT, calreticulin (CALR)
or MPL proto-oncogene thrombopoietin receptor (MPL) genes. Bone
marrow cell classification showed that the proportion of
lymphocytes was notably increased, and atypical lymphocytes were
easily seen (73.5%) (Fig. 2A). Bone
marrow tissue biopsies showed highly active myelodysplasia (60–70%)
and reticular fiber staining was grade 2 (Fig. 2B-T) (21). Immunohistochemistry of cutaneous,
lymph node and bone marrow tissue biopsies supported a diagnosis of
a BPDCN (Figs. 1F-K, 2 and 3).
Immunohistochemical examination of the bone marrow and lymph nodes
was positive for TET2, NF-κB, NRAS and phospho (p)-ERK, and
negative for PDGFRA, IL-6, IL-10, JAK2, STAT3 and TGF-β1. Flow
cytometry analysis of the bone marrow showed abnormal cells, with
54.61% (Fig. S1B) expressing CD56
(Fig. S1K), CD45RA (Fig. S2A), CD2 (Fig. S2B), CD303 (Fig. S3D), human leukocyte antigen DR
(Fig. S3E) and CD36 (Fig. S4B), partially expressing CD4
(Fig. S2C), CD103 (Fig. S2D), CD10 (Fig. S2E) and CD123 (Fig. S4A), weakly expressing CD7 (Fig. S2F), TCL1 (Fig. S4C) and T-cell restricted
intracellular antigen 1 (Fig.
S4D), and lacking expression of other myeloid or lymphoid
markers. Additional detailed results of flow cytometry are shown in
Fig. S1, Fig. S2, Fig.
S3, Fig. S4, Fig. S5, Fig.
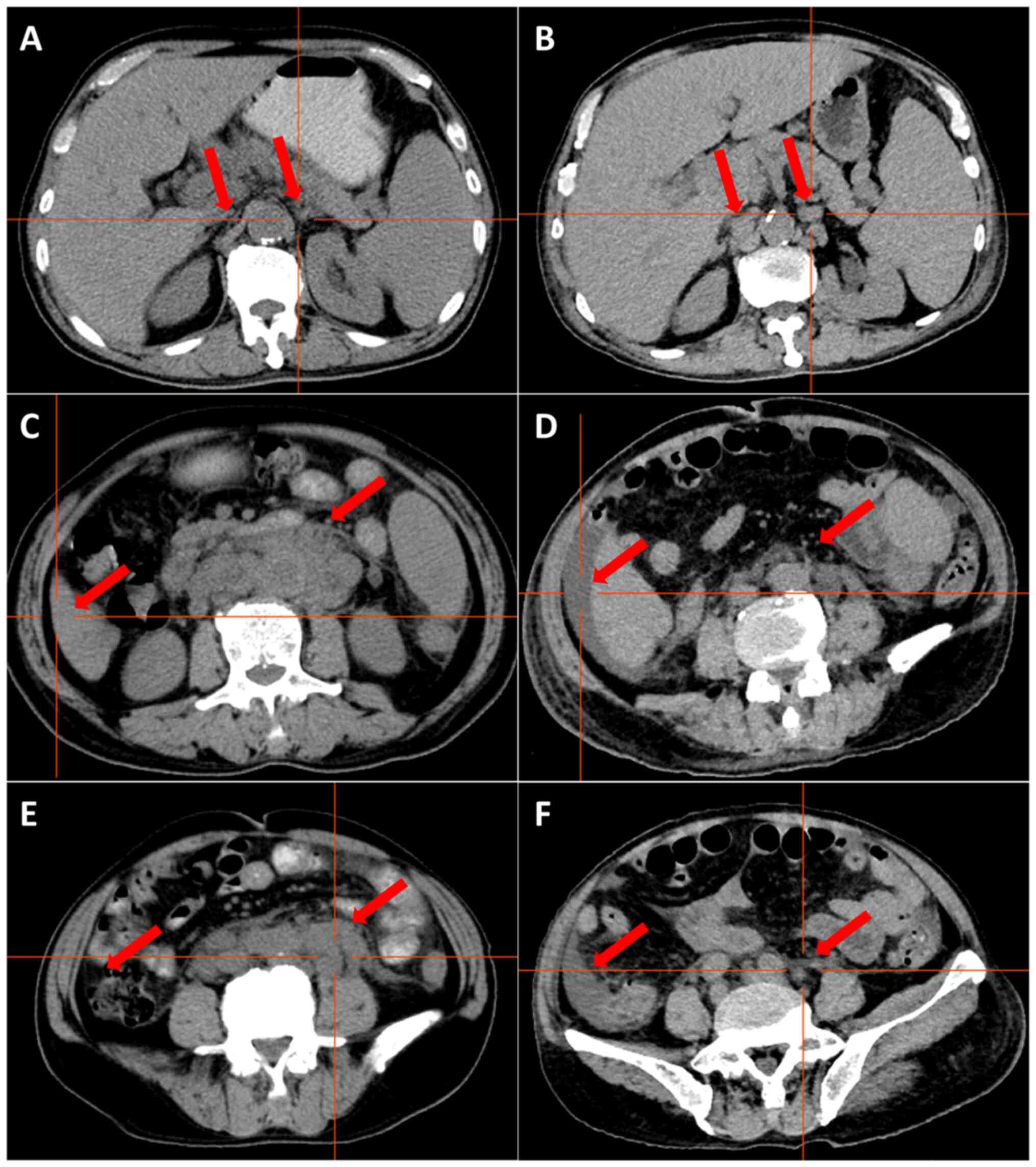
S6, Fig. S7. CT demonstrated
multiple enlarged lymph nodes above and below the transverse septum
and splenomegaly with slight hypermetabolism. The largest
retroperitoneal para-abdominal aortic lymph node was 30×20 mm
(Fig. 4A, C and E). On the basis of
these findings, the patient was diagnosed with BPDCN complicated by
MF. The patient received a course of VA regimen (venetoclax 100 mg,
D1; 200 mg, D2-28; azacytidine 0.1 g subcutaneously, D1-7). On day
6 of chemotherapy, the skin lesions had almost subsided (Fig. 1D and E) and the lymphocyte count had
gradually decreased during this treatment period (Table III). On day 10 of chemotherapy,
the patient was discharged.
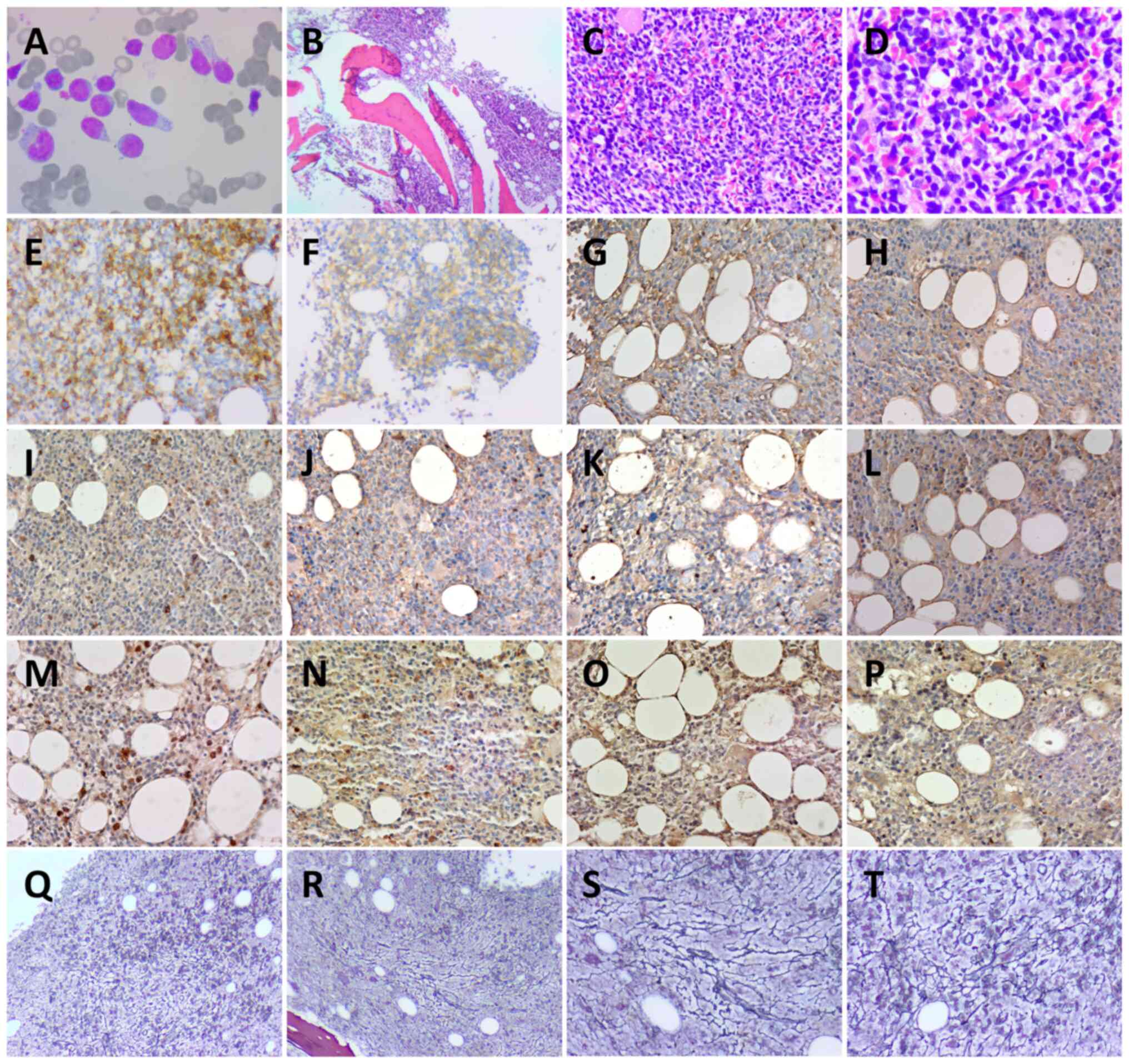 | Figure 2.Bone marrow smear and pathology. (A)
Wright-Giemsa staining of bone marrow smear (magnification,
×1,000). Hematoxylin and eosin staining of bone marrow at (B) ×40
magnification, (C) ×200 magnification and (D) ×400 magnification.
(E) CD56+ (magnification, ×200), (F) CD123+;
(magnification, ×200), (G) IL-6− (magnification, ×200),
(H) IL-10− (magnification, ×200), (I) JAK2−
(magnification, ×200), (J) STAT3- (magnification, ×200), (K)
TGF-β1- (magnification, ×200), (L) PDGFRA- (magnification, ×200),
(M) NF-κB+ (magnification, ×200), (N) TET2+
(magnification, ×200), (O) NRAS+ (magnification, ×200)
and (P) p-ERK+ (magnification, ×200) immunohistochemistry staining
results. Reticular fiber staining to assess myelofibrosis-2 at (Q
and R) ×40 and (S and T) ×200 magnification. (Q and R) and (S and
T) Different positions at the same magnification. Bone marrow cells
were round or sub-round. Cytoplasm was abundant and part of the
cytoplasm was trailing. Nuclei were round or sub-round with rough
chromatin. |
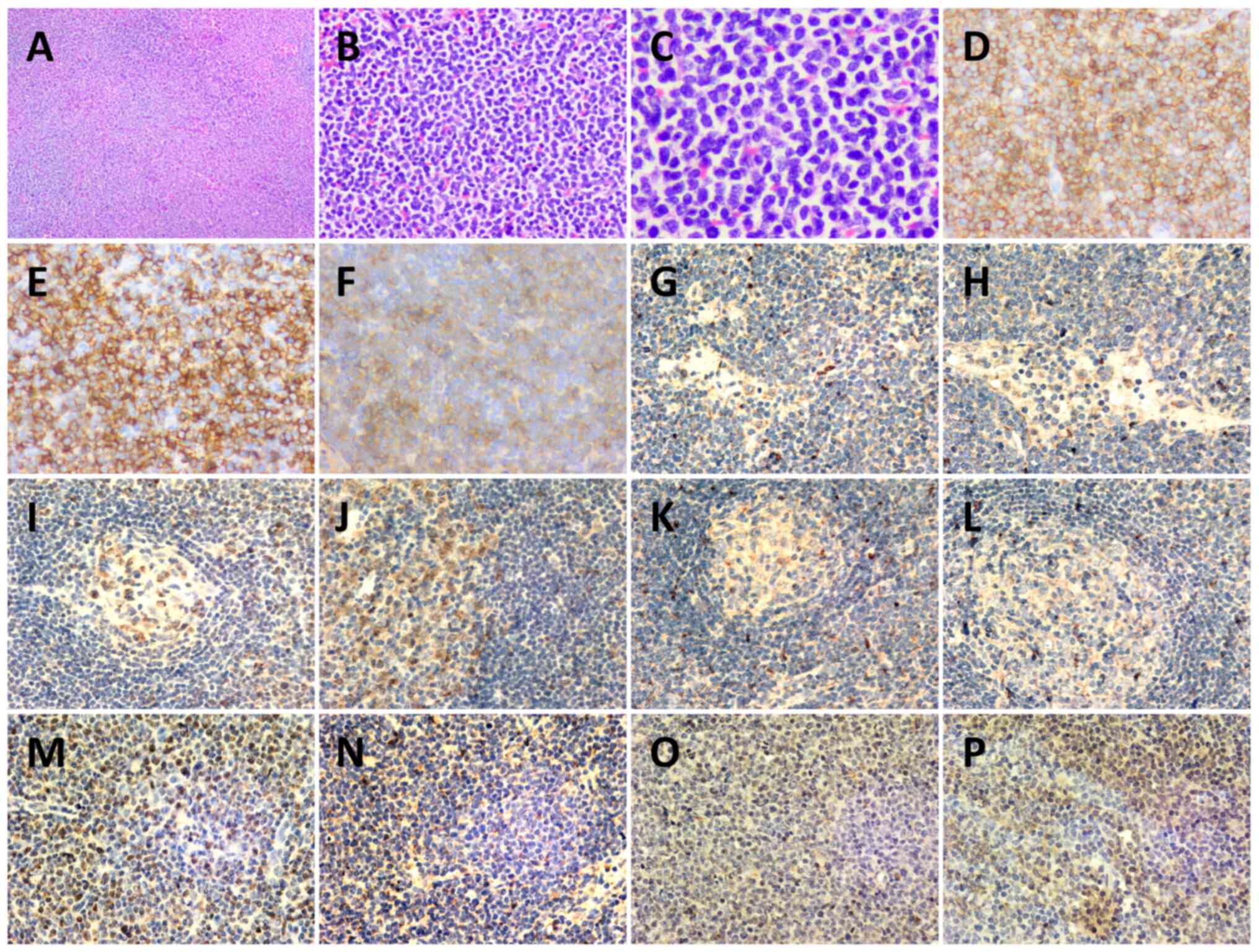 | Figure 3.Histopathology of a left cervical
lymph node biopsy. Hematoxylin-eosin staining of lymph node at (A)
×40, (B) ×200 and (C) ×400 magnification. (D) CD43+
(magnification, ×200), (E) CD56+ (magnification, ×200),
(F) CD123+ (magnification, ×200), (G) IL-6−
(magnification, ×200), (H) IL-10− (magnification, ×200),
(I) JAK2− (magnification, ×200) (J) STAT3−
(magnification, ×200), (K) TGF-β1− (magnification,
×200), (L) PDGFRA− (magnification, ×200), (M)
NF-κB+ (magnification, ×200), (N) TET2+
(magnification, ×200), (O) NRAS+ (magnification, ×200)
and (P) p-ERK+ (magnification, ×200)
immunohistochemistry staining results. The structure of the lymph
node was destroyed, and atypical proliferative blast-like cells had
diffusely proliferated and were mainly infiltrating the
interlobular area and medullary area. Cells were medium in size
with a uniform morphology, round or ovoid nucleus, fine and
granular chromatin, with one or more nucleoli visible, and mitosis
was common. |
 | Table I.Laboratory findings. |
Table I.
Laboratory findings.
| Parameter | On admission | Second day after
the first course of chemotherapy | Normal
range/limit |
|---|
| Complete blood
count |
|
|
|
| White
blood cells, 109/l | 10.53 | 2.33 | 3.50–9.50 |
|
Neutrophils,
109/l | 1.81 | 2.02 | 1.80–6.30 |
|
Lymphocytes,
109/l | 7.60 | 0.24 | 1.10–3.20 |
|
Eosinophils,
109/l | 1.60 | 0.00 | 0.02–0.52 |
| Red
blood cells, 1012/l | 3.10 | 3.10 | 3.80–5.10 |
|
Hemoglobin, g/l | 91.00 | 92.00 | 115.00–150.00 |
|
Platelets,
109/l | 70.00 | 104.00 | 125.00–350.00 |
| C-reactive protein,
mg/l | 31.19 | 61.83 | 0.00–6.00 |
| Coagulation |
|
|
|
|
Fibrinogen, g/l | 3.14 | 0.69 | 2.00–4.00 |
|
D-dimer, mg/l FEU | 1.65 | 5.12 | 0.00–0.55 |
| β2-microglobulin,
µg/ml | 6.29 | Untested | 0.90–2.70 |
| Biochemistry |
|
|
|
| Total
protein, g/l | 59.30 | 67.40 | 65.00–85.00 |
| Alanine
transaminase, U/l | 11.20 | 12.80 | 9.00–50.00 |
|
Aspartate transaminase,
U/l | 16.30 | 17.40 | 15.00–40.00 |
|
Alkaline phosphatase, U/l | 78.50 | 138.30 | 45.00–125.00 |
| Blood
urea nitrogen, mmol/l | 6.50 | 7.40 | 3.60–9.50 |
| Serum
creatinine, µmol/l | 90.60 | 118.30 | 57.00–111.00 |
| Uric
acid, µmol/l | 407.40 | 297.50 | 208.00–428.00 |
|
Glomerular filtration rate,
ml/min | 73.24 | 53.00 |
|
| Lactate
dehydrogenase, U/l | 259.10 | 203.00 | 120.00–250.00 |
| Immunoglobulin |
|
|
|
| IgG,
g/l | 11.69 | Untested | 8.60–17.40 |
| IgA,
g/l | 1.26 | Untested | 1.00–4.20 |
| IgM,
g/l | 0.63 | Untested | 0.50–2.80 |
| Cytokine |
|
|
|
| IL-6,
pg/ml | 47.51 | 13.92 | 0.00–5.30 |
| IL-10,
pg/ml | 98.43 | 2.42 | 0-4.91 |
|
Vascular endothelial growth
factor, pg/ml | 55.39 | Untested | 0-142.20 |
| Serum amyloid A,
mg/l | 87.22 | 233.46 | <10.00 |
| Procalcitonin,
ng/ml | 0.25 | 2.04 | <0.06 |
| Table II.Next-generation sequencing of the
patient's peripheral blood. |
Table II.
Next-generation sequencing of the
patient's peripheral blood.
| Mutant gene | Chromosome | Mutation location,
exon | Nucleotide
alteration | Amino acid
alteration | dbSNP reference
no. | Mutation
frequency | Sequencing depth,
X |
|---|
| DIS3 | 13q22.1 | 17 | c.2339G>C | p.R780T | - | 0.166 | 1,661 |
| NRAS | 1p13.2 | 4 | c.436G>A | p.A146T | - | 0.045 | 1,474 |
| NRAS | 1p13.2 | 2 | c.35G>A | p.G12D | rs121913237 | 0.017 | 2,051 |
| NRAS | 1p13.2 | 2 | c.34G>A | p.G12S | rs121913250 | 0.035 | 2,044 |
| TET2 | 4q24 | 3 | c.2245C>T | p.Q749* | - | 0.011 | 1,881 |
| TET2 | 4q24 | 5 | c.3578G>A | p.C1193Y | - | 0.580 | 1,573 |
| TET2 | 4q24 | 11 | c.4794T>G | p.Y1598* | - | 0.182 | 1,994 |
| CSMD1 | 8p23.2 | 14 | c.2012G>A | p.R671H | rs376413480 | 0.137 | 1,576 |
| TET2 | 4q24 | 9 | c.4160A>G | p.N1387S | - | 0.039 | 1,272 |
| BCORL1 | Xq26.1 | 4 | c.469G>A | p.A157T | rs147775035 | 0.998 | 996 |
| PDGFRA | 4q12 | 15 | c.2153G>A | p.R718Q | rs367722824 | 0.667 | 1,443 |
| Table III.Analysis of the peripheral blood. |
Table III.
Analysis of the peripheral blood.
|
|
| Day after the
first | Day of chemotherapy
course of chemotherapy |
|---|
|
|
|
|
|
|---|
| Parameter | On admission | 2 | 5 | 7 | 9 | 2 | 3 | 5 | 6 |
|---|
| White blood cells,
109/l | 10.53 | 5.71 | 3.52 | 2.99 | 2.76 | 2.33 | 2.12 | 2.1 | 3.36 |
| Neutrophils,
109/l | 1.81 | 0.88 | 0.93 | 0.79 | 0.98 | 2.02 | 1.81 | 1.39 | 2.14 |
| Lymphocytes,
109/l | 7.60 | 4.40 | 2.04 | 1.70 | 1.28 | 0.24 | 0.23 | 0.49 | 1.10 |
| Red blood cells,
1012/l | 3.10 | 2.75 | 2.79 | 2.85 | 3.01 | 3.10 | 3.06 | 3.26 | 2.96 |
| Hemoglobin,
g/l | 91.00 | 79.00 | 80.00 | 81.00 | 85.00 | 92.00 | 89.00 | 94.00 | 86.00 |
| Platelets,
109/l | 70.00 | 44.00 | 91.00 | 114.00 | 112.00 | 104.00 | 73.00 | 17.00 | 8.00 |
The second day after the first course of
chemotherapy, the patient was admitted to the hospital again and
the laboratory assessment results at readmission are shown in
Table I. The patient presented with
granulocyte deficiency with a fever, and CT demonstrated
splenomegaly, ascites and multiple lymph nodes that had increased
and enlarged, with few lymph nodes that were smaller than before
(Fig. 4B, D and F). This indicated
that the effect of the treatment was poor. Subsequently, the
condition of the patient progressed and included respiratory
failure, acute renal injury and delirium. A series of treatments
were administered, such as anti-infection medication [meropenem, 1
g intravenous (IV), 5 days] and a red blood transfusion
(recombinant human thrombopoietin, 1.5 WU, subcutaneous, 2 days;
human albumin, 20 g IV, 1 time; human fibrinogen, 4 g IV, 1 time;
leukocyte-depleted frozen plasma, 400 ml IV, 1 time). An
intermittent fever persisted; however, the neutrophil count on day
5 after the first course of treatment was slightly low and the rest
of the neutrophil counts after the first course of chemotherapy
were within the normal range (Table
III), no causative organisms were detected in the sputum, blood
or urine, and no lung infection was seen on the chest CT. The site
of infection could not be determined and the condition of the
patient continued to progress. The general condition of the patient
was so poor that the next course of chemotherapy was not
administered. Finally, the patient requested to be discharged from
the hospital due to financial reasons. In July 2022, a follow-up
phone call established that the patient had died.
Next-generation sequencing of
peripheral blood
Wet experiment part
Nucleic acid extraction was performed using the
Blood Genomic DNA Extraction Kit (0.1–1 ml; cat. no. YDP348-03;
Tiangen Biotech Co., Ltd.). Nucleic acid quality inspection was
performed using a NANODROP ONE (Thermo Fisher Scientific, Inc.) to
measure the preliminary DNA concentration, A260/280 and A260/230,
where A260 is the absorption wavelength of the highest absorption
peak of nucleic acid and A280 is the absorption wavelength of the
highest absorption peak of protein. A230 is the absorption
wavelength of the highest absorption peak of carbohydrates.
A260/280 and A260/230 are indicative values of nucleic acid purity.
An A260/280 ratio of 1.8–2.0 and an A260/230 ratio of 2.0–2.2
indicate that the purity of DNA is good. Qubit 4.0 fluorometer
(Q33238; Thermo Fisher Scientific, Inc.) detection was used as the
standard for library construction input. Raw samples were analyzed
by electrophoresis (2.5% agarose gels) using DNA marker (BM401-01;
TransGen Biotech Co., Ltd.) to initially confirm DNA integrity. The
DNA library was constructed with a commercial kit (cat. no.
20025524; Illumina DNA Prep with Enrichment) and the length of the
library was set to 220 bp. Library quality control was performed
using a Qubit 4.0 to detect the library concentration, and Agilent
DNF-915 Reagent Kits (Agilent Technologies, Inc.) were used to
perform fragment analysis quality control on the Agilent 5200
platform (Agilent Technologies, Inc.). The average fragments ranged
between 330 and 390 bp [dilution conversion formula for the
concentration of the library: 1.25 nm × average molar mass of bases
(660) × average fragment length of library (330–390
bp)/106=0.27–0.32 ng/µl (0.4–0.48 nM)]. Hybridization
was performed using the Nextera DNA Flex Pre-Enrichment Library
Prep and Enrichment Reagents 96 samples, and targeted capture
amplification of the target region was performed with probes
manufactured by Tianjin Xiehe Bojing Medical Diagnostic Technology
Co., Ltd. The sequencing platform was Illumina novaseq6000
(Illumina, Inc.) with the NovaSeq6000 S1 Reagents Kit v1.5 (300
cycles; cat. no. 20028318). The sequencing input protocol was as
follows: Samples to be uploaded were prepared and their molar
concentrations were calculated, then the libraries were denatured
and samples were detected at a concentration of 250 pM (1.25 nM).
The molarity was calculated as follows: (ng/µl
×105)/[660 g/mol × average library size (bp)]=Molarity
(nM). The sequencing protocol was paired-end 150 bp sequencing.
Next-generation sequencing
analysis
The quality control process Fastp (22) (version 0.23.2; http://github.com/OpenGene/fastp) was applied for
FASTQ data by removing the terminal adaptor sequences and
low-quality reads from the raw data. Dragen (version 3.10.4;
Illumina, Inc.), a local installation hardware-accelerated
sequencing processing pipeline, was used to perform data alignment
and mutation calling. The lower limit of detection for variant
allele frequency was 0.5%, and then an in-house algorithm was used
to review hotspot variants. The final candidate variants were all
manually verified in the Integrative Genomics Viewer (23) (IGV; http://www.igv.org/). Copy number alterations were
identified using CNVkit (24)
(version 0.9.10; http://cnvkit.readthedocs.io/en/stable/) with default
parameters. The insertion and deletion caller Pindel (25) (version v0.2.5b8; http://gmt.genome.wustl.edu/packages/pindel/) and
FLT3_ITD_ext (26) (version 1.1;
http://github.com/ht50/FLT3_ITD_ext)
were run on exon 13–15 to identify FMS-like tyrosine kinase 3
internal tandem duplication alleles. Next-generation sequencing
materials are shown in Table
IV.
| Table IV.Materials for next-generation
sequencing. |
Table IV.
Materials for next-generation
sequencing.
| Reagent name | Supplier | Step |
|---|
| No nuclease
water | Tiangen Biotech
Co., Ltd. | Overall
process |
| Blood Genomic DNA
Extraction Kit (0.1–1 ml) | Tiangen Biotech
Co., Ltd. | Extraction |
| Illumina DNA Prep
with Enrichment kits | Illumina, Inc. | Library
construction |
| AMPure XP
Beads | Beckman Coulter,
Inc. | Library
construction |
| Qubit™ dsDNA HS
Assay Kits | Thermo Fisher
Scientific, Inc. | Quality
testing |
| Nextera DNA Flex
Pre-Enrichment Library Prep and | Illumina, Inc. | Hybridization |
| Enrichment Reagents
96 samples |
|
|
| Agilent DNF-915
Reagent Kits | Agilent
Technologies, Inc. | Fragment
analysis |
| NovaSeq6000 S1
Reagents Kit v1.5 (300 cycles) | Illumina, Inc. | Machine
sequencing |
Flow cytometry
Experimental procedure
For cell membrane antibodies, antibody was added to
the specimen, followed by mixing by gentle oscillation and
incubation for 15–30 min at 20°C away from light. Red blood cell
lysis reagents (1.5 ml) were added into samples, which were gently
shaken and mixed evenly, and the process of lysing red blood cells
lasted 10 min at 20°C. Samples were centrifuged at 188 × g for 5
min at 20°C, the supernatant was discarded (an appropriate amount
of PBS was added before centrifugation). PBS was added for washing,
and samples were mixed with gentle shaking and then centrifuged at
188 × g for 5 min at 20°C, and the supernatant was discarded. PBS
(300 µl) was added for resuspension, followed by mixing with gentle
vibration and testing.
For cytoplasmic antibodies, cytoplasmic antibodies
and specimens were mixed with gentle shaking, and incubated for
15–30 min at 20°C away from light. The fixation reagent (100 µl)
was added, and samples were mixed with gentle shaking and incubated
for 5 min in the dark at 20°C. Hemolysin (1.5 ml) was added,
samples were mixed with gentle shaking, hemolysis was performed for
10 min at 20°C, centrifugation was performed at 282 × g for 5 min
at 20°C and the supernatant was discarded. The permeabilization
reagent (50 µl) and plasma antibody were added, samples were mixed
with gentle shaking and incubated for 15 min in the dark at 20°C.
An appropriate amount of PBS was added for washing, samples were
centrifuged at 282 × g for 5 min at 20°C, and the supernatant was
discarded. PBS (300 µl) was added for resuspension, samples were
mixed with gentle shaking, and then tested.
All experimental steps of flow cytometry were
performed at room temperature (~20°C).
Reagents
Red blood cell lysis reagents for flow cytometry
analysis (item no. 349202; BD Biosciences) were used. Fixation and
permeation reagents were included in the BD INTRASURE KIT RUO (item
no. 641776; BD Biosciences). The fluorescence reagents are shown in
Table V.
| Table V.Antibodies. |
Table V.
Antibodies.
| Antibody | Manufacturer | Product no. |
|---|
| CD56 | Beckman Coulter,
Inc. | B49189 |
| CD303 | BioLegend,
Inc. | 354204 |
| CD45RA | BD Biosciences | 663496 |
| HLA-DR | BioLegend,
Inc. | 307618 |
| CD36 | Beckman Coulter,
Inc. | IM0766U |
| CD2 | Beckman Coulter,
Inc. | A21689 |
| CD103 | Beckman Coulter,
Inc. | IM1856U |
| CD123 | BioLegend,
Inc. | 306010 |
| CD7 | Beijing
Kuangbo | A6005R12 |
|
| Biotechnology Co.,
Ltd. |
|
| TCL-1 | BioLegend,
Inc. | 330506 |
| TIA-1 | Beckman Coulter,
Inc. | IM3293 |
| CD4 | BD Biosciences | 341654 |
| CD10 | Beckman Coulter,
Inc. | A07760 |
| CD1a | BD Biosciences | 560945 |
| CD99 | BD Biosciences | 555689 |
| CD33 | BD Biosciences | 664937 |
| CD13 | BD Biosciences | 557454 |
| TDT | Dako; Agilent | F713950 |
|
| Technologies,
Inc |
|
| CD5 | BD Biosciences | 665001 |
| CD117 | BD Biosciences | 664936 |
| CD34 | BioLegend,
Inc. | 343522 |
| CD38 | BioLegend,
Inc. | 303516 |
| mCD3 | BD Biosciences | 663490 |
| cCD3 | BD Biosciences | 558117 |
| CD94 | BD Biosciences | 559876 |
| CD8 | BD Biosciences | 641400 |
| CD26 | BD Biosciences | 340426 |
| CD30 | Beckman Coulter,
Inc. | IM2033U |
| CD25 | BD Biosciences | 560503 |
| GranzymeB | BioLegend,
Inc. | 515408 |
| Perforin | BioLegend,
Inc. | 308106 |
| CD161 | BioLegend,
Inc. | 339906 |
| CD304 | BioLegend,
Inc. | 354510 |
| CD45RO | BD Biosciences | 340438 |
Instrument and analysis
The flow cytometer used was the FACSCantoII
(488/633/405 nm triple laser 8 colors) flow cytometer from BD
Biosciences. Kaluza Analysis software version number
2.1.00000.20651 (Beckman Coulter, Inc.) was used. A
FSC-H-Line/FSC-A-Line dual-parameter scatter was drawn to remove
adherent cells. A FSC-A-Line/SSC-A-Log dual-parameter scatter was
drawn to remove debris. A CD45-A-Log/SSC-A-Log dual-parameter
scatter was drawn to set up a gate to circle out the nucleated
cells, and to distinguish between the normal cell population and
the abnormal cell population. According to the normal cell
population as an internal control, whether the abnormal cells
expressed the target antigen and the degree of expression were
determined. Immunophenotyping of the abnormal cell population was
performed.
Immunohistochemistry
Reagents and instruments
Reagents and instruments used are shown in Tables VI and VII.
| Table VI.Reagents for
immunohistochemistry. |
Table VI.
Reagents for
immunohistochemistry.
| Main experimental
reagents | Manufacturer | Product number |
|---|
| Anti-CD123
Polyclonal Antibody | OriGene
Technologies, Inc. | ZM-0423 |
| Anti-CD56
Polyclonal Antibody | OriGene
Technologies, Inc. | ZM-0057 |
| Anti-CD43
Polyclonal Antibody | OriGene
Technologies, Inc. | ZM-0048 |
| Anti-PDGFRA
Polyclonal Antibody | BIOSS | bs-10989R |
| Anti-IL-6
Polyclonal Antibody | BIOSS | bs-4539R |
| Anti-IL-10
Antibody | Boster Biological
Technology | BA1201-1 |
| Anti-JAK2
Polyclonal Antibody | BIOSS | bs-0908R |
| Anti-NFKB P65
Polyclonal Antibody | BIOSS | bs-0465R |
| Anti-STAT3
Polyclonal Antibody | BIOSS | bs-1141R |
| Anti-TET2
Polyclonal Antibody | BIOSS | bs-9449R |
| Anti-TGF-β1/TGFB1
Antibody | Boster Biological
Technology | BA0290 |
| Secondary
Antibody | OriGene
Technologies, Inc. | Pv-6001 |
| Wright-Giemsa Stain
composite Stain kit | BIOSS | S0217 |
| Xylene | Tianjin Yongda
Chemical Reagent Co., Ltd. | YD20220904 |
| Alcohol | Tianjin Yongda
Chemical Reagent Co., Ltd. | YD20220720 |
| Hematoxylin
Staining Solution | OriGene
Technologies, Inc. | ZLI-9610 |
| Differentiation
solution | Shanghai Yuanye
Biotechnology Co., Ltd. | R33065 |
| Bluing
Solution | Shanghai Yuanye
Biotechnology Co., Ltd. | R22272 |
| Eosin staining
solution | OriGene
Technologies, Inc. | ZLI-9613 |
| Neutral resin | OriGene
Technologies, Inc. | ZLI-9555 |
| Paraffin wax,
melting point 56–58°C | Beijing BHKT
Clinical Reagent Co., Ltd. | 007003 |
| Table VII.Instruments for
immunohistochemistry. |
Table VII.
Instruments for
immunohistochemistry.
| Main experimental
instruments | Manufacturer | Product model |
|---|
| Microscope | Olympus
Corporation | BX51T-PHD-J11 |
| CMOS | Olympus
Corporation | DP22 |
| Multi-functional
True Color Cell Image Analysis | Media Cybernetics,
Inc. | Image-Pro Plus
6.0 |
| Management
System |
|
|
| Paraffin
Slicer | Leica Microsystems
GmbH | RM2015 |
| Centrifuge | Eppendorf SE | 5430 |
| Cryogenic
refrigerator | Sanyo | MDF-382E |
| Pipette | Eppendorf SE | 3123000 |
| Constant
temperature water bath | Taicang
HuadaExperimental | DSHZ-300 |
|
| Instrument
Technology Co., Ltd. |
|
|
| (Huamei Biochemical
Instrument Factory) |
|
Methods
Bone marrow cell smear staining was performed
according to the Wright-Giemsa Composite Stain Kit instructions.
The smear was covered with Wright-Giemsa Stain and stained for 1–2
min at 20°C. A light microscope was used (BX51T-PHD-J11; Olympus
Corporation).
Immunohistochemistry
The protein expression levels of CD123, CD56, CD43,
IL-6, IL-10, JAK2, NF-κB, TET2, TGF-β1, NRAS and p-ERK in
pathological tissues were detected using immunohistochemical
staining, and the results were interpreted by senior pathologists.
The cell positivity rate was observed at a high magnification in
five fields of view.
Fixation was performed using 4% paraformaldehyde at
20°C for 1–3 days. For intracellular antigens or membrane proteins
with an internal epitope, the permeabilization reagent was 0.1%
Triton X-100. The required pathological tissue wax block was
identified in the pathology specimen bank of Hebei General Hospital
(Shijiazhuang, China), according to the pathology number of the
patient. The wax block was cooled and fixed on the slicer. For
slicing, the thickness of the tissue section was 3 µm, the cut
tissue sections were spread, taken out, attached to the poly-lysine
attached slides and baked at 60°C for 12 h. The sections were
sequentially immersed in xylene, 100, 95, 90, 85 and 75% ethanol,
and this operation was performed in each reagent two times each for
5 min each. After the sections were removed, tap water and PBS were
used to rinse the sections two times consecutively for 5 min each.
The sections were incubated in 1% methanol hydrogen peroxide,
placed at room temperature for 10 min, washed once with distilled
water and three times with 0.1 M PBS for 5 min each. The sections
were placed in 0.01 M citrate buffer (pH 6.0) and underwent
microwave radiation in a microwave oven for 10 min. After the
citrate buffer was reduced to 20°C, the sections were washed three
times with 0.1 M PBS for 5 min each. Drops of normal goat serum
sealing solution (undiluted working solution; OriGene Technologies,
Inc.) were added to the sections and the sections were left at room
temperature for 20 min. Excess liquid was shaken off without
washing. Primary antibodies were diluted 1:200. Pre-diluted primary
antibody was added dropwise to the slices, uniformly covering the
pathological tissue. The secondary antibody was ready-to-use and
did not require dilution. The sections were incubated for 12 h at
4°C, rewarmed and washed three times with 0.1 M PBS for 5 min.
Biotinylated secondary antibody (IgG) was added dropwise to the
slices. Slices were incubated at 37°C for 20 min, washed three
times with 0.1 M PBS for 5 min. Horseradish enzyme-labeled
streptavidin working solution was added dropwise to the slices.
Slices were incubated at 37°C for 20 min and washed three times
with 0.1 M PBS for 5 min. One drop each of color developer A, B and
C of the DAB color development kit (cat. no. DA1016; Beijing
Solarbio Science & Technology Co., Ltd.) was added to 1 ml
distilled water and the liquid was mixed well, then the mixed
liquid was added dropwise to the specimen and left to stand for 6
min, then the specimens were washed well with water. Cell nuclei
were re-stained with hematoxylin at 20°C for 1 min, sections were
washed thoroughly with water, 1% hydrochloric acid alcohol was
added for differentiation and 1% amine water was added for bluing,
and sections were washed thoroughly with water, dehydrated with 70%
ethanol for 5 min, 80% ethanol for 5 min, 90% ethanol for 5 min
twice, 95% ethanol for 5 min twice, 100% ethanol for 5 min twice,
cleared with xylene for 5 min twice, sealed with neutral resin and
air-dried. For light microscopy observation, five high
magnification fields of view (magnification, ×400) were selected,
the positive cells were counted, the average of the five fields of
view was taken as the average positive rate and images were
captured. Myelofibrosis was graded according to the World Health
Organization (2016) myelofibrosis grading criteria (21).
Silver nitrate methods
Fixation was performed using 4% paraformaldehyde at
20°C for 1–3 days. The thickness of the tissue section was 3 µm.
Sections were dewaxed to water and washed with distilled water.
Sections were immersed in 1% silver nitrate solution at 20°C for
30–60 min. After the slice was removed, the excess silver nitrate
solution on the edge of the slide was absorbed using filter paper,
and the sections were washed with 50% ethanol at 20°C for 5–10 sec,
and then with distilled water. The sections were placed in aqueous
gold chloride solution at 20°C for 5 min until the yellowish brown
color was removed, and then the staining was intensified by
dropping aniline oil ethanol on the sections at 20°C for ~15 sec.
After rinsing the sections with running water, the sections were
treated with 2% sodium thiosulfate solution at 20°C for 2 min.
After rinsing the sections in running water, the sections were
dehydrated, cleared with xylene and sealed with neutral resin. A
CMOS light microscope (DP22; Olympus Corporation) was used, as
shown in Table VII.
Discussion
BPDCN is an aggressive hematopoietic neoplasm
derived from plasmacytoid dendritic cells and was first reported in
1994 (27). BPDCN mainly occurs in
elderly patients aged 60–70 years with a high male proportion.
BPDCN most commonly presents with skin lesions, and bone marrow
involvement is also frequent. The clinical presentation varies
widely, and there can be either single or multiple skin lesions.
Skin lesions appear as brown or purplish-red rashes, plaques or
nodules. This is often associated with involvement of peripheral
blood, bone marrow and lymph nodes, which usually show diffuse
proliferation or infiltration of abnormal matricellular-like cells
(28). Tissue biopsies of the bone
marrow, lymph nodes and skin of the patient in the present case
report showed abnormal blastocyte-like cells. Only one case report
has been reported that found conversion of PMF to BPDCN (20), but there is no relevant literature
on MF secondary to BPDCN. Therefore, it is rare for BPDCN to occur
simultaneously with MF. MF is a Philadelphia chromosome-negative
MPN with increased deposition of collagen fibers in bone marrow
hematopoietic tissue under certain conditions, causing abnormal
hematopoiesis. The pathogenesis of MPN is unclear and is mainly
associated with three clonal mutations, JAK2, CALR and MPL, and can
lead to constitutive activation of the JAK/STAT signaling pathway.
Conversely, triple-negative PMF is not associated with mutations in
the JAK2, CALR and MPL genes, but >50% of patients with MF have
with mutations in the TET2, additional sex combs like
1-transcriptional regulator and DNA (cytosine-5-)-methyltransferase
3α genes (29). A case of
follicular lymphoma with MF was previously diagnosed at Hebei
General Hospital (Shijiazhuang, China). Several cytokines were
detected, such as basic fibroblast growth factor, TNF-α, TGF-β,
PDGF, IL-1β, IL-2, IL-6 and IL-10, which promoted the development
of non-Hodgkin's lymphoma with MF. Moreover, the results indicated
that activation of the JAK/STAT pathway may be a common mechanism
in the development of B-cell non-Hodgkin's lymphoma and secondary
MF (30).
The bone marrow biopsy pathology in the present case
revealed that reticular fiber staining was grade 2, and
next-generation sequencing of the peripheral blood demonstrated
mutations in the PDGFRA, TET2 and NRAS genes, but no mutations in
JAK2, STAT, CALR or MPL. According to the diagnostic criteria for
autoimmune MF (AIMF), the presence of positive autoantibodies is
required for AIMF, and the negative autoantibodies in the present
case, combined with the absence of previous autoimmune disease
(AIMF is usually associated with a well-defined autoimmune
disease), ruled out AIMF (31).
Cytokine assays of the peripheral blood showed elevated levels of
IL-6 and IL-10. However, immunohistochemical examination of bone
marrow and lymph nodes revealed the expression of only TET2 and
NF-κB, with a negative result for the expression of PDGFRA, IL-6,
IL-10, JAK2, STAT3 and TGF-β1. These results indicate that the
occurrence of MF in the present case may not have been caused by
activation of the classical JAK/STAT pathway. Similar to these
results, Beird et al (32)
reported that JAK/STAT signaling in BPDCN, and the levels of the
upstream and downstream molecular serum proteins (colony
stimulating factor 3 receptor, STAT3 and STAT5B) in this pathway
were low, which may be related to the high level of IL-3Rα in
BPDCN, which inhibits IL-3/STAT3 signal transduction.
MF may occur before and/or after the development of
other hematological malignancies (30,33,34).
Although the patient in the present case did not have mutations in
JAK2, CALR or MPL, another clonal proliferative marker, TET2, was
present. When MF and other hematological malignancies occur
together, it is difficult to determine whether the disorder is
triple-negative primary MF or secondary MF (30,34–37).
In the present case, we hypothesize that secondary MF was more
likely and that the primary BPDCN had invaded the bone marrow and
caused secondary MF.
The mechanism of BPDCN combined with MF was further
assessed. Somatic point mutations of TET2 and NRAS are often found
in patients with BPDCN (3). TET2
encodes an enzyme that catalyzes 5-methyl cytosine oxidation to
5-hydroxymethylcytosine. When TET2 is mutated, it leads to loss of
function of this enzyme and DNA hypermethylation, which
subsequently amplifies precancerous clones of bone marrow and
abnormal hematopoiesis, driving MPN development (38–41).
Ostrander et al (42)
reported that the TET2 deletion mutation increases dendritic cell
production, increases hematopoietic stem cell self-renewal and
polarizes toward the myeloid lineage. NRAS is a member of the RAS
family, and mutations in NRAS facilitate GTP binding of RAS,
thereby promoting constitutive activation of the
RAS-RAF-MEK-ERK/MAPK signaling pathway and causing abnormal
proliferation of myeloid cells (41,43).
This may be involved in the occurrence of MF. As specimens from the
patient in the present case were limited, only the expression of
NRAS and p-ERK was assessed, and it was found that both NRAS and
p-ERK were expressed in the bone marrow and lymph nodes of the
patient. This indicates that the mutations in TET2 and NRAS may be
the bridge between BPDCN and MF, but whether RAS-related pathways
are involved requires further experiments for verification.
Although mutations in TET2 and NRAS may be the
potential pathological associating factors between BPDCN and MF, it
may also be a unique coincidence in the patient in the present
case. In addition to TET2 and NRAS, other mutated genes in the
patient appeared to be indirectly associated with the development
of BPDCN or MF. It has been reported that loss of function in DIS3
increases the RAS protein level in multiple myeloma cells, which
promotes the development of cancer cells (44). CSMD1 is a tumor suppressor gene, and
dysregulation of CSMD1 can drive the NF-κB pathway and promote
tumor progression (45). B-cell
lymphoma 6 corepressor (BCOR) mutations occur after mutations
affecting splicing mechanisms or epigenetically regulated genes
(46). Patients with adult acute
myeloid leukemia with BCOR mutations are predicted to have a
complete response to venetoclax plus hypomethylating agents
(47), and BCOR has been reported
to respond better to hypomethylating drugs when it is co-mutated
with other genes that regulate epigenetic inheritance, such as TET2
(48). PDGFRA mutations or
rearrangements serve an important role in the development of
hypereosinophilic syndrome and its mutation sites (H650Q, N659S,
R748G and Y849S). Additionally, FIP1L1-PDGFRA has been reported to
induce the activation of STAT5 and sensitivity to imatinib
(49). Abdulbaki et al
(50) reported a case of BPDCN
presenting with a PDGFRA mutation and suggested the possibility of
tyrosine kinase inhibitors for the treatment of BPDCN, but further
studies are lacking. Overall, all of these mutations may be
indirectly associated with the development of BPDCN or MF, and thus
more future cases are needed for further study.
Personalized treatment based on the immune phenotype
and mutation profile of a patient is important. CD123 is a
plasmacytoid dendritic cell-specific surface marker that is
prevalent in BPDCN, and tagraxofusp targets CD123. Macrophage
dendritic cell progenitors are common precursor cells of
plasmacytoid dendritic cells and monocytes (51), whereas tumorigenic monocyte-derived
fibroblasts contribute to myeloid cell fibrosis (52). A small number of circulating cells
have been reported to express CD123 in patients with MF (53), which can be targeted to treat BPDCN
in conjunction with MF. Clinical studies of tagraxofusp monotherapy
for MF are currently underway (trial registration nos. NCT0226825
and NCT05233618). Additionally, clinicians can target therapy in
accordance with other specific immunophenotypes of BPDCN, such as
targeting CD303 (litifilimab) (54), TCF4 (bromodomain and extra-terminal
domain inhibitors act by inhibiting TCF4) (55) and BCL2 (venetoclax) (56). BPDCN is closely related to DNA
hypermethylation (57). In the
present case, the patient presented with a TET2 mutation and was
treated with the DNA methyltransferase inhibitor, azacytidine, in
combination with venetoclax. Certain cases of BPDCN have been
treated with azacitidine in combination with venetoclax and it was
well tolerated (58,59). Moreover, azacitidine combined with
tagraxofusp has been reported to be effective for BPDCN treatment,
with azacitidine restoring sensitivity to tagraxofusp by restoring
diphthamide biosynthesis protein 1 expression (60). In terms of the NRAS mutation in the
patient in the present case, an RAF or an ERK inhibitor may have
been appropriate for use (61).
Furthermore, several retrospective clinical studies
have reported that achieving complete remission after first-line
therapy is the primary condition for long-term survival of patients
with BPDCN and that survival is prolonged by allogeneic
hematopoietic cell transplantation during the first complete
remission (4). Patients with MF
have reduced blood cell counts, and in the context of malignancy,
patients with MF are more (62)
likely to have myelosuppression after chemotherapy (63,64).
The patient in the present case had induced myelosuppression after
chemotherapy with poor efficacy, along with a severe infection,
which finally led to the poor outcome. Therefore, BPDCN combined
with MF may represent a poor prognosis with a limited therapeutic
effect. For such patients, physicians need to be particularly
mindful of infections associated with myelosuppression during
chemotherapy. While preventing or fighting infection, the selection
of reasonable chemotherapeutic drugs and doses should be focused
on.
In conclusion, patients with BPDCN and MF have a
poor prognosis, and mutations in TET2 and NRAS may be the bridge
between the two conditions. The findings of the present case
indicate the need for increased awareness of BPDCN among clinicians
in order to reduce misdiagnosis and improve the prognosis of
patients with BPDCN, to ensure appropriate and individualized
chemotherapeutic agents for the treatment of patients with BPDCN
and MF, and to increase awareness of potentially fatal infections
that may occur after myelosuppression during chemotherapy.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was financially supported by the Department of
Science and Technology of Hebei Province Plan Project (Priority
Research and Development Project; grant no. 18277720D).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the National Center for
Biotechnology Information database, https://www.ncbi.nlm.nih.gov/sra/SRR26895553.
Authors' contributions
YL made decisions regarding patient treatment. FL
and YL were accountable for all aspects of the work and contributed
to the analysis and interpretation of the data, and also
contributed to manuscript drafting and critical revisions of the
intellectual content. BL and JL analyzed and interpreted the data,
and contributed to the Discussion section. FL, BL, JL and YL
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent was obtained from the
patient for the publication of anonymized data and any accompanying
images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Guru Murthy GS, Pemmaraju N and Atallah E:
Epidemiology and survival of blastic plasmacytoid dendritic cell
neoplasm. Leuk Res. 73:21–23. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pagano L, Valentini CG, Pulsoni A, Fisogni
S, Carluccio P, Mannelli F, Lunghi M, Pica G, Onida F, Cattaneo C,
et al: Blastic plasmacytoid dendritic cell neoplasm with leukemic
presentation: An Italian multicenter study. Haematologica.
98:239–246. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jain A and Sweet K: Blastic plasmacytoid
dendritic cell neoplasm. J Natl Compr Canc Netw. 21:515–521. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garnache-Ottou F, Vidal C, Biichlé S,
Renosi F, Poret E, Pagadoy M, Desmarets M, Roggy A, Seilles E,
Soret L, et al: How should we diagnose and treat blastic
plasmacytoid dendritic cell neoplasm patients? Blood Adv.
3:4238–4251. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khoury JD, Solary E, Abla O, Akkari Y,
Alaggio R, Apperley JF, Bejar R, Berti E, Busque L, Chan JKC, et
al: The 5th edition of the World Health Organization classification
of haematolymphoid tumours: Myeloid and histiocytic/dendritic
neoplasms. Leukemia. 36:1703–1719. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Facchetti F, Cigognetti M, Fisogni S,
Rossi G, Lonardi S and Vermi W: Neoplasms derived from plasmacytoid
dendritic cells. Mod Pathol. 29:98–111. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pemmaraju N, Kantarjian H, Sweet K, Wang
E, Senapati J, Wilson NR, Konopleva M, Frankel AE, Gupta V, Mesa R,
et al: North American blastic plasmacytoid dendritic cell neoplasm
consortium: Position on standards of care and areas of need. Blood.
141:567–578. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yun S, Chan O, Kerr D, Vincelette ND,
Idrees A, Mo Q, Sweet K, Lancet JE, Kharfan-Dabaja MA, Zhang L and
Sokol L: Survival outcomes in blastic plasmacytoid dendritic cell
neoplasm by first-line treatment and stem cell transplant. Blood
Adv. 4:3435–3442. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Taylor J, Haddadin M, Upadhyay VA, Grussie
E, Mehta-Shah N, Brunner AM, Louissaint A Jr, Lovitch SB, Dogan A,
Fathi AT, et al: Multicenter analysis of outcomes in blastic
plasmacytoid dendritic cell neoplasm offers a pretargeted therapy
benchmark. Blood. 134:678–687. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang L and Wang F: Primary blastic
plasmacytoid dendritic cell neoplasm: A US population-based study.
Front Oncol. 13:11781472023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murthy HS, Zhang MJ, Chen K, Ahmed S,
Deotare U, Ganguly S, Kansagra A, Michelis FV, Nishihori T, Patnaik
M, et al: Allogeneic hematopoietic cell transplantation for blastic
plasmacytoid dendritic cell neoplasm: A CIBMTR analysis. Blood Adv.
7:7007–7016. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tefferi A: Primary myelofibrosis: 2023
Update on diagnosis, risk-stratification, and management. Am J
Hematol. 98:801–821. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Titmarsh GJ, Duncombe AS, McMullin MF,
O'Rorke M, Mesa R, De Vocht F, Horan S, Fritschi L, Clarke M and
Anderson LA: How common are myeloproliferative neoplasms? A
systematic review and meta-analysis. Am J Hematol. 89:581–587.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cervantes F, Dupriez B, Pereira A,
Passamonti F, Reilly JT, Morra E, Vannucchi AM, Mesa RA, Demory JL,
Barosi G, et al: New prognostic scoring system for primary
myelofibrosis based on a study of the international working group
for myelofibrosis research and treatment. Blood. 113:2895–2901.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McLornan DP, Psaila B, Ewing J, Innes A,
Arami S, Brady J, Butt NM, Cargo C, Cross NCP, Francis S, et al:
The management of myelofibrosis: A British society for haematology
guideline. Br J Haematol. 204:136–150. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tashi T, Yu J, Pandya S, Dieyi C, Scherber
R and Parasuraman S: Trends in overall mortality among US veterans
with primary myelofibrosis. BMC Cancer. 23:482023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thiele J and Kvasnicka HM:
Myelofibrosis-what's in a name? Consensus on definition and EUMNET
grading. Pathobiology. 74:89–96. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang N, Xu H, Li Q, Fang X, Liu J, Sui X,
Zhang L, Jiang Y and Wang X: Patients of myelodysplastic syndrome
with mild/moderate myelofibrosis and a monosomal karyotype are
independently associated with an adverse prognosis: Long-term
follow-up data. Cancer Manag Res. 12:5881–5891. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shehata M, Schwarzmeier JD, Hilgarth M,
Hubmann R, Duechler M and Gisslinger H: TGF-beta1 induces bone
marrow reticulin fibrosis in hairy cell leukemia. J Clin Invest.
113:676–685. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shenjere P, Chasty R, Chaturvedi A, Dennis
MW, Ong A, Wiseman DH and Menasce LP: E-cadherin expression in
blastic plasmacytoid dendritic cell neoplasms: An unrecognized
finding and potential diagnostic pitfall. Int J Surg Pathol.
29:289–293. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Arber DA, Orazi A, Hasserjian R, Thiele J,
Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M and Vardiman JW:
The 2016 revision to the World Health Organization classification
of myeloid neoplasms and acute leukemia. Blood. 127:2391–2405.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen S, Zhou Y, Chen Y and Gu J: Fastp: An
ultra-fast all-in-one fastq preprocessor. Bioinformatics.
34:i884–i890. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Thorvaldsdóttir H, Robinson JT and Mesirov
JP: Integrative genomics viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Talevich E, Shain AH, Botton T and Bastian
BC: CNVkit: Genome-wide copy number detection and visualization
from targeted DNA sequencing. PLoS Comput Biol. 12:e10048732016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ye K, Guo L, Yang X, Lamijer EW, Raine K
and Ning Z: Split-read indel and structural variant calling using
pindel. Methods Mol Biol. 1833:95–105. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tung JK, Suarez CJ, Chiang T, Zehnder JL
and Stehr H: Accurate detection and quantification of FLT3 internal
tandem duplications in clinical hybrid capture next-generation
sequencing data. J Mol Diagn. 23:1404–1413. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Adachi M, Maeda K, Takekawa M, Hinoda Y,
Imai K, Sugiyama S and Yachi A: High expression of CD56 (N-CAM) in
a patient with cutaneous CD4-positive lymphoma. Am J Hematol.
47:278–282. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Julia F, Dalle S, Duru G, Balme B, Vergier
B, Ortonne N, Vignon-Pennamen MD, Costes-Martineau V, Lamant L,
Dalac S, et al: Blastic plasmacytoid dendritic cell neoplasms:
Clinico-immunohistochemical correlations in a series of 91
patients. Am J Surg Pathol. 38:673–680. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Arber DA, Orazi A, Hasserjian RP, Borowitz
MJ, Calvo KR, Kvasnicka HM, Wang SA, Bagg A, Barbui T, Branford S,
et al: International consensus classification of myeloid neoplasms
and acute leukemias: Integrating morphologic, clinical, and genomic
data. Blood. 140:1200–1228. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kong LZ, Li J, Wang RC, Kang L, Wei Q and
Li Y: Simultaneous follicular lymphoma and myelofibrosis: Report of
a case with review of the literature. Onco Targets Ther.
14:4551–4559. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Amel Riazat-Kesh YJR, Maraveyas A, Martin
L and Tremblay D: An overlooked mimic? Autoimmune myelofibrosis-A
scoping review of the literature. Eur J Haematol. 111:706–714.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Beird HC, Khan M, Wang F, Alfayez M, Cai
T, Zhao L, Khoury J, Futreal PA, Konopleva M and Pemmaraju N:
Features of non-activation dendritic state and immune deficiency in
blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood Cancer
J. 9:992019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tabata R, Tabata C, Nagai T and Yasumizu
R: Follicular lymphoma with prominent fibrosis complicated by
peripheral eosinophilia. Ann Hematol. 91:965–967. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Scherber RM and Mesa RA: Managing
myelofibrosis (MF) that ‘blasts’ through: Advancements in the
treatment of relapsed/refractory and blast-phase MF. Hematology Am
Soc Hematol Educ Program. 2018:118–126. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Okabe S, Miyazawa K, Iguchi T, Sumi M,
Takaku T, Ito Y, Ito Y, Kimura Y, Serizawa H, Mukai K and Ohyashiki
K: Peripheral T-cell lymphoma together with myelofibrosis with
elevated plasma transforming growth factor-beta1. Leuk Lymphoma.
46:599–602. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tsutsui M, Yasuda H, Ota Y and Komatsu N:
Splenic marginal zone lymphoma with prominent myelofibrosis
mimicking triple-negative primary myelofibrosis. Case Rep Oncol.
12:834–837. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Matsunaga T, Takemoto N, Miyajima N, Okuda
T, Nagashima H, Sato T, Terui T, Sasaki H, Ohmi N, Hirayama Y, et
al: Splenic marginal zone lymphoma presenting as myelofibrosis
associated with bone marrow involvement of lymphoma cells which
secrete a large amount of TGF-beta. Ann Hematol. 83:322–325. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakajima H and Kunimoto H: TET2 as an
epigenetic master regulator for normal and malignant hematopoiesis.
Cancer Sci. 105:1093–1099. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Abdel-Wahab O, Mullally A, Hedvat C,
Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara
O, Bhat R, et al: Genetic characterization of TET1, TET2, and TET3
alterations in myeloid malignancies. Blood. 114:144–147. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Delhommeau F, Dupont S, Della Valle V,
James C, Trannoy S, Massé A, Kosmider O, Le Couedic JP, Robert F,
Alberdi A, et al: Mutation in Tet2 in myeloid cancers. N Eng J Med.
360:2289–2301. 2009. View Article : Google Scholar
|
|
41
|
Vainchenker W and Kralovics R: Genetic
basis and molecular pathophysiology of classical myeloproliferative
neoplasms. Blood. 129:667–679. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ostrander EL, Kramer AC, Mallaney C, Celik
H, Koh WK, Fairchild J, Haussler E, Zhang CRC and Challen GA:
Divergent effects of Dnmt3a and Tet2 mutations on hematopoietic
progenitor cell fitness. Stem Cell Reports. 14:551–560. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Schubbert S, Shannon K and Bollag G:
Hyperactive Ras in developmental disorders and cancer. Nat Rev
Cancer. 7:295–308. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ohguchi Y and Ohguchi H: Dis3: The
enigmatic gene in multiple myeloma. Int J Mol Sci. 24:40792023.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen XL, Hong LL, Wang KL, Liu X, Wang JL,
Lei L, Xu ZY, Cheng XD and Ling ZQ: Deregulation of CSMD1 targeted
by microRNA-10b drives gastric cancer progression through the NF-κB
pathway. Int J Biol Sci. 15:2075–2086. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Damm F, Chesnais V, Nagata Y, Yoshida K,
Scourzic L, Okuno Y, Itzykson R, Sanada M, Shiraishi Y, Gelsi-Boyer
V, et al: BCOR and BCORL1 mutations in myelodysplastic syndromes
and related disorders. Blood. 122:3169–3177. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sportoletti P, Sorcini D and Falini B:
BCOR gene alterations in hematologic diseases. Blood.
138:2455–2468. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Badaat I, Mirza S, Padron E, Sallman D,
Komrokji R, Song J and Hussaini MO: Concurrent mutations in other
epigenetic modulators portend better prognosis in BCOR-mutated
myelodysplastic syndrome. J Clin Pathol. 73:209–212. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Elling C, Erben P, Walz C, Frickenhaus M,
Schemionek M, Stehling M, Serve H, Cross NC, Hochhaus A, Hofmann
WK, et al: Novel imatinib-sensitive PDGFRA-activating point
mutations in hypereosinophilic syndrome induce growth factor
independence and leukemia-like disease. Blood. 117:2935–2943. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Abdulbaki R, DeRosa PA, Mobarek D, Liu ML
and Nava VE: Novel PDGFRA mutation in blastic plasmacytoid
dendritic cell neoplasm; possible therapeutic implications. Br J
Haematol. 197:82022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Collin M and Bigley V: Human dendritic
cell subsets: An update. Immunology. 154:3–20. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Verstovsek S, Manshouri T, Pilling D,
Bueso-Ramos CE, Newberry KJ, Prijic S, Knez L, Bozinovic K, Harris
DM, Spaeth EL, et al: Role of neoplastic monocyte-derived
fibrocytes in primary myelofibrosis. J Exp Med. 213:1723–1740.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tremblay D and Mascarenhas J: Next
generation therapeutics for the treatment of myelofibrosis. Cells.
10:10342021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Werth VP, Furie RA, Romero-Diaz J, Navarra
S, Kalunian K, van Vollenhoven RF, Nyberg F, Kaffenberger BH,
Sheikh SZ, Radunovic G, et al: Trial of anti-BDCA2 antibody
litifilimab for cutaneous lupus erythematosus. N Engl J Med.
387:321–331. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ceribelli M, Hou ZE, Kelly PN, Huang DW,
Wright G, Ganapathi K, Evbuomwan MO, Pittaluga S, Shaffer AL,
Marcucci G, et al: A druggable TCF4- and BRD4-dependent
transcriptional network sustains malignancy in blastic plasmacytoid
dendritic cell neoplasm. Cancer Cell. 30:764–778. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Montero J, Stephansky J, Cai T, Griffin
GK, Cabal-Hierro L, Togami K, Hogdal LJ, Galinsky I, Morgan EA,
Aster JC, et al: Blastic Plasmacytoid dendritic cell neoplasm is
dependent on BCL2 and sensitive to venetoclax. Cancer Discov.
7:156–164. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sapienza MR, Abate F, Melle F, Orecchioni
S, Fuligni F, Etebari M, Tabanelli V, Laginestra MA, Pileri A,
Motta G, et al: Blastic plasmacytoid dendritic cell neoplasm:
Genomics mark epigenetic dysregulation as a primary therapeutic
target. Haematologica. 104:729–737. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Azad F, Zhang J, Miranda CJ and Gravina M:
Venetoclax and Azacitidine in the treatment of blastic plasmacytoid
dendritic cell neoplasm refractory to conventional therapy. Cureus.
14:e331092022.PubMed/NCBI
|
|
59
|
Samhouri Y, Ursu S, Dutton N, Tanvi V and
Fazal S: Tagraxofusp followed by combined azacitidine and
venetoclax in blastic plasmacytoid dendritic cell neoplasm: A case
report and literature review. J Oncol Pharm Pract. 27:990–995.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Togami K, Pastika T, Stephansky J, Ghandi
M, Christie AL, Jones KL, Johnson CA, Lindsay RW, Brooks CL, Letai
A, et al: DNA methyltransferase inhibition overcomes diphthamide
pathway deficiencies underlying CD123-targeted treatment
resistance. J Clin Invest. 129:5005–5019. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Moore AR, Rosenberg SC, McCormick F and
Malek S: RAS-targeted therapies: Is the undruggable drugged? Nat
Rev Drug Discov. 19:533–552. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lyman GH, Poniewierski MS and Culakova E:
Risk of chemotherapy-induced neutropenic complications when
treating patients with non-Hodgkin lymphoma. Expert Opin Drug Saf.
15:483–492. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Dinan MA, Hirsch BR and Lyman GH:
Management of chemotherapy-induced neutropenia: Measuring quality,
cost, and value. J Natl Compr Canc Netw. 13:e1–e7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Epstein RS, Aapro MS, Basu Roy UK, Salimi
T, Krenitsky J, Leone-Perkins ML, Girman C, Schlusser C and
Crawford J: Patient burden and real-world management of
chemotherapy-induced myelosuppression: Results from an online
survey of patients with solid tumors. Adv Ther. 37:3606–3618. 2020.
View Article : Google Scholar : PubMed/NCBI
|