Introduction
Ferroptosis, first identified and reported by Dixon
et al in 2012 (1), is a cell
death process depending on iron and reactive oxygen species (ROS)
that differs from apoptosis, necrosis and autophagy. It is
characterized by a redox imbalance, elevated level of ROS, rupture
of the outer mitochondrial membrane, dense contents and the
decrease or loss of cristae of mitochondria (2). Previous studies have shown that
ferroptosis may be induced in both humans and animals under a
variety of physiological conditions and pathological stresses. A
number of cancer-related signaling pathways have been identified to
be associated with ferroptosis, and some cancer cells even relied
on ferroptosis defenses for survival. In recent years, the
discovery of new ferroptosis biomarkers, such as nicotinamide
adenine dinucleotide phosphate (NADPH), hyperoxidized PRDX3, 7-DHC,
has contributed to a clearer understanding of ferroptosis and the
application of ferroptosis in the prevention and treatment of
related diseases. Particularly, ferroptosis has been revealed to be
a congenital tumor suppressor mechanism that mediates the
anticancer activity of multiple tumor suppressors (3–5). Based
on such important findings, how to regulate cellular ferroptosis to
intervene in the occurrence and development of cancer has become a
current research focus which may provide new therapeutic clues to
combat this disease.
Ferroptosis mechanism
Iron metabolism
Iron is one of the essential trace minerals of the
body (6). It plays an essential
role in the oxidation-reduction catalysis and bioenergetics of the
cells, and is also involved in the formation of toxic oxygen
radicals. A previous study demonstrated that a high consumption of
iron raises the risk of several cancer forms, including
hepatocellular carcinoma and breast cancer (7). Compared with normal cells, cancer
cells require more iron for proliferation. Accordingly, iron uptake
is expedited, and their intracellular concentration is raised in
rapidly proliferating cancer cells (8). Although it has not yet been fully
understood as regard to how iron metabolism plays a role in the
process of ferroptosis, it is well known to be critical for
ferroptosis. According to Dixon et al (1), extracellular iron supplementation
rendered cells more vulnerable to ferroptosis inducers, and iron
complexes prevented the development of ferroptosis in cells both
in vivo and in vitro. Hou et al (9) reported a rise of intracellular active
iron concentration during the activation of ferroptosis in cells.
In 2017, Li et al (10)
found that both extra heme and non-heme iron could cause
ferroptosis in cells. Thus, triggering iron-dependent ferroptosis
by modulation of diverse active iron forms could provide a novel
cancer therapeutic strategy.
Ferroptosis is a type of cell death that is caused
by an increase in the labile iron pool in cells (11). The main mechanism of iron uptake in
cells is through the binding of Fe3+ to transferrin (TF)
and its recognition by the TF receptor (TFRC) to enter the cell.
Subsequently, Fe3+ is separated from TF in the nuclear
endosome, reduced to Fe2+ by six-transmembrane
epithelial antigen of prostate 3, and then released into the
cytoplasm via solute carrier family 39 member 8 (SLC39A8), SLC39A14
(zinc transporter) and divalent metal transporter 1. In addition,
two other mechanisms, endocytosis of lacto-TF and heme-mediated
iron absorption, provide additional iron sources for cells
(12). On the other hand,
SLC40A1-mediated iron export and exosome-mediated ferritin export
are primarily mechanisms of iron export (13). Ferroptosis could be induced through
modulation on multiple stages of iron metabolism, such as,
increasing iron uptake, decreasing iron storage, or restricting
iron ion efflux, mediated by the corresponding signaling pathways,
which all lead to the augmentation of intracellular iron ion
content and induce ferroptosis (14). At least two processes cause membrane
lipid peroxidation when there is excessive iron in cells. One of
them involves the Fenton reaction, which produces hydroxyl radicals
and takes part in the peroxidation of phospholipids, converting
non-toxic phosphatidyl alcohol into harmful phospholipid
hydroperoxides (PLOOHs). In the other process, extra iron activates
iron-containing enzymes such as lipoxygenase, to directly catalyze
the deoxygenation of free and esterified polyunsaturated fatty
alcohol and generate PLOOH (2,15).
Additionally, excess iron will catalyze the creation of ROS in
cells, triggering lipid peroxidation and eventually
ferroptosis.
Ferritin, a protein complex that stores iron,
prevents ROS from oxidizing Fe2+ and is essential for
maintaining iron levels in cells. Ferritin is primarily made up of
the ferritin heavy chain 1 (FTH1) and the ferritin light chain
(FTL); the former is primarily in charge of reducing
Fe3+ to Fe2+ at the iron oxidase active site,
whereas the latter serves to stabilize ferritin. In 2016, Hou et
al (9) demonstrated that
autophagy can induce ferroptosis by degrading ferritin in
fibroblasts and cancer cells. Gao et al (16) discovered that deleting
autophagy-associated proteins ATG3, ATG5, ATG7 and ATG13 prevented
ferroptosis. Although it was previously suggested that autophagy is
unrelated to ferroptosis, autophagy-dependent ferroptosis has been
observed in cancer cells. Furthermore, nuclear receptor coactivator
4-mediated ferritin autophagy raised intracellular Fe2+
levels and accelerated cellular ferroptosis. Kremer et al
(17) showed that inhibiting
glutamate-oxaloacetate transaminase 1 (GOT1) increased ferritin
autophagy, causing an increase in labile iron pools and promoting
ferroptosis in cells.
Iron-sulfur clusters (ISCs) are crucial cofactors in
the maintenance of redox and iron homeostasis. They are primarily
produced by a number of mitochondrial proteins involved in cysteine
desulfurase (NFS1) and ISC assembly, and their associated
regulatory pathways can assist cancer cells in avoiding ferroptosis
by lowering the level of labile iron pools. The iron-sulfur protein
family member CDGSH iron-sulfur domain 2 (CISD2) is highly
expressed in head and neck cancer. According to a previous study
(18), CISD2 may prevent
ferroptosis in cancer cells in vitro by controlling ISCs and
lowering the concentration of free iron. NFS1 is necessary for the
progression of lung cancer in vivo. The ability of NFS1 to
engage in the creation of ISCs, resulting in lower levels of labile
iron pools and protecting cancer cells from ferroptosis, may be
connected to its overexpression in lung cancer. Alvarez et
al (19) also demonstrated that
inhibition of NFS1 increases the intracellular level of labile iron
pools, making lung cancer cells susceptible to ferroptosis and thus
limiting the development of lung cancer in vivo. Moreover,
when under low ISC conditions, iron-sulfur regulatory proteins
(IRPs) such as IRP1 (also known as ACO1) and IRP2 (also known as
IREB2) (20), will translationally
regulate iron metabolism-related proteins, including TFRC, SLC11A2,
SLC40A1, FTH and FTL, raising iron levels in the cell and thus
promoting ferroptosis (14).
Lipid peroxidation
Ferroptosis is often characterized by high levels of
lipid peroxides. The accumulation of lipid peroxides in the cell
membrane change the membrane permeability and stability, and
provoke the cell membrane ruptures, finally leading to cell
ferroptosis (21). Acyl-CoA
synthase long chain family member 4 (ACSL4) and
lyso-phosphatidyl-choline acyltransferase 3 (LPCAT3), are
considered to be key mediators of PUFA-PL synthesis. The
accumulation of PLOOH relies on both enzymatic and non-enzymatic
processes. In enzymatic reactions, PLOOH production is mainly
mediated by lipoxygenases (LOXs) and cytochrome P450
oxidoreductases (POR) (11). LOXs
can directly catalyze the deoxygenation of free and esterified
PUFA-PL to PLOOH (22). Shah et
al (23) demonstrated that
LOX-5, LOX-12 and LOX-15 overexpression render cells more
susceptible to ferroptosis. It was also demonstrated that LOX
inhibitors effectively act as an antioxidant and protect cells from
lipid peroxidation. A previous study revealed that P450 can accept
electrons from POR and thus catalyze the peroxidation of PUFAs
(24). In 2020, Zou et al
(25) discovered that POR coupled
to cytochrome P450 generates enhanced levels of lipid peroxidation
as well as increased ROS. The aforementioned study provided
evidence that POR is necessary for ferroptosis to occur in cancer
cells. On the other hand, the non-enzymatic mechanism has been
reported that ASCL4 catalyzes the binding of free PUFAs to coenzyme
A (CoA) to produce PUFA-CoA, which is subsequently re-esterified by
LPCAT3 and bound to PLs to form PUFA-PLs, and finally catalyzed by
arachidonic LOX (ALOX) to produce PL-PUFA-OOH (26,27).
In addition, NADPH oxidases (NOXs), which catalyze the formation of
superoxide, contribute to the iron-dependent accumulation of lipid
peroxidation (1,28,29).
In the latest research report, 7-dehydrocholesterol (7-DHC), an
intermediate metabolite of distal cholesterol biosynthesis, plays
an important role in regulating ferroptosis. In a non-enzymatic
reaction, 7-DHC exerts its anti-phospholipid autooxidation function
through the use of conjugated diene, and protects plasma and
mitochondrial membrane from phospholipid autooxidation, inhibiting
the occurrence of ferroptosis in cells. Accordingly, when the
biosynthesis of endogenous 7-DHC is blocked by drug targeting, the
reduction of 7-DHC induced ferroptosis of the cells (30).
Ferroptosis defense mechanisms
Glutathione peroxidase 4 (GPX4)-glutathione
(GSH)
GPX4 is a member of the GPX family of proteins that
are found mainly in the cytoplasm, mitochondria and nucleus of
cells (31). A previous study
suggested that only cytoplasmic GPX4 plays a role in ferroptosis
(32), but a recent study found
that mitochondrial GPX4 also has an important role in ferroptosis
(33). GSH is a tripeptide composed
of glutamate, cysteine and glycine and is synthesized mainly by
glutamate-cysteine ligase (GCL) and glutathione synthase (GSS)
catalytic synthesis. Cysteine is the rate-limiting amino acid for
GSH synthesis. Cells mainly exchange intracellular glutamate for
extracellular cystine through System Xc at a ratio of 1:1, and then
reduce the imported cystine to cysteine, thus allowing the
synthesis of GSH (34). System Xc
is composed of the light chain of solute carrier family 7 member 11
(SLC7A11) and the heavy chain of solute carrier family 3 member 2
(SLC3A2). Inhibiting its expression can also induce ferroptosis
(35). In cells, GPX4 is able to
use glutathione as a substrate to reduce toxic PL-PUFA-OOH to
non-toxic PL-PUFA-OH, thereby inhibiting the occurrence of
ferroptosis (36). The inactivation
of GPX4 also leads to dysregulation of cellular REDOX and
ultimately results in the occurrence of cellular ferroptosis
(37). A recent study has
discovered that hyperoxidized PRDX3 serves as a specific marker for
ferroptosis and induces ferroptosis in cells by inhibiting cystine
uptake. During the process of ferroptosis, PRDX3 becomes
overoxidized due to exposure to mitochondrial lipid peroxides.
Subsequently, the over-oxidized PRDX3 translocates from
mitochondria to the plasma membrane, hindering cell uptake of
cystine, impacting GSH synthesis, and thereby promoting ferroptosis
(38). In cases where there is
cystine starvation or deficiency, certain tumor cells can prevent
ferroptosis by converting methionine into cysteine through the
transamination pathway (39).
However, it remains unclear whether metabolites produced by amino
acids are independently involved in ferroptosis apart from the
cysteine pathway. A recent study has found that tryptophan
metabolites serotonin (5-HT) and 3-hydroxyanthranilic acid (3-HA)
significantly inhibit ferroptosis in tumor cells. Mechanistically,
both 5-HT and 3-HA act as potent free radical trapping
antioxidants, effectively eliminating lipid peroxidation and thus
inhibiting the occurrence of ferroptosis (40).
Ferroptosis suppressor protein 1
(FSP1)-panthethine alcohol (CoQ10H2)
FSP1, also known as flavoprotein apoptosis-inducing
factor mitochondrial-associated 2, was originally considered to be
a pro-apoptotic gene (41), but it
was previously found that this gene is associated with ferroptosis
and that it can inhibit ferroptosis caused by GPX4 deletion
(42). The reduced form of coenzyme
Q (CoQ10), CoQ10H2, is a potent
lipophilic antioxidant that is considered to trap oxygen radicals
in phospholipids and lipoproteins (43). It was revealed that the plasma
membrane protein FSP1 may catalyze the conversion of
CoQ10 to CoQ10H2 using NAD(P)H to
trap free radicals of lipid peroxidation, alleviating cellular
lipid peroxidation and preventing ferroptosis (43). Although CoQ10 is mainly
synthesized in mitochondria, its presence can also be detected in
non-mitochondrial membranes, such as the plasma membrane (44,45).
Therefore, it has been hypothesized that FSP1 exerts its role in
inhibiting ferroptosis by generating CoQ10H2
in non-mitochondrial membranes, thereby trapping the free radicals
of lipid peroxidation (42,46). In conclusion, the
FSP1-CoQ10-NAD(P)H pathway can efficiently prevent the
occurrence of ferroptosis and may have a synergistic effect with
GPX4-GSH mechanism.
Dihydroorotate dehydrogenase
(DHODH)-CoQ10H2
In addition to the two major ferroptosis defense
systems aforementioned, the DHODH-CoQ10H2
system was identified by Mao et al (33) as the third one. By using metabolomic
research, the authors discovered that treatment of cancer cells
with GPX4 inhibitors, such as RSL3 or ML162, leads to the depletion
of N-carbamoyl-L-aspartate accompanied by uridine accumulation in
cancer cells. Supplementation of cells with dihydroorotate, the
substrate of mitochondrial DHODH, resists the inhibitory effect of
GPX4; whereas supplementation of its product, orotic acid, enhances
the sensitivity of cells to GPX4 inhibitors, suggesting that DHODH
may be involved in the regulation of cellular ferroptosis. It was
identified that the inhibition of DHODH sensitizes HT-1080
GPX4-high cancer cells to ferroptosis while significantly induces
ferroptosis in GPX4-low cancer cells. This research conclusively
demonstrated that DHODH inhibits ferroptosis and mitochondrial
lipid peroxidation in a CoQ10-dependent way. DHODH is an
enzyme that can take part in pyrimidine production and is primarily
present on the inner membrane surface of mitochondria (47). When GPX4 is inhibited or inactivated
in mitochondria, the level of DHODH increases significantly and is
able to reduce CoQ10 in the inner mitochondrial membrane
to CoQ10H2, thus neutralizing excessive lipid
peroxidation in mitochondria and protecting cells from ferroptosis
(33). Therefore, the discovery of
this pathway offers a fresh concept and method for cancer therapy,
with the inhibitors of DHODH as inducers of ferroptosis.
GTP cyclization hydrolase 1
(GCH1)-Tetrahydrobiopterin (BH4)
BH4 is a crucial cofactor for enzymes that
hydroxylate aromatic amino acids and has a redox function in the
processes of enzymes catalysis (48). Additionally, BH4 is a potent
antioxidant that may capture peroxyl radicals in lipids, but its
ability to inhibit ferroptosis is unrelated to its activity as an
aromatic amino acid hydroxylase cofactor (49). GCH1 is not only the initiating
enzyme in the GTP synthesis BH4 pathway, but also the rate-limiting
enzyme in the BH4 synthesis pathway, controlling the concentration
of intracellular BH4 (48,50). It was discovered that GCH1 was able
to prevent ferroptosis by starting the synthesis of
dihydrobiopterin and tetrahydrobiopterin, as well as by selectively
preventing two polyunsaturated fatty acyl tails from consuming
phospholipids. These actions trap lipid peroxidation radicals in
cells and shield some phospholipids from peroxidation (51). Meanwhile, BH4 can also promote the
production of CoQ10 by converting phenylalanine to
tyrosine, which is further converted into a precursor substance of
CoQ10, thus exerting an antioxidant function in another
way. In addition, the recycling of BH4 requires the involvement of
dihydrofolate reductase (DHFR). Therefore, blocking DHFR combined
with GPX4 inhibitor may also play a role in inducing
ferroptosis.
Membrane-bound O-acyltransferase
domain 2 (MBOAT1/2)-mediated cytosolic phospholipid remodeling
pathway
MBOAT2 is a lysine-Pl acyltransferase that
selectively transfers monounsaturated fatty acids (MUFA) to
lyso-phosphatidyl-ethanolamine, resulting in an increase of cell
PE-MUFA and a corresponding decrease of cell PE-PUFA. This process
effectively inhibits ferroptosis independent of the GPX4 and FSP1
pathways, as PE-PUFA is a major determinant of lipid peroxidation
and ferroptosis sensitivity (5,52).
MBOAT1, another member of this family, also inhibits ferroptosis
through similar mechanisms (53).
Furthermore, MBOAT2 and MBOAT1 are directly regulated by the
androgen receptor (AR) and estrogen receptor (ER), respectively,
which upregulate their gene expression. The use of anti-AR drugs
enzalutamide or ARV-110 can downregulate MBOAT2 expression in AR(+)
prostate cancer cells, making them sensitive to ferroptosis;
similarly, Ful, an ER depressant, can downregulate MBOAT1
expression in ER(+) breast cancer cells to achieve a similar
effect. These findings suggest that sex hormone signaling can
inhibit ferroptosis in cancer cells via phospholipid remodeling
mediated by MBOAT1/2 genes with potential implications for targeted
treatment (53).
Cancer-related pathways in ferroptosis
TP53
The tumor suppressor gene TP53, also known as P53,
is also referred to as the ‘genomic guardian’ because it is
essential for tumor inhibition and maintains genomic stability by
promoting cancer cell apoptosis and cell cycle arrest (54). TP53 is mainly divided into two
types: Wild type and mutant type. The wild type P53 can cause
apoptosis of tumor cells and prevent the cells from becoming
cancerous; whereas the mutant P53 gene will promote the cells to
become cancerous. It has been demonstrated that P53 deletions and
mutations are linked to ~50% of all human malignancies (55). In recent years, it has been
discovered that P53 can facilitate ferroptosis in cells by
controlling the expression of genes connected to ferroptosis. In
2015, Jiang et al (3)
discovered that P53 binds to the P53 response element in the
SLC7A11 promoter region, inhibiting the expression of SLC7A11 and
making cancer cells susceptible to ferroptosis. Furthermore, in
2019, Wang et al (56) found
that P53 reduces the level of monoubiquitination of histone H2B on
lysine 120 by promoting nuclear translocation of ubiquitin-specific
protease 7 (USP7), thereby epigenetically inhibiting the expression
of SLC7A11 (56). The newly
discovered impact of p53 on cell metabolism, oxidative responses
and ferroptotic cell death through regulation of
spermidine/spermine N1-acetyltransferase 1 (SAT1) has been a hot
research area (57). When SAT1 is
overexpressed, spermidine and spermine are rapidly depleted, which
leads to cellular mitochondria-mediated apoptosis and a decline in
cell growth (58). Ou et al
(59) discovered that P53 can
stimulate the expression of SAT1 at the transcriptional level,
hence enhancing the ability of arachidonate 15-LOX (ALOX15) to
sensitize cells to undergo ferroptosis. The aforementioned study
identified ALOX15 as a metabolic target of P53 involved in
ferroptotic cell death. However, the exact mechanism of how the
P53/SAT1/ALOX15 axis is activated remains unclear. Controversially,
P53 has been reported to have ferroptosis-suppressor activities,
for example by preventing the transcriptional activation of the
cyclin-dependent kinase inhibitor P21, thereby promoting GSH
production and reducing lipid peroxidation accumulation (60). Xie et al (28) demonstrated that TP53 restricts
erastin-induced ferroptosis by blocking dipeptidyl-peptidase-4
(DPP4) activity, thereby promoting plasma-membrane-associated
DPP4-dependent lipid peroxidation, which ultimately results in
ferroptosis (28). Furthermore, Hu
et al (61) identified
glutaminase 2 (GLS2) as a unique P53 target gene. In several cancer
cells, including human colorectal carcinoma HCT116 and
hepatocellular carcinoma HepG2, GLS2 expression can upregulate GSH
and NADH production, improving cellular antioxidant defense
capacity. Although GLS2 is suspected to be related to cellular
ferroptosis, more evidence is needed to determine whether GLS2
expression mediates P53-regulated ferroptosis. Taken together,
these studies suggested that P53 plays a dual role in the
occurrence of ferroptosis under various contexts. In order to
produce ferroptosis in cells, P53 can either decrease the
expression of SLC7A11 or increase the expression of specific target
genes, such as SAT1. However, P53 may also protect cells against
ferroptosis by inhibiting the activity of DPP4 or promoting the
transcriptional activation of P21.
Nuclear Factor erythroid 2-Related
Factor 2 (NRF2)
NRF2 is a key regulator of oxidative stress; when it
is activated, it encourages the iron storage, slows cells' iron
uptake and reduces the production of ROS in cells. NRF2 is
regulated by KELCH-like ECH-associated protein 1 (KEAP1). KEAP1 can
bind to NRF2 in the cytoplasm and be connected to the E3 ubiquitin
ligase (Cul3), where NRF2 can be degraded by the proteasome system
through ubiquitination. However, in some special cases, such as
mutations in NRF2 or KEAP1, a selective autophagy junction protein
SQSTM1, also known as P62, could bind and inactivate KEAP1,
preventing NRF2 degradation and enhancing subsequent NRF2 nuclear
accumulation. In such cases, the antioxidative transcriptional
factor NRF2 regulates the expression of numerous cytoprotective
genes involved in detoxification, antioxidant and drug metabolism
by binding to the antioxidant response element (62). For instance, as a target gene of
NRF2, SLC7A11 is essential for cell glutamate exchange and cysteine
production. It was found that SLC7A11 expression was upregulated
when NRF2 was activated (63). In
addition, GCL and GSS, which catalyze glutathione synthesis, are
also target genes of NRF2. Overexpression of NRF2 causes elevated
cellular GSH levels and promotes the expression of GPX4 (64). Heme-oxygenase 1 (HMOX-1) is also an
antioxidant gene targeted by NRF2. In cells, HMOX-1 catalyzes the
breakdown of heme to free iron, carbon monoxide and biliverdin. The
latter can be further converted to bilirubin, which acts as an
antioxidant and inhibits the production of ROS (65). It was discovered that upregulating
HMOX-1 has positive antioxidant benefits, whereas knocking down
HMOX-1 reduces the response of NRF2 to oxidative stress (66). NRF2 also controls the expression of
other genes to protect the cell from ferroptosis, such as GSTs,
GCLM and CHAC-1, which are related to ROS detoxifying enzymes and
GSH metabolism pathway (14,67),
as well as SLC40A1 and MT1G (Metallothionein-1G), which are engaged
in the iron metabolism route.
RAS
The main RAS family genes are HRAS, KRAS and NRAS,
which are collectively known as oncogenic RAS (68). KRAS mutations are among the most
prevalent in human cancers. Dolma et al (69) and Yang and Stockwell (70) demonstrated that the ferroptosis
inducers erastin and RSL3 were capable of selectively killing
RAS-mutated tumor cells long before the discovery of ferroptosis
(69,70). In a subsequent study, it was
revealed that numerous RAS-mutated cancer cells are vulnerable to
ferroptosis. This might be due to the fact that mutant RAS can
control the expression of genes involved in iron metabolism,
leading to higher iron levels, which in turn affects the process of
ferroptosis in cells. KRASG12D is the most common type of mutations
in KRAS. Dai et al (71)
found that, in pancreatic ductal adenocarcinoma cells, KRASG12D was
able to promote macrophage differentiation into the oncogenic M2
phenotype. KRASG12D was released by means of exosomes from cancer
cells undergoing ferroptosis and was taken up by macrophages
through advanced glycosylation end product-specific receptor. The
uptake of KRASG12D activates the STAT3 signaling pathway and
selectively upregulates the genes involved in the fatty acid
oxidation metabolic pathway, such as carnitine
palmitoyl-transferase 1A and acyl-CoA dehydrogenase medium chain,
leading to macrophage M2 polarization.
Hypoxia inducible factor (HIF)
Hypoxia is a hallmark of tumor formation and is
mainly regulated by HIF, which promotes the growth, survival and
metastasis of cancer cells (72).
HIF consists of an alpha subunit, such as HIF-1α, HIF-2α and
HIF-3α, and a beta subunit (HIF1β, also known as ARNT). Under
normal oxygen supply conditions, HIF-1α and HIF-2α are hydroxylated
by Egl nine homolog family (EGLN) family of HIFs and then
recognized by the E3 ubiquitin ligase (Von hippel-lindau, VHL) for
eventual degradation in the proteasome. However, under hypoxia,
prolyl hydroxylase is inactivated, HIF-1α and HIF-2α accumulate in
the cell and bind to ARNT to form the HIF complex, which is able to
bind to the hypoxia response element (HRE) of the hypoxia gene
promoter region, thereby inducing transcription of genes associated
with hypoxia adaptation and triggering a series of hypoxia
adaptation responses in tissue cells (73). Numerous studies have reported that
HIF is able to regulate the occurrence of ferroptosis in cells,
that a number of genes related to iron metabolism are regulated by
HRE, and that HIF has a dual role for ferroptosis in cancer cells.
Li et al (74) found that,
in human myeloma cells, carbonic anhydrase 9 (CA9) is overexpressed
as a result of hypoxia. Consequently, CA9 overexpression prevents
ferroptosis in cells via modulating TFR and FTH, leading to the
decreased iron intake and increased ferritin storage. Yang et
al (75) also discovered that
hypoxia stimulated the expression of HIF-1 in HT-1080 fibrosarcoma
cells and increased the expression of fatty acid binding proteins 3
and 7, which enhanced the ability of the cells to take in and store
fatty acids and prevented the development of ferroptosis. In
addition, HIF-2α activation upregulates hypoxia-inducible lipid.
Droplet associated protein overexpression in renal clear-cell
carcinoma, causes a rise in the synthesis of polyunsaturated fatty
acids and lipid peroxidation levels, which finally results in
ferroptosis (76).
Non-coding RNAs
Non-coding RNAs, including long non-coding RNA,
microRNA (miRNA or miR), circular RNA, and others, are RNA
molecules that are present in the gene transcriptome but do not
undergo translation to produce proteins. It was previously
suggested that certain non-coding RNAs could be involved in the
ferroptosis. For instance, ELAV-like RNA-binding protein 1 (ELAVL1)
is known to be able to bind and stabilize LINC00336. And it was
reported that P53 in lung cancer cells could bind to the ELAVL1
promoter region and suppress ELAVL1 expression. As a result,
LINC00336 was freed from its association with ELAVL1 and could
interact with miR-6852, which would then bind directly to
cystathionine-β-synthase (CBS) and suppress CBS expression,
ultimately promoting the occurrence of ferroptosis (77). Furthermore, another study by Wang
et al (78) identified
LINC00618 as a potential regulator of ferroptosis. It was found
that LINC00618 was able to increase the production of ROS and
affect iron metabolism. Moreover, LINC00618 could downregulate the
expression of SLC7A11, a gene involved in cellular antioxidant
defense. In the melanoma cell lines G-361 and A375, Zhang et
al (79) demonstrated that
miR-9 could prevent ferroptosis by targeting GOT1 which catabolizes
glutamine and eventually converts it to α-ketoglutarate, promoting
the accumulation of cellular ROS (79). On the other hand, Luo et al
(80) identified that miR-137
directly suppressed the expression of SLC1A5 in melanoma cells,
resulting in a slower uptake of glutamine and decreased levels of
malondialdehyde accumulation, which prevented the incidence of
ferroptosis in cells (80). In
addition, it has also been reported that miRNAs are also involved
in the regulation of other genes implicated in ferroptosis, such as
SLC7A11, TFR and FTH. These miRNAs target and regulate factors such
as NRF2 and ACSL4, which are known to promote or inhibit the
occurrence of ferroptosis in cells.
Ferroptosis in the combination therapy of
cancer
Lung cancer
Lung cancer, which accounted for 17.09% of new
cancer cases countrywide in China, was first in the prevalence of
malignant tumors and second most frequent disease worldwide
(81). Most prevalently in the
non-small cell lung cancer (NSCLC) form, lung cancer is
characterized by a poor survival rate (82). Chemotherapy drugs such as
acetaminophen (APAP) and cisplatin (CDDP) are currently used in the
treatment of lung cancer, but often cause drug resistance in tumor
cells, resulting in unsatisfactory therapeutic efficacy (82,83).
Recent studies suggested that the combined application of these
drugs with ferroptosis inducers might help to improve the
therapeutic efficacy of lung cancer (84,85).
APAP is a kind of analgesic and detoxification drug. When combined
with erastin, it could induce ferroptosis of cells by regulating
the nuclear translocation of Nrf2 and inhibit the growth of lung
cancer xenografts (84). Meanwhile,
it has also been reported that erastin or sorafenib combined with
low-dose CDDP can effectively inhibit the growth of CDDP-resistant
NSCLC cells in vivo and promote ferroptosis by inducing the
accumulation of intracellular lipid peroxides (85). Although few studies of these
combined drugs have yet been performed, and specific mechanisms of
action have not yet been fully understood, this allowed the opening
of new perspectives for lung cancer therapy in the future.
Gastric cancer
Gastric cancer, a prevalent malignancy in China,
ranks fifth in incidence and fourth in fatality (86). Adriamycin, platinum drugs,
5-fluorouracil and paclitaxel are widely used in clinical treatment
of gastric cancer (87,88); although these drugs demonstrated
antitumor effects in gastric cancer, they are often limited by drug
resistance. Cai et al (89)
detected that SIRT6 silencing could induce ferroptosis in gastric
cancer cells and overcome the resistance to sorafenib. Precisely,
inhibition of SIRT6 leads to the inactivation of Keap1/Nrf2
signaling pathway and downregulation of GPX4, while overexpression
of GPX4 or activation of Keap1/Nrf2 signaling pathway can reverse
the effect of downregulation of SIRT6 on sorafenib-induced
ferroptosis. Therefore, targeting the SIRT6/Keap1/Nrf2/GPX4
signaling pathway may be a potential strategy to overcome sorafenib
resistance in gastric cancer cells. A previous study showed that
during sorafenib-induced ferroptosis in gastric cancer cells,
Activating Transcription Factor 2 (ATF2) can inhibit SLC7A11
protein degradation by promoting the expression of heat shock
protein 110, thereby protecting gastric cancer cells from
sorafenib-mediated ferroptosis. Accordingly, ATF2 knockout promoted
sorafenib-induced ferroptosis (90). Therefore, ATF2 could be a potential
target for effective treatment of gastric cancer. Cancer-associated
fibroblasts (CAF) are the main stromal cell type in the tumor
microenvironment. Zhang et al (91) found that the exosome miR-522
secreted by CAF inhibited the occurrence of ferroptosis and
promoted acquired drug resistance in gastric cancer cells. This
occurs through the selective packaging of miR-522 into CAF-derived
exosomes by heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1)/
USP7 axis. Subsequently, miR-522 directly targets ALOX15,
preventing the accumulation of lipid peroxidation and inhibiting
ferroptosis in gastric cancer cells. Some chemotherapeutic drugs
may promote the secretion of miR-522 by CAF through activating the
expression of USP7 and hnRNPA1, making gastric cancer cells
resistant to these drugs.
Breast cancer
Breast cancer is one of the most common types of
cancer in women, second only to uterine cancer, and represents a
significant threat to the health of women (92). Tumor-infiltrating neutrophils (TIN)
are the population of immune cells most strongly associated with
poor prognosis for 25 cancer types, including breast cancer
(93). For breast cancer, the
TIN-rich tumor microenvironment is resistant to immune checkpoint
blocking therapy (94). When
combined with immunotherapy, clearing or targeting
immunosuppressive TIN is expected to produce synergistic effects
(95). Zhao et al (96) found through single-cell RNA
sequencing that aconite decarboxylase 1 (Acod1) was the metabolic
enzyme with the highest degree of upregulation in TIN. It was also
demonstrated that TIN inhibits the occurrence of ferroptosis and
promotes breast cancer metastasis through high expression of Acod1.
Knockout of Acod1 reduces the survival and accumulation of TIN and
improves the efficacy of immune checkpoint blocking therapy in the
treatment of breast cancer metastasis. The aforementioned study
showed that Acod1 is a promising immune-metabolic target that can
induce ferroptosis in TIN and protect neutrophils that do not
express Acod1 outside the tumor, thus addressing the safety issue
of targeting TIN in the treatment of tumor metastasis.
Hepatocellular carcinoma
Hepatocellular carcinoma is a common type of liver
cancer, with a five-year survival rate of only 12%. It ranks as the
second leading cause of cancer-related deaths worldwide (97). Sorafenib is currently used as a
first-line agent for hepatocellular carcinoma whose mechanism of
action has been associated to ferroptosis. However, it is unable to
induce extensive ferroptosis in all hepatocellular carcinomas
(98). Interestingly, it was found
that in the process of REDOX homeostasis maintenance, the
RNA-binding protein PNO1 promoted the occurrence of autophagy,
which enabled macromolecules and organelles to resynthesize new
amino acids, thereby increasing the content of glutamate in cells.
With the increase of glutamate content in the cell, system Xc is
activated, resulting in the increase of cystine uptake and the
promotion of GSH biosynthesis, thus protecting hepatocellular
cancer cells from ferroptosis. Accordingly, inhibition of PNO1
expression will inhibit the transcription of SLC7A11 and ultimately
promote the occurrence of ferroptosis (99). The discovery identified PNO1 as a
new candidate therapeutic target in the treatment of hepatocellular
carcinoma. The combination of PNO1 inhibitors with agents that
induce ferroptosis, such as sorafenib, would provide a potential
therapeutic strategy for antitumor therapy based on ferroptosis.
NADPH is an important cofactor regulating cystine reduction or
reducing/oxidizing GSH (GSH/GSSG) cycle reaction (100). Decreased NADPH abundance can be
used as a biomarker to detect ferroptosis in the liver (101,102). Fang et al (103) found in their study that
cytoplasmic NADPH provider malidase 1 (ME1) is a novel inhibitor of
ferroptosis in the liver. They specifically knocked out the ME1
gene in mouse liver cells and observed that this increased the
susceptibility to ferroptosis and aggravated liver damage after
liver ischemia/reperfusion surgery. By contrast, supplementation
with ME1 substrate L-malate increased NADPH abundance to protect
the liver from ferroptosis and tissue damage. The study suggested
that ME1 is a potential therapeutic target for the treatment of
liver ischemia-reperfusion injury or other diseases associated with
ferroptosis. Using drug screening, Tang et al (104) found that USP8 small molecule
inhibitors can significantly inhibit the activity of liver cancer
cells. Mechanism studies have shown that USP8 modified OGT protein
through deubiquitination to improve the stability of the protein,
which subsequently regulated SLC7A11 through glycosylation and
inhibited ferroptosis in hepatocellular carcinoma, thus promoting
the occurrence of liver cancer.
Ovarian cancer
Ovarian cancer is the third most common and second
most deadly female cancer globally, with ~240,000 new cases
diagnosed each year and 150,000 deaths (97,105).
Currently, the first-line chemotherapy for ovarian cancer is
platinum-based drugs combined with paclitaxel, while targeted drugs
bevacizumab and poly(adp-ribose) polymerase (PARP) inhibitors are
often used for maintenance therapy (106). However, because of the primary or
secondary resistance to these drugs developed in the patients
receiving the treatment, the therapeutic efficacy remains to be
desired. Recent studies have revealed that ferroptosis inducers,
such as erastin, in combination with CDDP or PARP inhibitors,
inhibit the growth and metastasis of ovarian cancer cells (107,108). Therefore, the development of new
drugs for the relevant targets of ferroptosis can help to overcome
the shortcomings of current first-line chemotherapy drugs and
provide a new strategy for the treatment of ovarian cancer.
A recent study has also shed light on the role of
miRNAs associated with ferroptosis in the development of ovarian
cancer. Ma et al (109)
discovered that the tumor suppressor miR-424-5p might target ACSL4
in ovarian cancer cells to regulate the initiation of ferroptosis.
When miR-424-5p was expressed, it could directly bind to the 3′-UTR
of ACSL4 and decrease its production, reducing the ferroptosis
triggered by erastin in ovarian cancer cells. Conversely, when
miR-424-5p expression was decreased, the ovarian cancer cells
became more sensitive to erastin induction. Another key enzyme
involved in malignant tumor cells, including ovarian cancer, is
Stearoyl CoA desaturase 1 (SCD1). It is a rate-limiting enzyme that
catalyzes the conversion of saturated fatty acids to
monounsaturated fatty acids (110). Tesfay et al (111) indicated that inhibiting SCD1
expression led to a decrease in CoQ10 expression,
resulting in lipid peroxidation and ferroptosis in ovarian cancer
cells.
The Hippo pathway, a major signaling pathway
regulating cell proliferation, differentiation and metastasis, has
recently been shown to be associated with ferroptosis in ovarian
cancer cells. Yes-associated protein (YAP) and transcriptional
coactivator with PDZ-binding motif (TAZ), the two key factors in
this pathway, are significant transcriptional coactivators and are
considered to act as receptors for cell density (111). When cells proliferate at high
densities, YAP and TAZ are phosphorylated and degraded by
proteasomes; conversely, when cells proliferate at low densities,
YAP and TAZ are dephosphorylated and translocated from the
cytoplasm to the nucleus, where they bind to the transcription
factor TEAD to drive gene expression and regulate cell
proliferation and differentiation (112). A number of studies revealed that
YAP and TAZ exerted regulatory functions in the initiation of
cellular ferroptosis. In 2020, Yang et al (29) discovered that TAZ directly promoted
the expression of angiopoietin-like 4, which enhanced the activity
of NOX2, leading to ferroptosis in ovarian cancer cells. Another
study revealed that YAP regulated the expression of its target
gene, S phase kinase-associated protein 2 (SKP2), contributing to
ferroptosis in ovarian cancer cells (113). When YAP was overexpressed, it
increased the expression of SKP2, resulting in increased lipid
peroxidation. Conversely, when YAP was silenced, SKP2 expression
was inhibited, resulting in a decrease in iron concentration and
protection from ferroptosis in ovarian cancer cells. Furthermore,
Li et al (114)
demonstrated that inhibiting acetyl-galactosyl-transferase 14
expression decreased mTOR activity by controlling EGFR
glycosylation, which in turn inhibited the expression levels of
SLC7A11 and GPX4, ultimately inducing ferroptosis in ovarian cancer
cells.
Conclusions and perspectives
Ferroptosis, a novel form of cell death that relies
on iron and ROS, offers new therapeutic perspectives for the
treatment of cancer.
Lipid peroxidation is hallmark of ferroptosis and
takes place in both enzymatic and non-enzymatic reactions. To date,
five principal ferroptosis-related antioxidant defense mechanisms
have been described in the literature, including the GPX4-GSH
pathway, the FSP1-CoQ10H2 pathway, the
DHODH-CoQ10H2 pathway, the GCH1-BH4 pathway
and the newly discovered MBOAT1/2-mediated cellular phospholipid
remodeling pathway (Fig. 1).
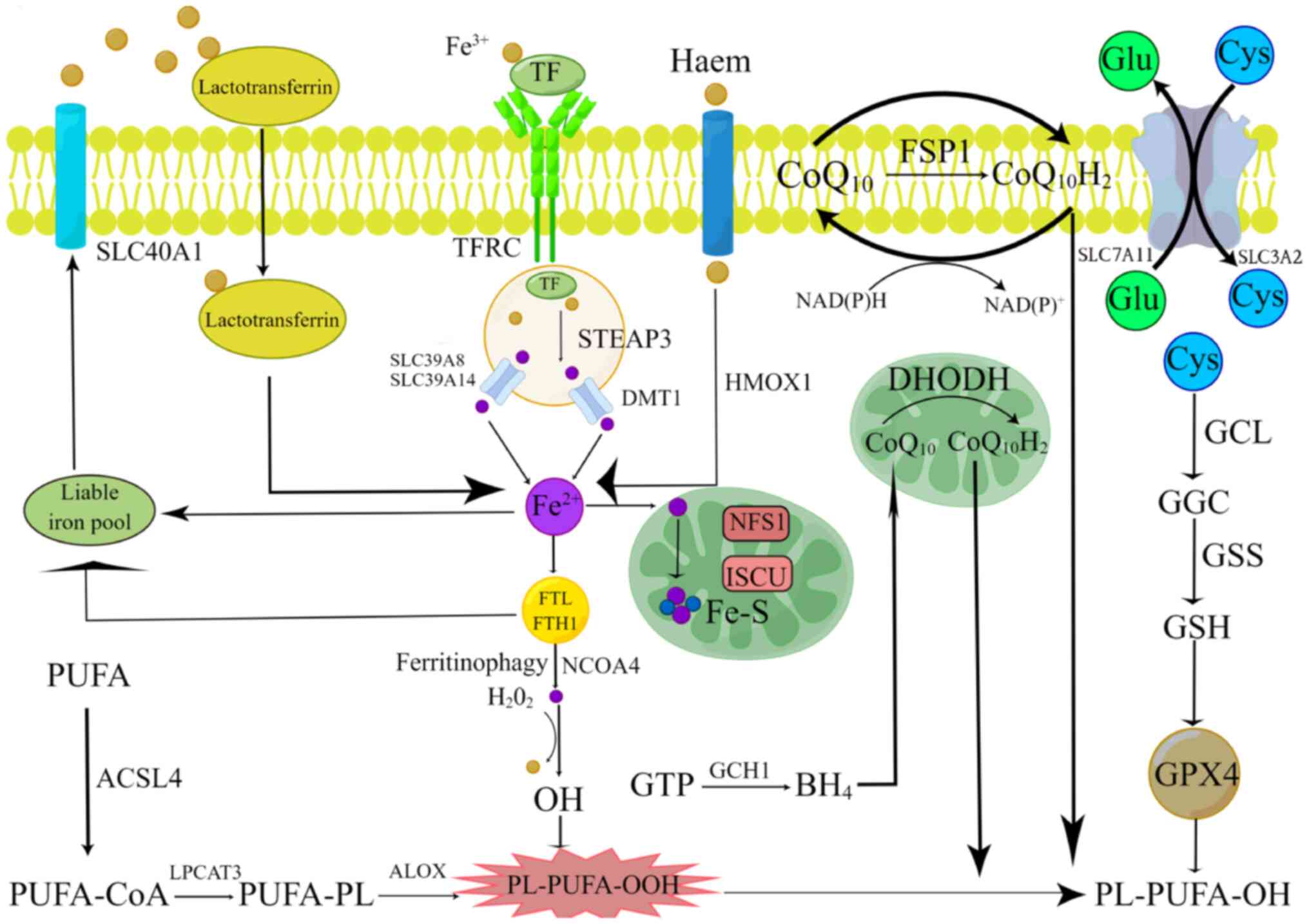 | Figure 1.Molecular mechanisms of ferroptosis.
Lipid peroxidation, iron metabolism and ferroptosis defense
mechanisms are the three primary mechanisms of ferroptosis. The
principal ferroptosis-related antioxidant defense mechanisms
include the GPX4-GSH pathway, the
FSP1-CoQ10H2 pathway, the
DHODH-CoQ10H2 pathway and the GCH1-BH4
pathway. The figure was created by Figdraw (2.0). GPX4, glutathione
peroxidase 4; GSH, glutathione; FSP1, ferroptosis suppressor
protein 1; CoQ10H2, pantethine alcohol;
DHODH, dihydroorotate dehydrogenase; GCH1, GTP cyclization
hydrolase 1; BH4, tetrahydrobiopterin. |
Iron metabolic processes in ferroptosis have been
widely explored, primarily involving the dynamic balance of labile
iron pools in cells, autophagic ferritin degradation and
mitochondrial ISC production.
Multiple key factors (and the related signaling
pathways), such as TP53, NRF2 and RAS, which have been
significantly investigated in the previous studies for their
critical roles and intricate mechanisms of action in various cancer
development and metastasis, have been revealed to be associated
with the process of cell ferroptosis. These findings, on one hand,
show that cell ferroptosis is, as well as other programmed cell
death processes previously discovered (such as apoptosis, autophagy
or pyroptosis), part of the essential regulating mechanisms of cell
growth, survival and response to external signaling; on the other
hand, they provide new clues for the understanding of cancer
physiopathology, and open new therapeutic perspectives for cancer,
particularly in the field of drug combination therapy.
Considerable advances in the research on ferroptosis
have been achieved in the past decade. A total of >10,000
published articles retrieved by Pubmed (https://pubmed.ncbi.nlm.nih.gov/) have been devoted to
ferroptosis. However, further research on this subject is still
required. Particularly, deeper comprehension of the roles of
ferroptosis in cancer development and drug resistance in
association with specific cancer types, would allow to devise more
efficient drugs and treatments based on the new concept.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural Science
Foundation of Guangdong (grant nos. 2022A1515140022 and
2023A1515010248) and the MOE Key Laboratory of Tumor Molecular
Biology (grant no. 202201).
Availability of data and materials
Not applicable.
Authors' contributions
LZ, XL and CG designed the project and drafted the
manuscript, XG and LL revised and edited the manuscript. All
authors read and approved the final manuscript. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Y, Shi J, Liu X, Feng L, Gong Z,
Koppula P, Sirohi K, Li X, Wei Y, Lee H, et al: BAP1 links
metabolic regulation of ferroptosis to tumour suppression. Nat Cell
Biol. 20:1181–1192. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao M, Yi J, Zhu J, Minikes AM, Monian P,
Thompson CB and Jiang X: Role of mitochondria in ferroptosis. Mol
Cell. 73:354–363. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lei G, Zhuang L and Gan B: Targeting
ferroptosis as a vulnerability in cancer. Nat Rev Cancer.
22:381–396. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fonseca-Nunes A, Jakszyn P and Agudo A:
Iron and cancer risk-a systematic review and meta-analysis of the
epidemiological evidence. Cancer Epidemiol Biomarkers Prev.
23:12–31. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo Q, Li L, Hou S, Yuan Z, Li C, Zhang W,
Zheng L and Li X: The Role of Iron in Cancer Progression. Front
Oncol. 10:7784922021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Q, Han X, Lan X, Gao Y, Wan J, Durham
F, Cheng T, Yang J, Wang Z, Jiang C, et al: Inhibition of neuronal
ferroptosis protects hemorrhagic brain. JCI Insight. 2:e907772017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang C, Liu X, Jin S, Chen Y and Guo R:
Ferroptosis in cancer therapy: A novel approach to reversing drug
resistance. Mol Cancer. 21:472022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Richardson DR and Ponka P: The molecular
mechanisms of the metabolism and transport of iron in normal and
neoplastic cells. Biochimica et biophysica acta. 1331:1–40. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brown CW, Amante JJ, Chhoy P, Elaimy AL,
Liu H, Zhu LJ, Baer CE, Dixon SJ and Mercurio AM: Prominin2 drives
ferroptosis resistance by stimulating iron export. Dev Cell.
51:575–586.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen X, Kang R, Kroemer G and Tang D:
Broadening horizons: The role of ferroptosis in cancer. Nat Rev
Clin Oncol. 18:280–296. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E4975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kremer DM, Nelson BS, Lin L, Yarosz EL,
Halbrook CJ, Kerk SA, Sajjakulnukit P, Myers A, Thurston G, Hou SW,
et al: GOT1 inhibition promotes pancreatic cancer cell death by
ferroptosis. Nat Commun. 12:48602021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim EH, Shin D, Lee J, Jung AR and Roh JL:
CISD2 inhibition overcomes resistance to sulfasalazine-induced
ferroptotic cell death in head and neck cancer. Cancer Lett.
432:180–190. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alvarez SW, Sviderskiy VO, Terzi EM,
Papagiannakopoulos T, Moreira AL, Adams S, Sabatini DM, Birsoy K
and Possemato R: NFS1 undergoes positive selection in lung tumours
and protects cells from ferroptosis. Nature. 551:639–643. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu T, Xiao Z, Yuan H, Tian H, Chen T,
Chen Q, Chen M, Yang J, Zhou Q, Guo W, et al: ACO1 and IREB2
downregulation confer poor prognosis and correlate with
autophagy-related ferroptosis and immune infiltration in KIRC.
Front Oncol. 12:929838. 2022.
|
|
21
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Porter NA, Caldwell SE and Mills KA:
Mechanisms of free radical oxidation of unsaturated lipids. Lipids.
30:277–290. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shah R, Shchepinov MS and Pratt DA:
Resolving the role of lipoxygenases in the initiation and execution
of ferroptosis. ACS Cent Sci. 4:387–396. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghosh MK, Mukhopadhyay M and Chatterjee
IB: NADPH-initiated cytochrome P450-dependent free iron-independent
microsomal lipid peroxidation: Specific prevention by ascorbic
acid. Mol Cell Biochem. 166:35–44. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zou Y, Li H, Graham ET, Deik AA, Eaton JK,
Wang W, Sandoval-Gomez G, Clish CB, Doench JG and Schreiber SL:
Cytochrome P450 oxidoreductase contributes to phospholipid
peroxidation in ferroptosis. Nat Chem Biol. 16:302–309. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dixon SJ, Winter GE, Musavi LS, Lee ED,
Snijder B, Rebsamen M, Superti-Furga G and Stockwell BR: Human
haploid cell genetics reveals roles for lipid metabolism genes in
nonapoptotic cell death. ACS Chem Biol. 10:1604–1609. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J,
Zhong M, Yuan H, Zhang L, Billiar TR, et al: The tumor suppressor
p53 limits ferroptosis by blocking DPP4 activity. Cell Rep.
20:1692–1704. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang WH, Huang Z, Wu J, Ding CC, Murphy SK
and Chi JT: A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell
death and chemoresistance in epithelial ovarian cancer. Mol Cancer
Res. 18:79–90. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li Y, Ran Q, Duan Q, Jin J, Wang Y, Yu L,
Wang C, Zhu Z, Chen X and Weng L: 7-Dehydrocholesterol dictates
ferroptosis sensitivity. Nature. 626:411–418. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maiorino M, Scapin M, Ursini F, Biasolo M,
Bosello V and Flohé L: Distinct promoters determine alternative
transcription of gpx-4 into phospholipid-hydroperoxide glutathione
peroxidase variants. J Biol Chem. 278:34286–34290. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yant LJ, Ran Q, Rao L, Van Remmen H,
Shibatani T, Belter JG, Motta L, Richardson A and Prolla TA: The
selenoprotein GPX4 is essential for mouse development and protects
from radiation and oxidative damage insults. Free Radic Biol Med.
34:496–502. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee
H, Koppula P, Wu S, Zhuang L, Fang B, et al: DHODH-mediated
ferroptosis defence is a targetable vulnerability in cancer.
Nature. 593:586–590. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu J, Liu M, Zhang H, Wei X, Wang J, Xian
M and Guo W: Exploring cysteine regulation in cancer cell survival
with a highly specific ‘Lock and Key’ fluorescent probe for
cysteine. Chem Sci. 10:10065–10071. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Roh JL, Kim EH, Jang HJ, Park JY and Shin
D: Induction of ferroptotic cell death for overcoming cisplatin
resistance of head and neck cancer. Cancer Lett. 381:96–103. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ingold I, Berndt C, Schmitt S, Doll S,
Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T,
et al: Selenium utilization by GPX4 is required to prevent
Hydroperoxide-Induced ferroptosis. Cell. 172:409–422.e21. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Seiler A, Schneider M, Förster H, Roth S,
Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W, et
al: Glutathione peroxidase 4 senses and translates oxidative stress
into 12/15-lipoxygenase dependent- and AIF-mediated cell death.
Cell Metab. 8:237–248. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cui S, Ghai A, Deng Y, Li S, Zhang R,
Egbulefu C, Liang G, Achilefu S and Ye J: Identification of
hyperoxidized PRDX3 as a ferroptosis marker reveals ferroptotic
damage in chronic liver diseases. Mol Cell. 83:3931–3939.e5. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Floros KV, Chawla AT, Johnson-Berro MO,
Khatri R, Stamatouli AM, Boikos SA, Dozmorov MG, Cowart LA and
Faber AC: MYCN upregulates the transsulfuration pathway to suppress
the ferroptotic vulnerability in MYCN-amplified neuroblastoma. Cell
Stress. 6:21–29. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu D, Liang CH, Huang B, Zhuang X, Cui W,
Yang L, Yang Y, Zhang Y, Fu X, Zhang X, et al: Tryptophan
metabolism acts as a new anti-ferroptotic pathway to mediate tumor
growth. Adv Sci (Weinh). 10:e22040062023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu M, Xu LG, Li X, Zhai Z and Shu HB:
AMID, an apoptosis-inducing factor-homologous
mitochondrion-associated protein, induces caspase-independent
apoptosis. J Biol Chem. 277:25617–25623. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Doll S, FreitasF P, Shah R, Aldrovandi M,
da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius
E, Scheel CH, et al: FSP1 is a glutathione-independent ferroptosis
suppressor. Nature. 575:693–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Frei B, Kim MC and Ames BN: Ubiquinol-10
is an effective lipid-soluble antioxidant at physiological
concentrations. Proc Natl Acad Sci USA. 87:4879–4883. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Turunen M, Olsson J and Dallner G:
Metabolism and function of coenzyme Q. Biochim Biophys Acta.
1660:171–199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Takahashi T, Okamoto T, Mori K, Sayo H and
Kishi T: Distribution of ubiquinone and ubiquinol homologues in rat
tissues and subcellular fractions. Lipids. 28:803–809. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bersuker K, Hendricks JM, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Vasan K, Werner M and Chandel NS:
Mitochondrial metabolism as a target for cancer therapy. Cell
Metab. 32:341–352. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Thöny B, Auerbach G and Blau N:
Tetrahydrobiopterin biosynthesis, regeneration and functions.
Biochem J. 347:1–16. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Soula M, Weber RA, Zilka O, Alwaseem H, La
K, Yen F, Molina H, Garcia-Bermudez J, Pratt DA and Birsoy K:
Metabolic determinants of cancer cell sensitivity to canonical
ferroptosis inducers. Nat Chem Biol. 16:1351–1360. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Werner ER, Blau N and Thöny B:
Tetrahydrobiopterin: Biochemistry and pathophysiology. Biochem J.
438:397–414. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kraft VAN, Bezjian CT, Pfeiffer S,
Ringelstetter L, Müller C, Zandkarimi F, Merl-Pham J, Bao X,
Anastasov N, Kössl J, et al: GTP Cyclohydrolase
1/Tetrahydrobiopterin counteract ferroptosis through lipid
remodeling. ACS Cent Sci. 6:41–53. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liang D, Feng Y, Zandkarimi F, Wang H,
Zhang Z, Kim J, Cai Y, Gu W, Stockwell BR and Jiang X: Ferroptosis
surveillance independent of GPX4 and differentially regulated by
sex hormones. Cell. 186:2748–2764.e22. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kaiser AM and Attardi LD: Deconstructing
networks of p53-mediated tumor suppression in vivo. Cell Death
Differ. 25:93–103. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bykov VJN, Eriksson SE, Bianchi J and
Wiman KG: Targeting mutant p53 for efficient cancer therapy. Nat
Rev Cancer. 18:89–102. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang Y, Yang L, Zhang X, Cui W, Liu Y, Sun
QR, He Q, Zhao S, Zhang GA, Wang Y, et al: Epigenetic regulation of
ferroptosis by H2B monoubiquitination and p53. EMBO Rep.
20:e475632019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Thomas T and Thomas TJ: Polyamine
metabolism and cancer. J Cell Mol Med. 7:113–126. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mandal S, Mandal A and Park MH: Depletion
of the polyamines spermidine and spermine by overexpression of
spermidine/spermine N¹-acetyltransferase 1 (SAT1) leads to
mitochondria-mediated apoptosis in mammalian cells. Biochem J.
468:435–447. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ou Y, Wang SJ, Li D, Chu B and Gu W:
Activation of SAT1 engages polyamine metabolism with p53-mediated
ferroptotic responses. Proc Natl Acad Sci USA. 113:E6806–E6812.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hu W, Zhang C, Wu R, Sun Y, Levine A and
Feng Z: Glutaminase 2, a novel p53 target gene regulating energy
metabolism and antioxidant function. Proc Natl Acad Sci USA.
107:7455–7460. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R
and Tang D: Activation of the p62-Keap1-NRF2 pathway protects
against ferroptosis in hepatocellular carcinoma cells. Hepatology.
63:173–184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M,
Buchfelder M and Savaskan N: Nrf2-Keap1 pathway promotes cell
proliferation and diminishes ferroptosis. Oncogenesis. 6:e3712017.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chan JY and Kwong M: Impaired expression
of glutathione synthetic enzyme genes in mice with targeted
deletion of the Nrf2 basic-leucine zipper protein. Biochim Biophys
Acta. 1517:19–26. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lu J, Zhao Y, Liu M, Lu J and Guan S:
Toward improved human health: Nrf2 plays a critical role in
regulating ferroptosis. Food Funct. 12:9583–9606. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lu C, Xu W, Zhang F, Shao J and Zheng S:
Nrf2 knockdown disrupts the protective effect of curcumin on
alcohol-induced hepatocyte necroptosis. Mol Pharm. 13:4043–4053.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Anandhan A, Dodson M, Schmidlin CJ, Liu P
and Zhang DD: Breakdown of an ironclad defense system: The critical
role of NRF2 in mediating ferroptosis. Cell Chem Biol. 27:436–447.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ryan MB and Corcoran RB: Therapeutic
strategies to target RAS-mutant cancers. Nat Rev Clin Oncol.
15:709–720. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Dolma S, Lessnick SL, Hahn WC and
Stockwell BR: Identification of genotype-selective antitumor agents
using synthetic lethal chemical screening in engineered human tumor
cells. Cancer Cell. 3:285–296. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yang WS and Stockwell BR: Synthetic lethal
screening identifies compounds activating iron-dependent,
nonapoptotic cell death in oncogenic-RAS-harboring cancer cells.
Chem Biol. 15:234–245. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Dai E, Han L, Liu J, Xie Y, Kroemer G,
Klionsky DJ, Zeh HJ, Kang R, Wang J and Tang D: Autophagy-dependent
ferroptosis drives tumor-associated macrophage polarization via
release and uptake of oncogenic KRAS protein. Autophagy.
16:2069–2083. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Singhal R, Mitta SR, Das NK, Kerk SA,
Sajjakulnukit P, Solanki S, Andren A, Kumar R, Olive KP, Banerjee
R, et al: HIF-2α activation potentiates oxidative cell death in
colorectal cancers by increasing cellular iron. J Clin Invest.
131:e1436912021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Keith B, Johnson RS and Simon MC: HIF1α
and HIF2α: Sibling rivalry in hypoxic tumour growth and
progression. Nat Rev Cancer. 12:9–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Li Z, Jiang L, Chew SH, Hirayama T, Sekido
Y and Toyokuni S: Carbonic anhydrase 9 confers resistance to
ferroptosis/apoptosis in malignant mesothelioma under hypoxia.
Redox Biol. 26:1012972019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yang M, Chen P, Liu J, Zhu S, Kroemer G,
Klionsky DJ, Lotze MT, Zeh HJ, Kang R and Tang D: Clockophagy is a
novel selective autophagy process favoring ferroptosis. Sci Adv.
5:eaaw22382019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zou Y, Palte MJ, Deik AA, Li H, Eaton JK,
Wang W, Tseng YY, Deasy R, Kost-Alimova M, Dančík V, et al: A
GPX4-dependent cancer cell state underlies the clear-cell
morphology and confers sensitivity to ferroptosis. Nat Commun.
10:16172019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu
N, Shi Y, Chen L, Xiao D, Yu F, et al: Long noncoding RNA LINC00336
inhibits ferroptosis in lung cancer by functioning as a competing
endogenous RNA. Cell Death Differ. 26:2329–2343. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wang Z, Chen X, Liu N, Shi Y, Liu Y,
Ouyang L, Tam S, Xiao D, Liu S, Wen F, et al: A nuclear long
Non-Coding RNA LINC00618 accelerates ferroptosis in a manner
dependent upon apoptosis. Mol Ther. 29:263–274. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhang K, Wu L, Zhang P, Luo M, Du J, Gao
T, O'Connell D, Wang G, Wang H and Yang Y: miR-9 regulates
ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in
melanoma. Mol Carcinog. 57:1566–1576. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Luo M, Wu L, Zhang K, Wang H, Zhang T,
Gutierrez L, O'Connell D, Zhang P, Li Y, Gao T, et al: miR-137
regulates ferroptosis by targeting glutamine transporter SLC1A5 in
melanoma. Cell Death Differ. 25:1457–1472. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kryczka J, Kryczka J, Czarnecka-Chrebelska
KH and Brzeziańska-Lasota E: Molecular mechanisms of
chemoresistance induced by cisplatin in NSCLC cancer therapy. Int J
Mol Sci. 22:88852021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gai C, Yu M, Li Z, Wang Y, Ding D, Zheng
J, Lv S, Zhang W and Li W: Acetaminophen sensitizing
erastin-induced ferroptosis via modulation of Nrf2/heme oxygenase-1
signaling pathway in non-small-cell lung cancer. J Cell Physiol.
235:3329–3339. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Li Y, Yan H, Xu X, Liu H, Wu C and Zhao L:
Erastin/sorafenib induces cisplatin-resistant non-small cell lung
cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway.
Oncol Lett. 19:323–333. 2020.PubMed/NCBI
|
|
86
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Gu R, Xia Y, Li P, Zou D, Lu K, Ren L,
Zhang H and Sun Z: Ferroptosis and its role in gastric cancer.
Front Cell Dev Biol. 10:8603442020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wei L, Sun J, Zhang N, Zheng Y, Wang X, Lv
L, Liu J, Xu Y, Shen Y and Yang M: Noncoding RNAs in gastric
cancer: Implications for drug resistance. Mol Cancer. 19:622020.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Cai S, Fu S, Zhang W, Yuan X, Cheng Y and
Fang J: SIRT6 silencing overcomes resistance to sorafenib by
promoting ferroptosis in gastric cancer. Biochem Biophys Res
Commun. 577:158–164. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Xu X and Li Y, Wu Y, Wang M, Lu Y, Fang Z,
Wang H and Li Y: Increased ATF2 expression predicts poor prognosis
and inhibits sorafenib-induced ferroptosis in gastric cancer. Redox
Biol. 59:1025642023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zhang H, Deng T, Liu R, Ning T, Yang H,
Liu D, Zhang Q, Lin D, Ge S, Bai M, et al: CAF secreted miR-522
suppresses ferroptosis and promotes acquired chemo-resistance in
gastric cancer. Mol Cancer. 19:432020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Waks AG and Winer EP: Breast cancer
treatment: A Review. JAMA. 321:288–300. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Gentles AJ, Newman AM, Liu CL, Bratman SV,
Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al: The
prognostic landscape of genes and infiltrating immune cells across
human cancers. Nat Med. 21:938–945. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kim IS, Gao Y, Welte T, Wang H, Liu J,
Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, et al:
Immuno-subtyping of breast cancer reveals distinct myeloid cell
profiles and immunotherapy resistance mechanisms. Nat Cell Biol.
21:1113–1126. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Lu X and Lu X: Enhancing immune checkpoint
blockade therapy of genitourinary malignancies by co-targeting
PMN-MDSCs. Biochim Biophys Acta Rev Cancer. 1877:1887022022.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhao Y, Liu Z, Liu G, Zhang Y, Liu S, Gan
D, Chang W, Peng X, Sung ES, Gilbert K, et al: Neutrophils resist
ferroptosis and promote breast cancer metastasis through aconitate
decarboxylase 1. Cell Metab. 35:1688–1703.e10. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Louandre C, Marcq I, Bouhlal H, Lachaier
E, Godin C, Saidak Z, François C, Chatelain D, Debuysscher V,
Barbare JC, et al: The retinoblastoma (Rb) protein regulates
ferroptosis induced by sorafenib in human hepatocellular carcinoma
cells. Cancer Lett. 356:971–977. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hu X, He Y, Han Z, Liu W, Liu D, Zhang X,
Chen L, Qi L, Chen L, Luo Y, et al: PNO1 inhibits
autophagy-mediated ferroptosis by GSH metabolic reprogramming in
hepatocellular carcinoma. Cell Death Dis. 13:10102022. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Shimada K, Hayano M, Pagano NC and
Stockwell BR: Cell-line selectivity improves the predictive power
of pharmacogenomic analyses and helps identify NADPH as biomarker
for ferroptosis sensitivity. Cell Chem Biol. 23:225–235. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Wang H, An P, Xie E, Wu Q, Fang X, Gao H,
Zhang Z, Li Y, Wang X, Zhang J, et al: Characterization of
ferroptosis in murine models of hemochromatosis. Hepatology.
66:449–465. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Fang X, Zhang J, Li Y, Song Y, Yu Y, Cai
Z, Lian F, Yang J, Min J and Wang F: Malic Enzyme 1 as a novel
Anti-Ferroptotic regulator in hepatic Ischemia/Reperfusion injury.
Adv Sci (Weinh). 10:e22054362023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Tang J, Long G, Hu K, Xiao D, Liu S, Xiao
L, Zhou L and Tao Y: Targeting USP8 Inhibits O-GlcNAcylation of
SLC7A11 to promote ferroptosis of hepatocellular carcinoma via
stabilization of OGT. Adv Sci (Weinh). 10:e23029532023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Lheureux S, Gourley C, Vergote I and Oza
AM: Epithelial ovarian cancer. Lancet. 393:1240–1253. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Yao Y, Wang B, Jiang Y, Guo H and Li Y:
The mechanisms crosstalk and therapeutic opportunities between
ferroptosis and ovary diseases. Front Endocrinol (Lausanne).
14:11940892023. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Cheng Q, Bao L, Li M, Chang K and Yi X:
Erastin synergizes with cisplatin via ferroptosis to inhibit
ovarian cancer growth in vitro and in vivo. J Obstet Gynaecol Res.
47:2481–2491. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Hong T, Lei G, Chen X, Li H, Zhang X, Wu
N, Zhao Y, Zhang Y and Wang J: PARP inhibition promotes ferroptosis
via repressing SLC7A11 and synergizes with ferroptosis inducers in
BRCA-proficient ovarian cancer. Redox Biol. 42:1019282021.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Ma LL, Liang L, Zhou D and Wang SW: Tumor
suppressor miR-424-5p abrogates ferroptosis in ovarian cancer
through targeting ACSL4. Neoplasma. 68:165–173. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Igal RA: Stearoyl CoA desaturase-1: New
insights into a central regulator of cancer metabolism. Biochim
Biophys Acta. 1861:1865–1880. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Tesfay L, Paul BT, Konstorum A, Deng Z,
Cox AO, Lee J, Furdui CM, Hegde P, Torti FM and Torti SV:
Stearoyl-CoA Desaturase 1 protects ovarian cancer cells from
ferroptotic cell death. Cancer Res. 79:5355–5366. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Hsiao C, Lampe M, Nillasithanukroh S, Han
W, Lian X and Palecek SP: Human pluripotent stem cell culture
density modulates YAP signaling. Biotechnol J. 11:662–675. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Yang WH, Lin CC, Wu J, Chao PY, Chen K,
Chen PH and Chi JT: The hippo pathway effector YAP promotes
ferroptosis via the E3 ligase SKP2. Mol Cancer Re. 19:1005–1014.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Li HW, Liu MB, Jiang X, Song T, Feng SX,
Wu JY, Deng PF and Wang XY: GALNT14 regulates ferroptosis and
apoptosis of ovarian cancer through the EGFR/mTOR pathway. Future
Oncol. 18:149–161. 2022. View Article : Google Scholar : PubMed/NCBI
|














