Introduction
Cholangiocarcinoma (CCA) is a rare malignancy that
originates in the epithelial cells within the biliary tract,
occurring either within (intrahepatic) or outside (extrahepatic)
the liver. CCA is the second most prevalent form of liver cancer,
with its highest incidence observed in northeast Thailand (1,2). The
major risk factors associated with CCA often occur from chronic
biliary inflammation, due to conditions such as liver fluke
infection, hepatitis B and C viruses, and primary sclerosing
cholangitis (3–5). Diagnosis at advanced stages and the
challenging prognosis greatly complicate treatment strategies.
While surgical resection remains the primary therapeutic option,
its applicability is limited, depending upon the site and stage of
the cancer (6,7). To enhance the treatment choices,
chemotherapeutic regimens are crucial in patients with CCA.
Notably, drugs such as 5-fluorouracil, gemcitabine and cisplatin,
either individually or in combination, are frequently employed in
the treatment of patients with cancer (8–10).
However, long-term drug treatment outcomes remain insufficient,
emphasizing the need for innovative treatment approaches in
patients with CCA.
Previously published research from Chai saingmongkol
et al (11) identified
molecular subtypes of intrahepatic CCA from Thai patients through
the systematic integration of genomics, transcriptomics and
metabolomics data derived from paired tumor and non-tumor tissue
specimens. This previous study discovered the C1 subtype, which is
associated with a poor prognosis and shows defects of mitotic
checkpoint signaling. Importantly, the authors discovered the
polo-like kinase 1 (PLK1) gene as a subtype biomarker and also as a
molecular target for CCA (11).
PLK1 is a serine/threonine protein kinase involved
in various steps in mitotic events. Among other PLK family proteins
(PLK1-5), PLK1 has been extensively examined for its role in the
control of the cell cycle and cancer (12). PLKs comprise an N-terminal kinase
domain, which is responsible for the binding of ATP and activating
the enzyme, and a C-terminal polo box domain, involved in
phosphopeptide binding (13). PLK1
regulates a number of steps in mitosis, including maturation of the
centrosome, initiation of mitosis, establishment of
kinetochore-microtubule attachments, spindle orientation and the
process of cytokinesis (14,15).
PLK1 is upregulated in numerous types of cancer, including
hepatocellular carcinoma (16),
breast cancer (17), colorectal
cancer (18), gastric cancer
(19), ovarian carcinoma (20), chronic myeloid leukemia (21) and CCA (11). On the other hand, PLK1 expression is
mostly absent in normal cells and non-dividing healthy cells
(22). This makes PLK1 an appealing
target for the enhancement of cancer therapeutic drugs.
A number of reports have revealed that inhibiting
PLK1 favors antitumor effects in numerous types of cancer.
Depleting PLK1 levels in cancer cells reduces cell proliferation
and perturbs the spindle assembly, triggering activation of the
mitotic checkpoint, resulting in mitotic arrest and ultimately
leading to cell death (23,24). Several PLK1 inhibitors have been
developed as cancer therapeutic agents. BI2536
(dihydropteridinone), a highly selective and potent inhibitor, was
first developed and has been shown to exert an antitumor effect in
several types of cancer (25,26).
BI6727, another dihydropteridinone derivate, emerged as an
ATP-competitive inhibitor of PLK1 that was later developed as a
drug (27). Small molecule
inhibitors targeting PLK1 can lead to a decrease in cell
proliferation, disruption in cell cycle progression (specifically
G2/M-phase arrest) and the induction of cell apoptosis
in a number of cancers, including oral cancer cells (28), melanoma cells (29), lymphoma cells (30) and non-small lung cancer cells
(31). To date, there are only a
few studies exploring the consequences of inhibiting PLK1 in CCA
(32–34).
The present study aimed to explore the effects of
inhibiting PLK1 in four distinct CCA cell lines derived from Thai
patients. It revealed the antiproliferative effects,
G2/M-phase arrest and apoptosis in CCA cells following
the inhibition of PLK1 using both small molecule inhibitors and
small interfering (si)RNA.
Materials and methods
Cell culture
In the present study, four different CCA cell lines
derived from Thai patients with CCA, HuCCA1, KKU055, KKU100 and
KKU213A, from the Japanese Collection of Research Bioresources Cell
Bank (JCRB) were employed. KKU100 (cat. no. JCRB1568, RRID:
CVCL_3996), KKU055 (JCRB cat. no. JCRB1551, RRID: CVCL_M258) and
KKU213A (JCRB cat. no. JCRB1557, RRID: CVCL_M261). The cell passage
numbers for KKU055, KKU100, and KKU213A upon purchase were P4, P9,
and P9, respectively. HuCCA1 (RRID: CVCL_M255; cell passage 108)
was established and generously provided by Dr Stitaya Sirisinha,
Department of Microbiology, Faculty of Science, Mahidol University,
Thailand (35). The attributes of
the CCA cell lines used in this investigation are detailed in
Table SI. HuCCA1 originated from a
patient with intrahepatic CCA, and the serum of the patient was
positive for Opisthorchis viverrini (Ov) antigen.
KKU055 and KKU213A were also established from patients with
intrahepatic CCA (36,37). KKU100 was isolated from the tissue
of a patient with extrahepatic CCA (38). KKU100 and KKU213A cell lines were
both from patients with Ov in stool samples. The doubling
times of the CCA cell lines are as follows: HuCCA1, 55 h (35); KKU100, 72 h (38); KKU055, 23.6 h (39); and KKU213A, 23.4 h (40). Genetic alterations of genes
frequently mutated in cancer were also shown in Table SI (41–43).
KKU100, KKU055 and KKU213A cells were cultured in DMEM (cat. no.
SH3024302; HyClone; Cytiva), and HuCCA1 cells were maintained in
Ham's F12 media (cat. no. SH3002602; HyClone; Cytiva). All cell
culture media were supplemented with 10% FBS (cat. no.
1IVG3-10270-106; Invitrogen; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (cat. no. 1TFS-1CC-15140122; Invitrogen;
Thermo Fisher Scientific, Inc.) and all cells were cultured at 37°C
in a 5% CO2 humidified incubator. All four CCA cell
lines used in the present study were sent to CLS Cell Lines Service
GmbH (cat. no. 900154) for cell line authentication (report date:
August 2023) using highly polymorphic short tandem repeat loci. The
analyzed data of the samples perfectly aligned with the DNA
profiles of the cell lines HuCCA1, KKU100 and KKU213A with a 100%
match. The score for the KKU055 cell line, however, was slightly
lower at 93.8%.
Agents and antibodies
PLK1 inhibitors, BI2536 (cat. no. HY-50698) and
BI6727 (cat. no. HY12137) were purchased from MedChemExpress. The
primary antibodies for various proteins were obtained from Cell
Signaling Technology, Inc., unless specified otherwise:
Phosphorylated (p)-PLK1 (Thr210; cat. no. 9062; RRID: AB_11127447),
aurora kinase A (aurora A/AIK; cat. no. 3092; RRID: AB_2061342),
cyclin B1 (cat. no. 4138; RRID: AB_2072132), poly (ADP-ribose)
polymerase (PARP; cat. no. 9542; RRID: AB_2160739), p-Histone H2AX
(Ser139; γH2AX; cat. no. 9718; RRID: AB_2118009), Histone H2A.X
(cat. no. 2595; RRID: AB_10694556), GAPDH (cat. no. 8884; RRID:
AB_11129865), α-tubulin (cat. no. 2125; RRID: AB_2619646) and PLK1
(anti-PLK1; clone 35–206; cat. no. 05-844; RRID: AB_310836;
Millipore). The secondary antibodies used were HRP-conjugated goat
anti-rabbit (cat. no. 7074) and horse anti-mouse (cat. no. 7076).
The dilutions of primary antibodies and secondary antibodies were
1:1,000 and 1:2,000, respectively.
Gene expression analysis by reverse
transcription-quantitative (RT-q) PCR
Cells (1×106) were initially seeded into
10-cm cell culture plates. The following day, the cells were
collected by trypsinization and washed with PBS. The extraction of
total RNA was carried out using the RNAeasy mini kit (cat. no.
75144; Qiagen GmbH), and its quantification was assessed using the
NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Inc.),
following the manufacturer's instructions. Subsequently, cDNA
synthesis was accomplished using 1 µg total RNA with the
SuperScript III First-strand synthesis system (cat. no.
1IV02-18080-051; Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. The expression of the
PLK1 gene was conducted through qPCR, utilizing the Power SYBR
Green PCR Master Mix (cat. no. 4367659, Applied Biosystems; Thermo
Fisher Scientific, Inc.) and was analyzed using the Applied
Biosystem StepOnePlus Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
instructions. The PCR cycling conditions were as follows: Initial
activation at 95°C for 10 min, followed by 40 cycles consisting of
denaturation at 95°C for 15 sec, and annealing and extension at
60°C for 30 sec. GAPDH expression served as the endogenous control.
The primer sequences were as follows: GAPDH forward,
5′-ACAGTCAGCCGCATCTTCTT-3′ and reverse, 5′-ACGACCAAATCCGTTGACTC-3′;
PLK1 forward, 5′-CGTGACCTACATCGACGAGA-3′ and reverse,
5′-AGCAGCTTGTCCACCATAGT-3′. The relative mRNA expression levels
were analyzed by normalization to the endogenous gene using the
2−ΔCq method, where
ΔCq=CqPLK1-CqGAPDH (44). The experiments were repeated three
times.
Cell viability and colony formation
assays
Cell proliferation was evaluated by analyzing cell
viability and the ability to form colonies. Cell viability was
assessed using MTT assay, a colorimetric method that measures
metabolically active cells. In this experiment, 10,000 cells were
placed in duplicate into 24-well plates and treated with different
concentrations of PLK1 inhibitors dissolved in DMSO for 24, 48 and
72 h. After treatment, the cell culture medium was replaced with a
solution containing 5 mg/ml MTT (cat. no. M2128; MilliporeSigma)
and the cells were incubated at 37°C for 1.5 h. The medium was then
substituted with DMSO, and the absorbance of the resultant solution
was measured at 540 nm using the PerkinElmer Victor Nivo plate
reader (PerkinElmer, Inc.). The absorbance is directly proportional
to the number of viable cells. Cell viability was compared with
that of cells treated with a vehicle control, and these experiments
were replicated three times.
In the colony formation assay, 3,000 cells were
placed in 6-well plates and exposed to PLK1 inhibitors for 48 h at
37°C in a 5% CO2 humidified incubator. Following this
treatment, the culture medium was substituted with drug-free
medium, and the cells were allowed to grow for 1 additional week.
The resulting colonies were rinsed twice with 1X PBS, fixed with
cold methanol for 2 min, air-dried and then stained with a 0.25%
aqueous crystal violet solution (cat. no. V5265; MilliporeSigma) at
room temperature for 30 min. Subsequently, the plates were rinsed
with tap water and left to air dry.
PLK1 gene silencing by small
interfering RNA (siRNA)
A synthetic siRNA targeting PLK1 (cat. no. 6292;
sense: 5′-CCCUCACAGUCCUCAAUAA-3′, antisense:
5′-UUAUUGAGGACUGUGAGGG-3′) and control siRNA (cat. no. 6568; sense:
5′-CGUACGCGGAAUACUUCGA-3′, antisense: 5′-UCGAAGUAUUCCGCGUACG-3′)
were purchased from Cell Signaling Technology, Inc. CCA cell lines
were transfected with 100 nM PLK1 siRNA or control siRNA (negative
control) using Lipofectamine® 3000 reagent (cat. no.
L300015; Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions. Following a 48-h transfection at
37°C in a 5% CO2 humidified incubator, the transfected
cells were analyzed for cell viability, cell cycle distribution,
cell apoptosis and protein detection. PLK1 protein depletion was
confirmed through western blotting.
Cell cycle analysis
Cell cycle distribution was examined using a flow
cytometer and the Muse cell cycle assay kit (cat. no. MCH100106;
Luminex Corporation) following the manufacturer's guidelines. In
summary, treated cells were harvested through trypsinization,
rinsed with PBS and fixed overnight at −20°C with ice-cold 70%
ethanol. After washing with PBS, the cells were centrifuged at 300
× g for 5 min at room temperature, suspended in Muse cell cycle
reagent, and then incubated in the dark at room temperature for 30
min. Subsequently, the samples were analyzed using the Muse cell
analyzer (MUSE 0500-3115B; Luminex Corporation) to determine the
percentage of cells in each cell cycle phase
(G0/G1, S and G2/M) based on their
varying DNA content. These experiments were conducted in biological
triplicates.
Detection of cell apoptosis
To evaluate the impact of PLK1 inhibition on
apoptotic cell death, flow cytometric analysis was performed using
the Muse Annexin V and dead cell kit (cat. no. MCH100106; Luminex
Corporation), following the manufacturer's guidelines. The assay is
based on the binding of Annexin V to phosphatidylserine (PS) on the
surface of apoptotic cells. The kit uses a premixed reagent
containing fluorescently labeled Annexin V and a dead cell marker
known as 7-Aminoactinomycin D (7-AAD) as the second dye. 7-AAD is a
fluorescent dye used for evaluating cell viability by binding to
DNA through intercalation. Live cells typically exclude 7-AAD due
to intact cell membranes, while it readily enters cells with
compromised membranes, such as late apoptotic and dead cells. Cells
(15×104) were cultured in 6-well plates with 2 ml
culture media. After an overnight incubation, the cells were
treated with 10 and 100 nM BI2536 or BI6727. Following a 48-h
treatment at 37°C in a 5% CO2 humidified incubator,
floating cells were collected and adherent cells were detached
through trypsinization. The harvested cells were then centrifuged
at 300 × g for 5 min at room temperature, and cell pellets were
suspended in culture media with 1% FBS. Equal volumes of cell
suspension and Muse Annexin V reagent were mixed and incubated for
20 min at room temperature. The percentages of live, early
apoptotic, late apoptotic, total apoptotic and dead cells were
determined using the Muse cell analyzer (MUSE 0500-3115B; Luminex
Corporation). The experiments were repeated three times.
Western blot analysis
The treated cells were rinsed with cold PBS, and
then scraped and collected. They were lysed in a cold buffer
(composed of 50 mM Tris-HCl, pH 7.4; 150 mM NaCl; 1% NP40; 0.25%
Na-deoxycholate; 1 mM EDTA) containing the Halt protease and
phosphatase inhibitor cocktail (cat. no. 78440; MilliporeSigma).
After centrifugation of the cell lysates at 13,000 × g for 15 min
at 4°C, the protein concentration was determined using Bradford
reagent (cat. no. B6916; MilliporeSigma) according to the
manufacturer's protocols. Equal amounts of protein (20–30 µg) were
separated using 12% polyacrylamide gels and were then transferred
onto Immobilon PVDF transfer membranes (MilliporeSigma). The
membrane was cut into two or three strips based on the molecular
weight of the targeted proteins. Subsequently, the membrane strips
were blocked with 5% non-fat dry milk in Tris-buffered saline (TBS)
with 0.1% Tween-20 for 1 h at room temperature. Each strip was then
probed separately with the specific antibodies. Proteins were
identified by incubating with primary antibodies, appropriately
diluted in 4% BSA (cat. no. sc-2323; Santa Cruz Biotechnology,
Inc.) or 5% dried milk in TBS with 0.1% Tween-20, overnight at 4°C,
followed by a 2-h incubation with secondary antibodies at room
temperature. Ultimately, the proteins were visualized using the ECL
plus western blotting detection system (Cytiva) and a gel western
blot imaging system (G: BOX; Syngene Europe). The relative
expression of the protein bands was semi-quantified using ImageJ
version 1.53 software (National Institutes of Health).
Statistical analysis
The bar graphs show the mean ± standard deviation of
three independent experiments. The Student's two-tailed unpaired
t-test for independent samples was used to analyze the
statistically significant differences between treated and control
cells. Comparisons among treatment groups were performed by one-way
analysis of variance, followed by the Bonferroni post hoc test
using SPSS version 26.0 (IBM Corp.). P<0.05 was considered to
indicate a statistically significant difference.
Results
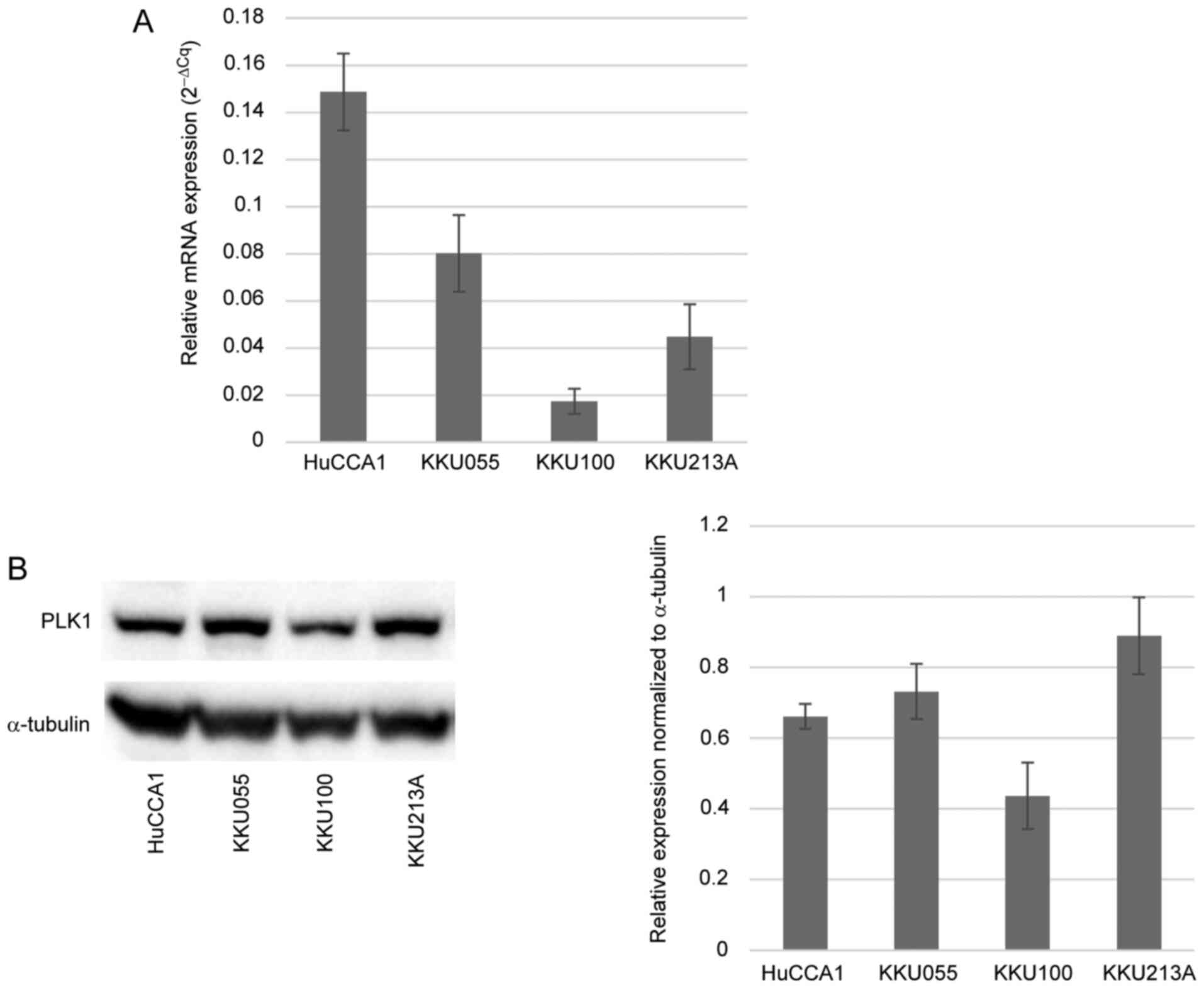
PLK1 is upregulated in CCA cell
lines
the level of PLK1 was assessed in four different CCA
cell lines: HuCCA1, KKU055, KKU100 and KKU213A, by detecting the
gene and protein expression levels. These CCA cell lines are widely
used in CCA research. PLK1 mRNA expression was quantified by
RT-qPCR, with relative mRNA expression normalized to a reference
gene (Fig. 1A). PLK1 protein
expression was detected by western blotting and the results
revealed a substantial presence of PLK1 protein in CCA cell lines
(Fig. 1B). Notably, among the four
CCA cell lines, KKU100 exhibited the lowest expression of PLK1 at
both the mRNA and protein levels.
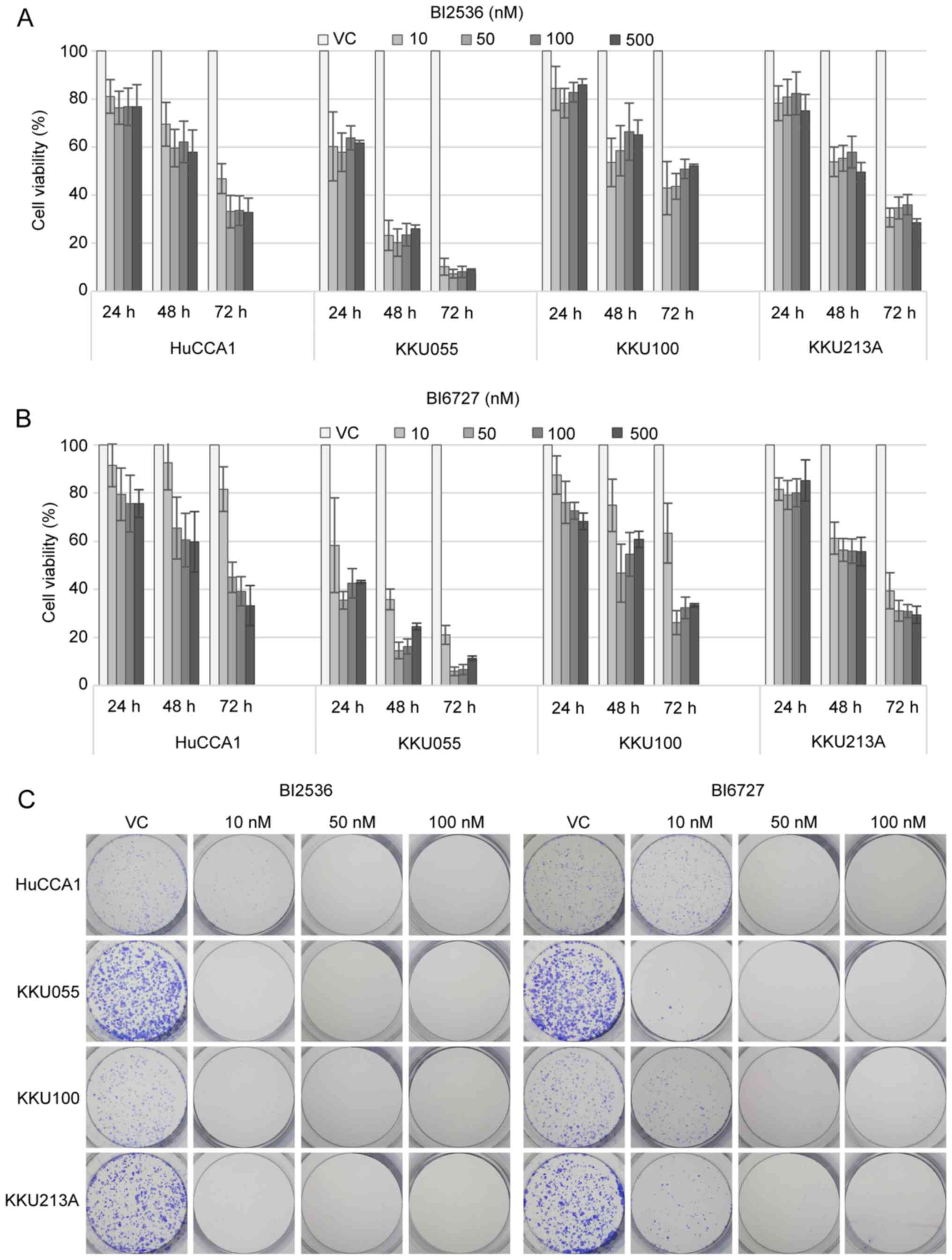
BI2536 and BI6727 suppress the
proliferation of CCA cell lines
To determine the effects of PLK1 inhibition on CCA
cells, BI2536 and BI6727, two potent PLK1 inhibitors, were used to
treat four CCA cell lines, HuCCA1, KKU055, KKU100 and KKU213A,
across a range of increasing concentrations (0–500 nM) and
treatment durations (24–72 h). BI2536 (Fig. 2A) and BI6727 (Fig. 2B) exhibited a time-dependent
reduction in cell viability among CCA cell lines. A summary of the
half maximal inhibitory concentration (IC50) values of
both BI2536 and BI6727 in all cell lines tested at each treatment
time (24–72 h) is shown in Table
SII. Among the four cell lines, KKU055 displayed the highest
sensitivity, while HuCCA1 exhibited the lowest sensitivity. The
concentrations of PLK1 inhibitors at 10 and 100 nM were selected
for subsequent experiments based on the observation that the
concentrations higher than 100 nM for both BI2536 and BI6727 did
not yield a more pronounced inhibitory effect. Moreover,
considering the varying sensitivity to PLK1 inhibitors across the
four distinct cell lines, it was decided to employ a broad range of
concentrations (10-fold), to capture and analyze the various
responses. Additionally, to further investigate the
antiproliferative effect of PLK1 inhibitors, the ability of BI2536-
and BI6727-treated cells to form colonies was examined. The
results, as shown in Fig. 2C,
demonstrated an inhibition in the colony-forming capability of the
treated cells across all four CCA cell lines.
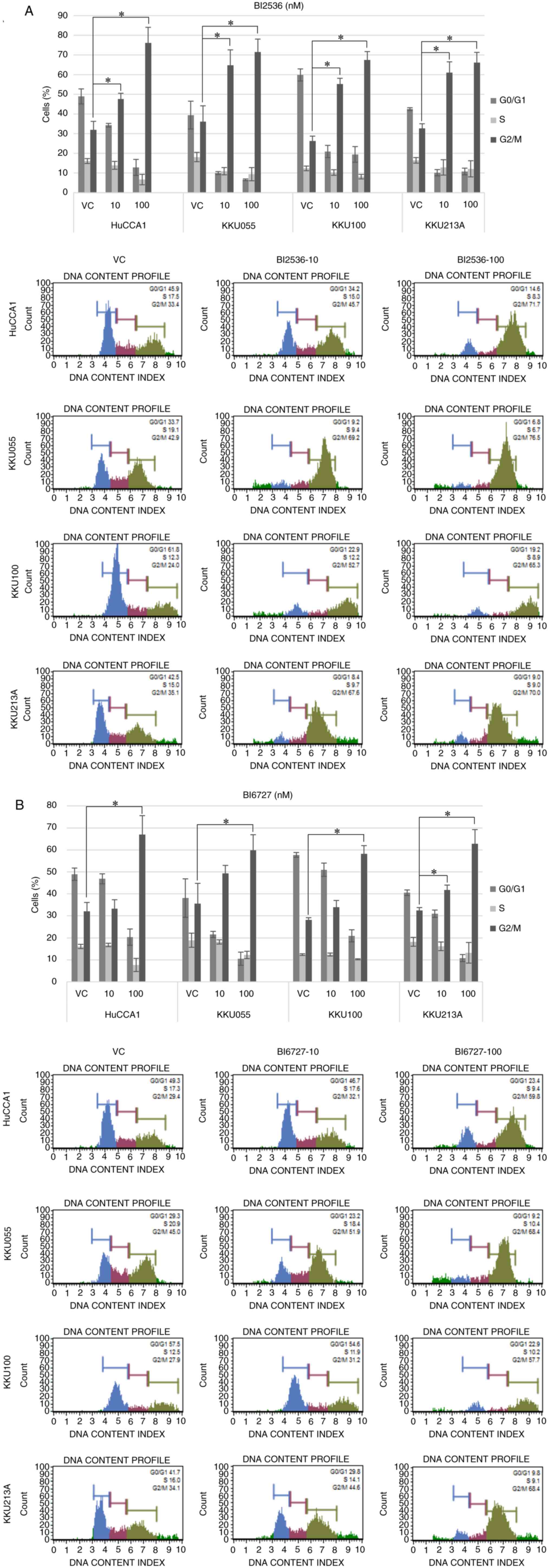
BI2536 and BI6727 induce
G2/M-phase arrest in CCA cells
To investigate the effects of PLK1 inhibition on
cell cycle distribution, cells were treated with BI2536 and BI6727
for 24 h followed by cell cycle analysis. The population of cells
in G2/M phase was significantly increased in CCA cells
treated with both BI2536 (Fig. 3A)
and BI6727 (Fig. 3B), as compared
to cells treated with the vehicle control. The induction of
G2/M-phase arrest was significantly pronounced in all
four CCA cell lines. Furthermore, it was observed that BI6727 at a
concentration of 10 nM displayed a lesser capacity to induce cell
cycle arrest when compared with BI2536 at the same
concentration.
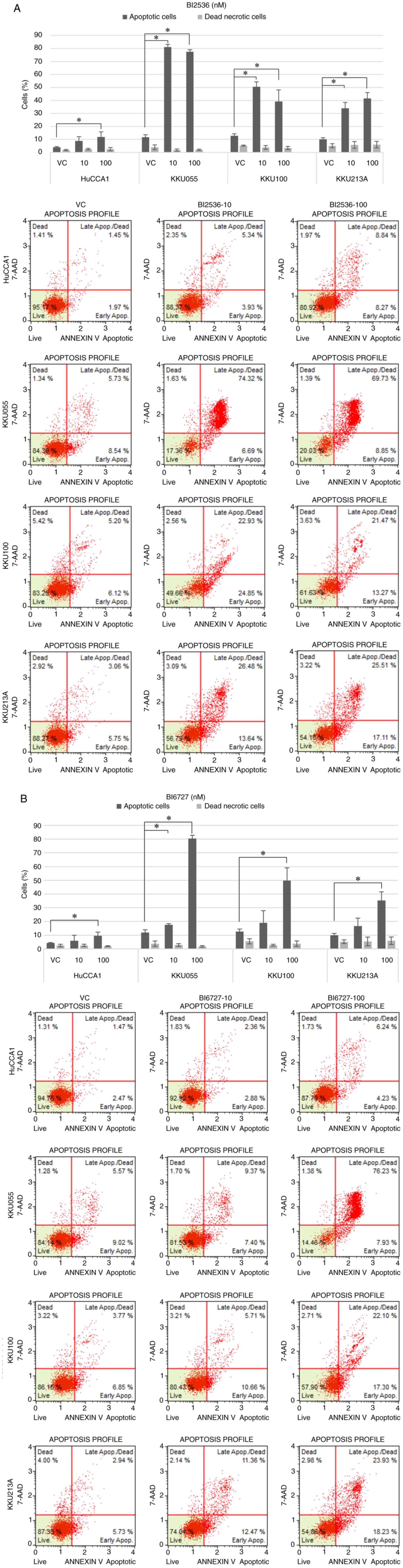
BI2536- and BI6727-treated CCA cells
undergo apoptosis
Next, the effect of PLK1 inhibition on cell
apoptosis was determined. The effect of PLK1 inhibitors on cell
cycle arrest observed after a 24-h treatment suggested that the
inhibitors were able to halt cell cycle progression, the present
study thus aimed to determine whether cell apoptosis manifested at
a later time point, at 48 h post-treatment. CCA cells were treated
with 10 and 100 nM BI2536 (Fig. 4A)
and BI6727 (Fig. 4B), followed by
Annexin V/7-AAD double staining followed by flow cytometry to
detect apoptotic cells. Cells treated with both inhibitors
exhibited an increase in total apoptotic cells (early and late
apoptotic cells). Furthermore, there was no increase in dead
necrotic cells observed with either of the PLK1 inhibitors, as
indicated by the absence of significant differences in dead
necrotic cells across all treatment conditions. However, it was
revealed that the percentages of total apoptotic cells were notably
lower in the HuCCA1 cell line in comparison to the other CCA cell
lines.
Inhibiting PLK1 causes alterations in
mitotic proteins, and induces DNA damage and PARP cleavage
The present study proceeded to evaluate the levels
of mitotic proteins in CCA cells subjected to PLK1 inhibition,
since CCA cells treated with BI2536 and BI6727 displayed
G2/M arrest. Treatment with BI2536 and BI6727 led to the
upregulation of PLK1-related proteins, including aurora kinase A,
p-PLK1 (T210), PLK1 and cyclin B1, in CCA cell lines (Fig. 5A). However, treatment with PLK1
inhibitors did not result in an elevation in the expression levels
of cyclin B1 in KKU213A cells. The upregulation of these proteins
signified the activation of the spindle assembly checkpoint. This
confirmed that inhibition of PLK1 initiated a state of mitotic
arrest and subsequently induced apoptotic cell death.
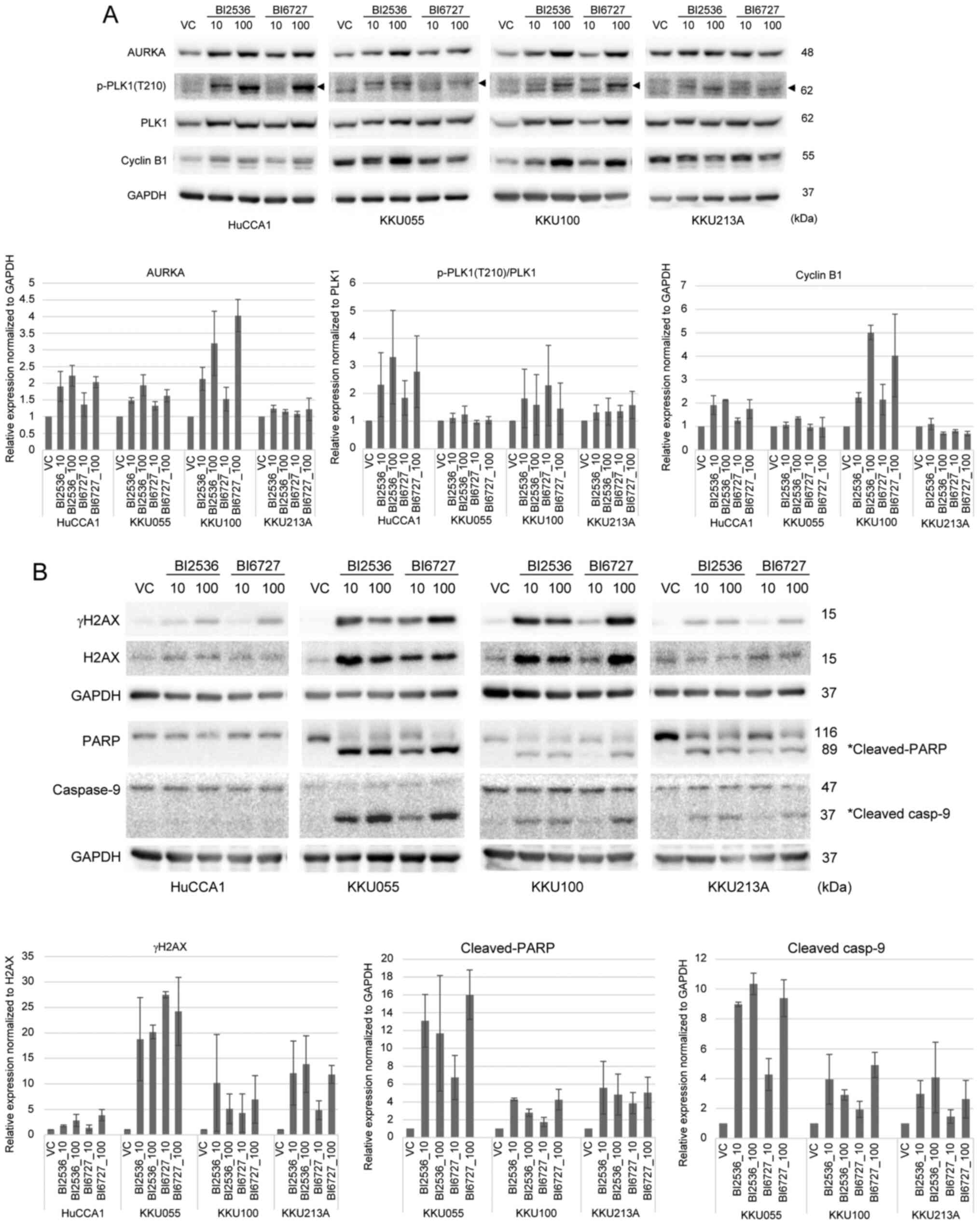 | Figure 5.BI2536 and BI6727 upregulate the
expression of (A) mitotic proteins and (B) induce DNA damage, as
well as cleavage of PARP and caspase-9. CCA cells were treated with
VC, or 10 and 100 nM BI2536 and BI6727 for 24 h. The expression of
AURKA, p-PLK1 (T210), PLK1, cyclin B1, gH2AX, H2AX, PARP, caspase-9
and GAPDH proteins were detected by western blot analysis.
Representative western blot images are shown. The bar graphs
demonstrate the relative expression of the proteins normalized to
GAPDH and VC-treated cells. The relative expression of p-PLK1
(T210) was normalized to total PLK1 and VC-treated cells. The
relative expression of gH2AX was normalized to total H2AX and
VC-treated cells. The relative protein expression was calculated
from three independent experiments. PARP, poly (ADP-ribose)
polymerase; CCA, cholangiocarcinoma; VC, vehicle control; AURKA,
aurora kinase A; p-, phosphorylated; PLK1, polo-like kinase 1;
H2AX, histone H2AX. |
Additionally, the effect of PLK1 inhibition on the
induction of DNA damage leading to cell death has been previously
reported (45). To verify the
possible mechanisms of PLK1 inhibitor-induced cell death, the
levels of DNA strand breaks were assessed via the detection of
phosphorylated histone H2AX at S139 (called γH2AX), as well as the
detection of apoptotic markers, PARP and caspase-9 cleavage,
following BI2536 and BI6727 treatment. The present study observed
the upregulation of gH2AX across all four CCA cell lines, thus
indicating an increase in DNA damage after PLK1 inhibition
(Fig. 5B). Furthermore, both BI2536
and BI6727 caused PARP and caspase-9 cleavage in KKU055, KKU100 and
KKU213A cell lines. These results are in line with the percentages
of total apoptotic cells shown in Fig.
4B, whereby HuCCA1 cells displayed a comparatively lower count
of apoptotic cells in relation to the other cell lines. Similar
trends were observed upon treatment with BI6727.
Depletion of the PLK1 gene exhibits
antiproliferative effects on CCA cell lines
To further validate the antiproliferative effects of
PLK1 inhibitors, PLK1 gene silencing by siRNA was performed in all
four CCA cell lines. Cells were transfected with PLK1 siRNA or
control siRNA for 48 h, followed by the analysis of cell viability,
cell cycle distribution and cell apoptosis. As shown in Fig. 6A, cells transfected with PLK1 siRNA
exhibited diminished cell viability in comparison to cells
transfected with control siRNA. Silencing of PLK1 induced
G2/M arrest, as demonstrated in Fig. 6B, where representative histograms
depicted the distribution of cells across
G0/G1, S, and G2/M phases
(Fig. S1A). Although the histogram
for siPLK1-transfected KKU055 cells displayed a lower peak in the
G2/M phase, possibly due to the lower number of cells
counted by the analyzer compared to siSC-transfected KKU055 cells,
the percentage of cells in the G2/M phase was higher in
siPLK1-transfected cells compared to siSC-transfected cells, as
illustrated in Fig. 6B. Moreover,
silencing of PLK1 triggered cell apoptosis, particularly notable in
KKU055 and KKU213A, as shown in Fig.
6C. The representative dot plots showing Annexin V/7-AAD double
staining of CCA cells transfected with siSC or siPLK1 were shown in
Fig. S1B. There was no significant
increase in dead necrotic cells in siPLK1- and siSC-transfected
cells. However, KKU100 cells transfected with both control siRNA
and PLK1 siRNA displayed a substantial population of total
apoptotic cells, potentially attributed to the toxicity of the
transfection reagent. PLK1 gene depletion was confirmed by the
reduction in PLK1 protein expression (Fig. 6D). Moreover, the upregulation of
mitotic proteins, including aurora kinase A and cyclin B1, was
consistent with the results obtained in response to PLK1
inhibitors. The silencing of PLK1 induced DNA damage and cell
apoptosis, as shown by increased γH2AX and PARP cleavage,
respectively. Overall, these results agreed with the effects of
BI2536 and BI6727 treatment on CCA cells.
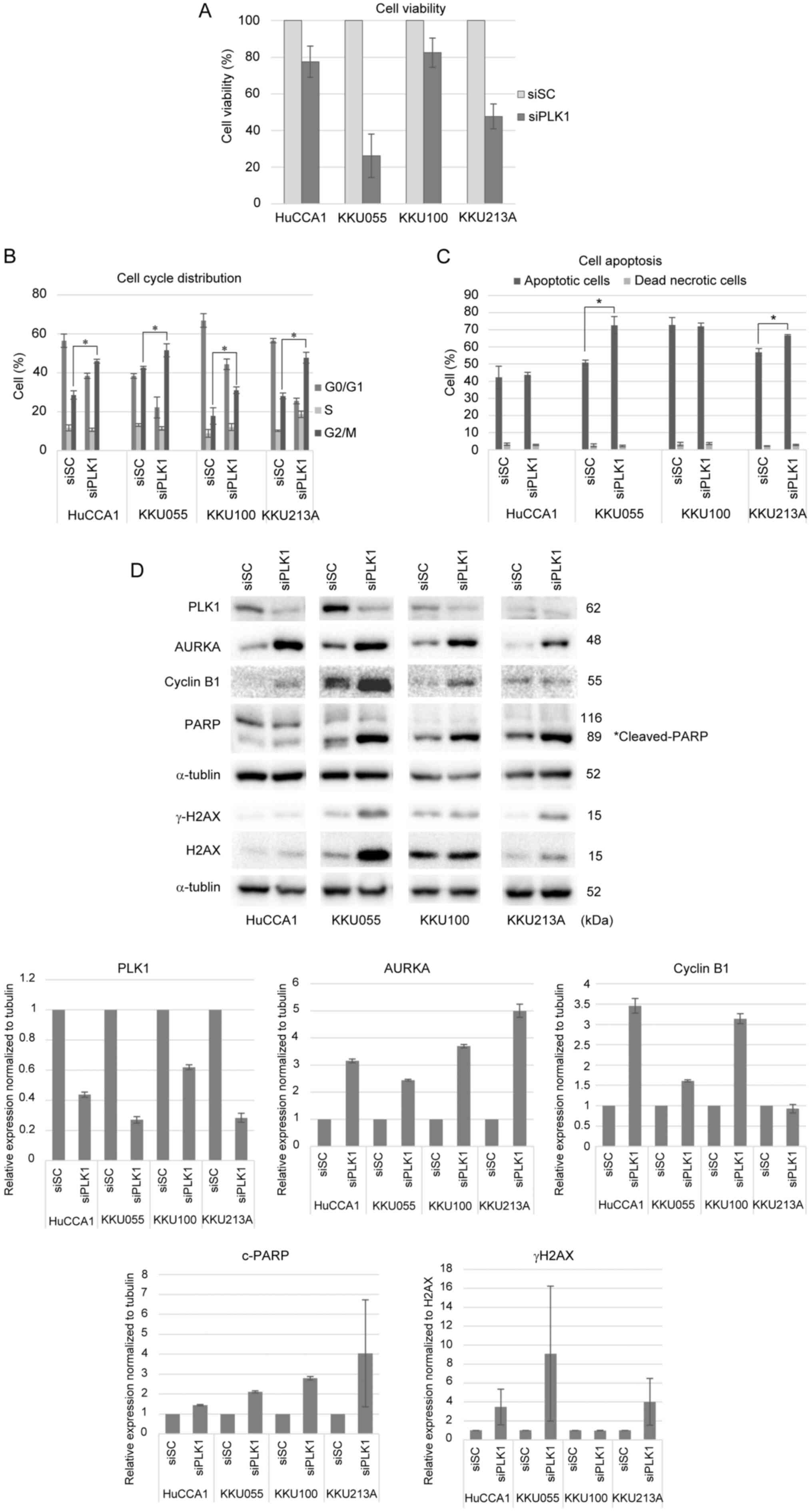 | Figure 6.Silencing PLK1 shows
antiproliferative effects on CCA cell lines. (A) CCA cells were
transfected with 100 nM siSC or siPLK1 for 48 h and cell viability
was measured by MTT assay. The bar graphs show the normalization to
siSC-transfected cells. (B) Cell cycle analysis was performed by
flow cytometry using the Muse cell cycle assay kit. The percentages
of cells in each phase of the cell cycle are shown. Representative
cell cycle distribution of CCA cells transfected with siSC or
siPLK1 were shown in Fig. S1A. (C)
Total apoptotic cells were determined by flow cytometry using the
Muse Annexin-V and Dead cell kit. The percentages of total
apoptotic and dead necrotic cells are shown. The results are
presented as the mean ± SD of at least three independent
experiments. Representative plots showing Annexin V/7-AAD double
staining of CCA cells transfected with siSC or siPLK1 are shown in
Fig. SIB. A significant difference
was observed when comparing siPLK1-transfected cells with
siSC-transfected cells (*P<0.05). (D) Protein levels of AURKA,
PLK1, cyclin B1, gH2AX, H2AX, PARP and a-tubulin were examined by
western blotting. Representative western blot images are shown.
PLK1, polo-like kinase 1; CCA, cholangiocarcinoma; si, short
interfering; SC, negative control; AURKA, aurora kinase A; PLK1,
polo-like kinase 1; H2AX, histone H2AX; PARP, poly (ADP-ribose)
polymerase. |
Discussion
Alterations in the expression of genes related to
the regulation of the cell cycle are frequently observed in various
types of cancer. PLK1, a protein kinase that is involved in
multiple stages of cell cycle progression, is often overexpressed
and is correlated with aggressiveness and unfavorable prognosis
across various types of cancer (12,14).
Therefore, inhibiting PLK1 is a potential therapeutic approach for
cancer treatment. The present study examined the inhibitory impacts
of PLK1 inhibition on four CCA cell lines derived from Thai
patients with CCA: HuCCA1, KKU055, KKU100 and KKU213A. These cell
lines represented both intrahepatic (HuCCA1, KKU055, KKU213A) and
extrahepatic (KKU100) CCA, ensuring a comprehensive examination of
different subtypes of the disease within the Thai population.
The expression of PLK1 was initially detected in the
four CCA cell lines. The analysis revealed that three out of the
four cell lines exhibited substantial PLK1 expression. KKU100
demonstrated the lowest levels of PLK1 expression at both the mRNA
and protein levels. BI2536 and BI6727, two potent PLK1 inhibitors,
inhibited cell viability and colony formation in all four CCA cell
lines, with slightly different sensitivity. The most sensitive cell
line was KKU055, which agreed with the findings of Tepsiri et
al (37) showing that KKU055
was the most sensitive cell toward chemotherapeutic drugs, such as
cisplatin, gemcitabine and 5-fluorouracil. Notably, it was
discovered that the expression levels of PLK1 did not determine the
sensitivity to PLK1 inhibition. Despite KKU100 cells exhibiting the
lowest PLK1 expression, they were not the most sensitive to PLK1
inhibition. This observation suggested the involvement of other
factors, such as DNA damage response and DNA repair mechanisms, in
modulating the response to PLK1 inhibition. Investigation into
these factors should be extended.
Following the cell proliferation assay, 10 and 100
nM concentrations of PLK1 inhibitors were chosen for subsequent
experiments. The IC50 values were not directly used in
subsequent experiments because the present study aimed to explore
the concentrations and IC50 values to capture a broader
range of responses and consider the different sensitivities to PLK1
inhibitions among cell lines; using IC50 might overlook
potential differences in their responsiveness. While
IC50 values provide valuable insights into the
inhibitory potency, they may not be consistent with each experiment
setup. The present study opted to conduct subsequent experiments,
at either 24 or 48 h post-treatment due to significant cell death
occurring at 72 h after treatment, especially in the case of KKU055
cells. PLK1 inhibition via BI2536, BI6727 and siRNA markedly
induced G2/M-phase arrest. These results are similar to
those previously reported in several types of cancer, such as
glioblastoma, non-small cell lung cancer and cervical cancer
(31,46,47).
Several studies have explored the effects of a 100 nM concentration
of PLK1 inhibitor (BI2536) across different cell types. Steegmaier
et al (25) demonstrated
that this concentration arrested HeLa cells with monopolar
spindles, characteristic of PLK1 inhibition. Similarly, Pezuk et
al (46) and Lu et al
(48) found that 100 nM BI2536
induced G2/M arrest of glioblastoma cells and rat
cardiac fibroblast, respectively. However, it remains difficult to
entirely dismiss the possibility of off-target effects at this
concentration. Similarly, the present study was unable to
definitively exclude the potential for off-target effects. Mitotic
arrest is one of the effects of PLK1 inhibition and this event
leads to increased DNA damage followed by cell death. The present
study also observed increasing DNA damage, as revealed by
upregulation of γH2AX, a marker of DNA double-strand breaks
(49), in all cell lines. Moreover,
both PLK1 inhibitor treatments and PLK1 silencing induced apoptotic
cell death. Cleavage of PARP by caspase enzymes serves as a
hallmark of apoptosis, whereas depletion of NAD+ and ATP
leads to induction of necrosis (50). The present study observed the
cleavage of both PARP and caspase 9 following PLK1 inhibition,
using both PLK1 inhibitors and PLK1 gene depletion. Notably, Herceg
and Wang (50) demonstrated that
PARP cleavage, mediated by caspase activation, prevents necrosis
induction during apoptosis. Although changes in necrosis markers,
NAD+ and ATP depletion, were not measured, the findings of the
present study, including the cleavage of caspase 9 and PARP, along
with the results from Annexin V/7-AAD double staining via flow
cytometry and the absence of an increase in dead necrotic cells,
suggested that PLK1 inhibition leads to apoptosis rather than
necrosis. However, it may not be possible to confirm the effect of
PLK1 silencing by siRNA on cell apoptosis of the KKU100 cell line,
due to the toxicity of the transfection procedure in this cell
type. Among the four CCA cell lines, HuCCA1 cells did not show a
markedly increased population of apoptotic cells and cleavage of
PARP protein compared with the other cell lines, despite mitotic
arrest and DNA damage being observed. This result indicated that
increasing DNA damage can occur due to prolonged mitotic arrest,
irrespective of the apoptotic pathway. A deeper study of the
molecular mechanism should be conducted to pursue a more
comprehensive understanding of these effects. The varying responses
to PLK1 inhibition across the four CCA cell lines were studied,
particularly the induction of apoptosis alongside the observed
mitotic arrest in all cell lines. It is planned to explore the
mechanisms involved in these differences in sensitivity following
PLK1 inhibition in CCA cells, for instance, the involvement of DNA
damage response and DNA repair machinery that could play a role in
this matter.
The spindle assembly checkpoint (SAC), also referred
to as the mitotic checkpoint, is recognized for stopping mitotic
progression during cell cycle dysfunction, ensuring the accurate
alignment of chromosomes before segregation. The SAC primarily
functions to postpone the transition to anaphase until there is
proper attachment of kinetochores to microtubules (51). PLK1 participates in this process,
with its activity notably elevated on unattached kinetochores,
indicating a potential role for PLK1 in the regulation of
kinetochore attachment or SAC (52). In the present study, the detection
of mitotic proteins following treatment with BI2536 and BI6727
showed upregulated aurora kinase A, p-PLK1 (T210), PLK1 and cyclin
B1 in the CCA cell lines. These proteins have been found to be
stimulated in mitotic-arrested cells and SAC-activated cells upon
PLK1 inhibition (28,31,53).
The CDK1-cyclin B1 complex is an important component of the SAC
(54,55). Aurora kinase A is an upstream
regulator of PLK1, which phosphorylates and activates PLK1 at
Thr210 (56). The present study
agreed with the findings from Choi et al (31), showing that inhibition of PLK1
activity with BI2536 preserves the absence of kinetochore tension,
leading to prolonged activation of the SAC. This event activates
phosphorylation of PLK1 at Thr210 by aurora kinase A. Choi et
al (31) also show that
BI2536-treated non-small lung cancer cells experience arrest at
prometaphase and monopolar spindles are formed. Furthermore, BI2536
treatment induces mitotic arrest by impeding the attachment of
kinetochores to microtubules, a process dependent on PLK1 activity
(31). In addition, a study from
Steegmaier et al (25)
demonstrates that BI2536-treated cells are arrested in prometaphase
and contain aberrant mitotic spindles. BI6727 has also been shown
to accumulate mitotic cells with monopolar spindles (27). On the other hand, the accumulation
of PLK1 protein levels in BI2536- and BI6727-treated cells observed
in the present study was in contrast to the reduced accumulation of
PLK1 observed in BI6727-treated chronic myeloid leukemia (57) and Burkitt lymphoma cells (30). These studies report that BI6727 can
reduce activated PLK1.
PLK1 is primarily recognized for its pivotal role in
several events during mitosis (14). However, emerging evidence suggests
its involvement in the DNA damage response. DNA damage occurring
during mitosis triggers the induction of mitotic arrest to avoid
the generation of abnormal daughter cells (58). Studies have demonstrated that PLK1
kinase regulates several proteins involved in DNA damage response,
particularly the ATM/ATR/Chk pathway (58,59).
PLK1 can phosphorylate and inhibit Chk2, compromising the DNA
damage checkpoint and facilitating cell cycle progression despite
the presence of DNA damage (60).
Additionally, PLK1 has been shown to interact with and
phosphorylate p53, influencing its activity (61). Tamura et al (62) revealed the function of PLK1 in
phosphorylating and modulating the activity of Bcl-2 family
proteins, key regulators of apoptosis. These findings underscore
the role of PLK1 in cellular processes, including its effect on the
response to various cellular stresses, such as DNA damage.
While PLK1 inhibition has demonstrated
anti-proliferative effects on cancer cells, concerns have been
raised regarding its potential side effects on normal cells. In a
study by Liu et al (63),
the depletion of PLK1 in normal hTERT-RPE1 and MCF10A cell lines
did not affect cell proliferation or cell cycle progression. Lu
(48) et al investigated the
effects of BI2536 on primary cardiac fibroblast and cardiomyocytes.
While BI2536 did not generate adverse effects in cardiomyocytes,
which are differentiated cells, it affected primary fibroblast by
inducing mitotic arrest with monopolar spindles, leading to cell
death after prolonged arrest. A number of PLK1 inhibitors have been
developed and tested in clinical trials (12). BI2536, for instance, has undergone
evaluation in NSCLC patients, both as a monotherapy and in
combination therapy (64,65). BI6727 has reached phase III clinical
trial for AML (66). However, in a
number of cases, these inhibitors have shown limited efficacy and
high toxicity. Efforts to explore strategies for limiting PLK1
activity in cancer cells continue. The combined use of PLK1
inhibitors with conventional chemotherapeutics has shown promise
and warrants further investigation. These aspects of study remain
relatively limited in cholangiocarcinoma research.
While preparing the present manuscript, a recent
study by Riantana et al (34) was published regarding the effects of
two potent PLK1 inhibitors, BI6727 and GSK461364A, on inducing
G2/M arrest and apoptosis in two CCA cell lines (KKU100
and KKU213A). Some of the results presented in the present study
agree with this recent publication. The levels of PLK1 protein in
KKU100 and KKU213A cells are consistent with the present findings.
Furthermore, similar effects of BI6727 on inhibiting cell
proliferation were observed, inducing G2/M cell arrest
and triggering apoptosis (as indicated by PARP cleavage). However,
there is enhanced support for our current findings by using siRNA
to inhibit PLK1 expression and by detecting alterations in mitotic
proteins following PLK1 inhibition.
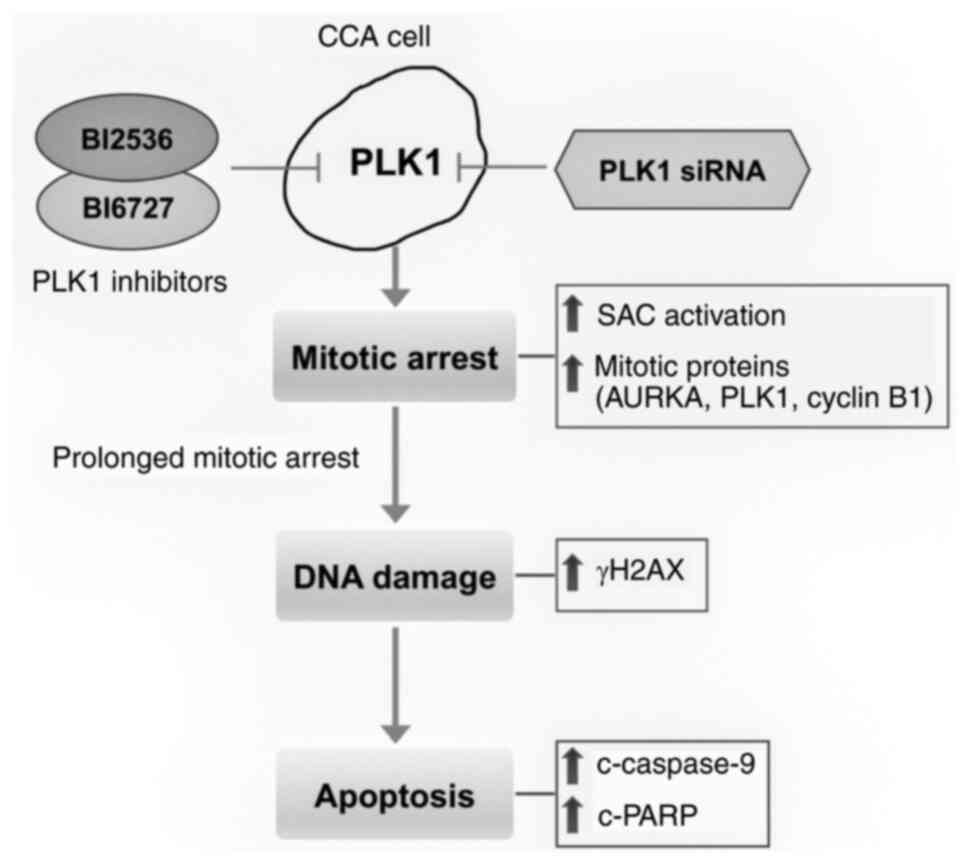
In conclusion, the results of the present study
indicated that inhibition of PLK1 by small molecule inhibitors,
BI2536 and BI6727, and via the siRNA method, can suppress cell
proliferation, induce mitotic arrest and DNA damage, and trigger
cell apoptosis in CCA cells (Fig.
7). Regarding CCA, the present study demonstrated convincing
results by using both inhibitors and siRNA to explore the effects
of PLK1 inhibition. The strong induction of apoptotic cell death
did not occur in all cell lines. While the present study used four
different CCA cell lines, their representation of the heterogeneity
found in human CCA is a valid concern. The present study recognized
that the results obtained from different cell lines may vary,
emphasizing the need to validate across a broader range of cases.
Further research is needed to understand the specific mechanisms
involved in determining the response to PLK1 inhibition and to
identify potential biomarkers that can predict sensitivity to PLK1
inhibitors. The present study identified PLK1 as a promising
treatment target for CCA cells. To validate its effectiveness,
future plans will involve both animal experiments and clinical
research.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
We would like to thank Dr Stitaya Sirisinha
(Department of Microbiology, Faculty of Science, Mahidol
University, Thailand) for kindly providing HuCCA1 cell line, and Dr
Banchob Sripa (Khon Kaen University, Thailand) for establishing and
commercializing KKU100 cell line through the JCRB cell bank.
Funding
The present study was supported by Thailand Science Research and
Innovation, Chulabhorn Research Institute Thailand (grant nos.
2536699/42117 and 48291/4691912).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
BM and MR conceptualized and designed the study. BM
developed the methodology, conducted the experiments and drafted
the manuscript. BM, JC and PS analyzed and interpreted the data.
BM, JC, PS and MR reviewed and revised the manuscript. BM and PS
confirm the authenticity of all the raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Parkin DM, Ohshima H, Srivatanakul P and
Vatanasapt V: Cholangiocarcinoma: Epidemiology, mechanisms of
carcinogenesis and prevention. Cancer Epidemiol Biomarkers Prev.
2:537–544. 1993.PubMed/NCBI
|
|
2
|
Patel T: Worldwide trends in mortality
from biliary tract malignancies. BMC Cancer. 2:102002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shin HR, Oh JK, Masuyer E, Curado MP,
Bouvard V, Fang YY, Wiangnon S, Sripa B and Hong ST: Epidemiology
of cholangiocarcinoma: An update focusing on risk factors. Cancer
Sci. 101:579–585. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gores GJ: Cholangiocarcinoma: Current
concepts and insights. Hepatology. 37:961–969. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lazaridis KN and Gores GJ:
Cholangiocarcinoma. Gastroenterology. 128:1655–1667. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Khan SA, Davidson BR, Goldin RD, Heaton N,
Karani J, Pereira SP, Rosenberg WM, Tait P, Taylor-Robinson SD,
Thillainayagam AV, et al: Guidelines for the diagnosis and
treatment of cholangiocarcinoma: An update. Gut. 61:1657–1669.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Friman S: Cholangiocarcinoma-current
treatment options. Scand J Surg. 100:30–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hezel AF and Zhu AX: Systemic therapy for
biliary tract cancers. Oncologist. 13:415–423. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eckel F and Schmid RM: Chemotherapy in
advanced biliary tract carcinoma: A pooled analysis of clinical
trials. Br J Cancer. 96:896–902. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Valle JW, Wasan H, Johnson P, Jones E,
Dixon L, Swindell R, Baka S, Maraveyas A, Corrie P, Falk S, et al:
Gemcitabine alone or in combination with cisplatin in patients with
advanced or metastatic cholangiocarcinomas or other biliary tract
tumours: A multicentre randomised phase II study-the UK ABC-01
study. Br J Cancer. 101:621–627. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chaisaingmongkol J, Budhu A, Dang H,
Rabibhadana S, Pupacdi B, Kwon SM, Forgues M, Pomyen Y,
Bhudhisawasdi V, Lertprasertsuke N, et al: Common molecular
subtypes among asian hepatocellular carcinoma and
cholangiocarcinoma. Cancer Cell. 32:57–70.e53. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Iliaki S, Beyaert R and Afonina IS:
Polo-like kinase 1 (PLK1) signaling in cancer and beyond. Biochem
Pharmacol. 193:1147472021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weiß L and Efferth T: Polo-like kinase 1
as target for cancer therapy. Exp Hematol Oncol. 1:382012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schmucker S and Sumara I: Molecular
dynamics of PLK1 during mitosis. Mol Cell Oncol. 1:e9545072014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumar S, Sharma AR, Sharma G, Chakraborty
C and Kim J: PLK-1: Angel or devil for cell cycle progression.
Biochim Biophys Acta. 1865:190–203. 2016.PubMed/NCBI
|
|
16
|
He ZL, Zheng H, Lin H, Miao XY and Zhong
DW: Overexpression of polo-like kinase1 predicts a poor prognosis
in hepatocellular carcinoma patients. World J Gastroenterol.
15:4177–4182. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weichert W, Kristiansen G, Winzer KJ,
Schmidt M, Gekeler V, Noske A, Müller BM, Niesporek S, Dietel M and
Denkert C: Polo-like kinase isoforms in breast cancer: Expression
patterns and prognostic implications. Virchows Arch. 446:442–450.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Takahashi T, Sano B, Nagata T, Kato H,
Sugiyama Y, Kunieda K, Kimura M, Okano Y and Saji S: Polo-like
kinase 1 (PLK1) is overexpressed in primary colorectal cancers.
Cancer Sci. 94:148–152. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jang YJ, Kim YS and Kim WH: Oncogenic
effect of Polo-like kinase 1 expression in human gastric
carcinomas. Int J Oncol. 29:589–594. 2006.PubMed/NCBI
|
|
20
|
Weichert W, Denkert C, Schmidt M, Gekeler
V, Wolf G, Köbel M, Dietel M and Hauptmann S: Polo-like kinase
isoform expression is a prognostic factor in ovarian carcinoma. Br
J Cancer. 90:815–821. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gleixner KV, Ferenc V, Peter B, Gruze A,
Meyer RA, Hadzijusufovic E, Cerny-Reiterer S, Mayerhofer M, Pickl
WF, Sillaber C and Valent P: Polo-like kinase 1 (Plk1) as a novel
drug target in chronic myeloid leukemia: Overriding imatinib
resistance with the Plk1 inhibitor BI 2536. Cancer Res.
70:1513–1523. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schöffski P: Polo-like kinase (PLK)
inhibitors in preclinical and early clinical development in
oncology. Oncologist. 14:559–570. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu X and Erikson RL: Polo-like kinase
(Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad
Sci USA. 100:5789–5794. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barr FA, Silljé HH and Nigg EA: Polo-like
kinases and the orchestration of cell division. Nat Rev Mol Cell
Biol. 5:429–440. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Steegmaier M, Hoffmann M, Baum A, Lénárt
P, Petronczki M, Krssák M, Gürtler U, Garin-Chesa P, Lieb S, Quant
J, et al: BI 2536, a potent and selective inhibitor of polo-like
kinase 1, inhibits tumor growth in vivo. Curr Biol. 17:316–322.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lénárt P, Petronczki M, Steegmaier M, Di
Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N and Peters JM: The
small-molecule inhibitor BI 2536 reveals novel insights into
mitotic roles of polo-like kinase 1. Curr Biol. 17:304–315. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rudolph D, Steegmaier M, Hoffmann M,
Grauert M, Baum A, Quant J, Haslinger C, Garin-Chesa P and Adolf
GR: BI 6727, a Polo-like kinase inhibitor with improved
pharmacokinetic profile and broad antitumor activity. Clin Cancer
Res. 15:3094–3102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cheng CY, Liu CJ, Huang YC, Wu SH, Fang HW
and Chen YJ: BI2536 induces mitotic catastrophe and
radiosensitization in human oral cancer cells. Oncotarget.
9:21231–21243. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schmit TL, Zhong W, Setaluri V, Spiegelman
VS and Ahmad N: Targeted depletion of Polo-like kinase (Plk) 1
through lentiviral shRNA or a small-molecule inhibitor causes
mitotic catastrophe and induction of apoptosis in human melanoma
cells. J Invest Dermatol. 129:2843–2853. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen E and Pei R: BI6727, a polo-like
kinase 1 inhibitor with promising efficacy on Burkitt lymphoma
cells. J Int Med Res. 48:3000605209260932020.PubMed/NCBI
|
|
31
|
Choi M, Kim W, Cheon MG, Lee CW and Kim
JE: Polo-like kinase 1 inhibitor BI2536 causes mitotic catastrophe
following activation of the spindle assembly checkpoint in
non-small cell lung cancer cells. Cancer Lett. 357:591–601. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thrum S, Lorenz J, Mössner J and Wiedmann
M: Polo-like kinase 1 inhibition as a new therapeutic modality in
therapy of cholangiocarcinoma. Anticancer Res. 31:3289–3299.
2011.PubMed/NCBI
|
|
33
|
Zhou Y, Xu L, Wang Z, Liu H, Zhang X, Shu
C, Zhang M, Wang T, Xu X, Pu X, et al: Sequentially targeting and
intervening mutual Polo-like Kinase 1 on CAFs and tumor cells by
dual targeting nano-platform for cholangiocarcinoma treatment.
Theranostics. 12:3911–3927. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Riantana H, Waenphimai O, Mahalapbutr P,
Karnchanapandh K, Vaeteewoottacharn K, Wongkham S and Sawanyawisuth
K: BI6727 and GSK461364A, potent PLK1 inhibitors induce G2/M arrest
and apoptosis against cholangiocarcinoma cell lines. Pathol Res
Pract. 248:1546782023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sirisinha S, Tengchaisri T, Boonpucknavig
S, Prempracha N, Ratanarapee S and Pausawasdi A: Establishment and
characterization of a cholangiocarcinoma cell line from a Thai
patient with intrahepatic bile duct cancer. Asian Pac J Allergy
Immunol. 9:153–157. 1991.PubMed/NCBI
|
|
36
|
Sripa B, Seubwai W, Vaeteewoottacharn K,
Sawanyawisuth K, Silsirivanit A, Kaewkong W, Muisuk K, Dana P,
Phoomak C, Lert-Itthiporn W, et al: Functional and genetic
characterization of three cell lines derived from a single tumor of
an Opisthorchis viverrini-associated cholangiocarcinoma
patient. Hum Cell. 33:695–708. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tepsiri N, Chaturat L, Sripa B, Namwat W,
Wongkham S, Bhudhisawasdi V and Tassaneeyakul W: Drug sensitivity
and drug resistance profiles of human intrahepatic
cholangiocarcinoma cell lines. World J Gastroenterol. 11:2748–2753.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sripa B, Leungwattanawanit S, Nitta T,
Wongkham C, Bhudhisawasdi V, Puapairoj A, Sripa C and Miwa M:
Establishment and characterization of an opisthorchiasis-associated
cholangiocarcinoma cell line (KKU-100). World J Gastroenterol.
11:3392–3397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fehling SC, Miller AL, Garcia PL, Vance RB
and Yoon KJ: The combination of BET and PARP inhibitors is
synergistic in models of cholangiocarcinoma. Cancer Lett.
468:48–58. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Uthaisar K, Vaeteewoottacharn K, Seubwai
W, Talabnin C, Sawanyawisuth K, Obchoei S, Kraiklang R, Okada S and
Wongkham S: Establishment and characterization of a novel human
cholangiocarcinoma cell line with high metastatic activity. Oncol
Rep. 36:1435–1446. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jamnongsong S, Kueanjinda P, Buraphat P,
Sakornsakolpat P, Vaeteewoottacharn K, Okada S, Jirawatnotai S and
Sampattavanich S: Comprehensive drug response profiling and
pan-omic analysis identified therapeutic candidates and prognostic
biomarkers for Asian cholangiocarcinoma. iScience. 25:1051822022.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Saensa-Ard S, Leuangwattanawanit S,
Senggunprai L, Namwat N, Kongpetch S, Chamgramol Y, Loilome W,
Khansaard W, Jusakul A, Prawan A, et al: Establishment of
cholangiocarcinoma cell lines from patients in the endemic area of
liver fluke infection in Thailand. Tumour Biol.
39:10104283177259252017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lau DK, Mouradov D, Wasenang W, Luk IY,
Scott CM, Williams DS, Yeung YH, Limpaiboon T, Iatropoulos GF,
Jenkins LJ, et al: Genomic profiling of biliary tract cancer cell
lines reveals molecular subtypes and actionable drug targets.
iScience. 21:624–637. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Driscoll DL, Chakravarty A, Bowman D,
Shinde V, Lasky K, Shi J, Vos T, Stringer B, Amidon B, D'Amore N
and Hyer ML: Plk1 inhibition causes post-mitotic DNA damage and
senescence in a range of human tumor cell lines. PLoS One.
9:e1110602014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pezuk JA, Brassesco MS, Morales AG, de
Oliveira JC, de Paula Queiroz RG, Machado HR, Carlotti CG Jr, Neder
L, Scrideli CA and Tone LG: Polo-like kinase 1 inhibition causes
decreased proliferation by cell cycle arrest, leading to cell death
in glioblastoma. Cancer Gene Ther. 20:499–506. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xie FF, Pan SS, Ou RY, Zheng ZZ, Huang XX,
Jian MT, Qiu JG, Zhang WJ, Jiang QW, Yang Y, et al: Volasertib
suppresses tumor growth and potentiates the activity of cisplatin
in cervical cancer. Am J Cancer Res. 5:3548–3559. 2015.PubMed/NCBI
|
|
48
|
Lu B, Mahmud H, Maass AH, Yu B, van Gilst
WH, de Boer RA and Silljé HH: The Plk1 inhibitor BI 2536
temporarily arrests primary cardiac fibroblasts in mitosis and
generates aneuploidy in vitro. PLoS One. 5:e129632010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shiloh Y: ATM and related protein kinases:
Safeguarding genome integrity. Nat Rev Cancer. 3:155–168. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Herceg Z and Wang ZQ: Failure of
poly(ADP-ribose) polymerase cleavage by caspases leads to induction
of necrosis and enhanced apoptosis. Mol Cell Biol. 19:5124–5133.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sinha D, Duijf PHG and Khanna KK: Mitotic
slippage: An old tale with a new twist. Cell Cycle. 18:7–15. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ahonen LJ, Kallio MJ, Daum JR, Bolton M,
Manke IA, Yaffe MB, Stukenberg PT and Gorbsky GJ: Polo-like kinase
1 creates the tension-sensing 3F3/2 phosphoepitope and modulates
the association of spindle-checkpoint proteins at kinetochores.
Curr Biol. 15:1078–1089. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sanhaji M, Kreis NN, Zimmer B, Berg T,
Louwen F and Yuan J: p53 is not directly relevant to the response
of Polo-like kinase 1 inhibitors. Cell Cycle. 11:543–553. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
D'Angiolella V, Mari C, Nocera D, Rametti
L and Grieco D: The spindle checkpoint requires cyclin-dependent
kinase activity. Genes Dev. 17:2520–2525. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Penna LS, Henriques JAP and Bonatto D:
Anti-mitotic agents: Are they emerging molecules for cancer
treatment? Pharmacol Ther. 173:67–82. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Macůrek L, Lindqvist A, Lim D, Lampson MA,
Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB and Medema
RH: Polo-like kinase-1 is activated by aurora A to promote
checkpoint recovery. Nature. 455:119–123. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mancini M, De Santis S, Monaldi C,
Castagnetti F, Lonetti A, Bruno S, Dan E, Sinigaglia B, Rosti G,
Cavo M, et al: Polo-like kinase-1, Aurora kinase A and WEE1 kinase
are promising druggable targets in CML cells displaying
BCR::ABL1-independent resistance to tyrosine kinase inhibitors.
Front Oncol. 12:9011322022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jang YJ, Ji JH, Choi YC, Ryu CJ and Ko SY:
Regulation of Polo-like kinase 1 by DNA damage in mitosis.
Inhibition of mitotic PLK-1 by protein phosphatase 2A. J Biol Chem.
282:2473–2482. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hyun SY, Hwang HI and Jang YJ: Polo-like
kinase-1 in DNA damage response. BMB Rep. 47:249–255. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tsvetkov L, Xu X, Li J and Stern DF:
Polo-like kinase 1 and Chk2 interact and co-localize to centrosomes
and the midbody. J Biol Chem. 278:8468–8475. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ando K, Ozaki T, Yamamoto H, Furuya K,
Hosoda M, Hayashi S, Fukuzawa M and Nakagawara A: Polo-like kinase
1 (Plk1) inhibits p53 function by physical interaction and
phosphorylation. J Biol Chem. 279:25549–25561. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tamura Y, Simizu S, Muroi M, Takagi S,
Kawatani M, Watanabe N and Osada H: Polo-like kinase 1
phosphorylates and regulates Bcl-x(L) during pironetin-induced
apoptosis. Oncogene. 28:107–116. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu X, Lei M and Erikson RL: Normal cells,
but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol.
26:2093–2108. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sebastian M, Reck M, Waller CF, Kortsik C,
Frickhofen N, Schuler M, Fritsch H, Gaschler-Markefski B, Hanft G,
Munzert G and von Pawel J: The efficacy and safety of BI 2536, a
novel Plk-1 inhibitor, in patients with stage IIIB/IV non-small
cell lung cancer who had relapsed after, or failed, chemotherapy:
Results from an open-label, randomized phase II clinical trial. J
Thorac Oncol. 5:1060–1067. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ellis PM, Chu QS, Leighl N, Laurie SA,
Fritsch H, Gaschler-Markefski B, Gyorffy S and Munzert G: A phase I
open-label dose-escalation study of intravenous BI 2536 together
with pemetrexed in previously treated patients with non-small-cell
lung cancer. Clin Lung Cancer. 14:19–27. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Cortes J, Podoltsev N, Kantarjian H,
Borthakur G, Zeidan AM, Stahl M, Taube T, Fagan N, Rajeswari S and
Uy GL: Phase 1 dose escalation trial of volasertib in combination
with decitabine in patients with acute myeloid leukemia. Int J
Hematol. 113:92–99. 2021. View Article : Google Scholar : PubMed/NCBI
|