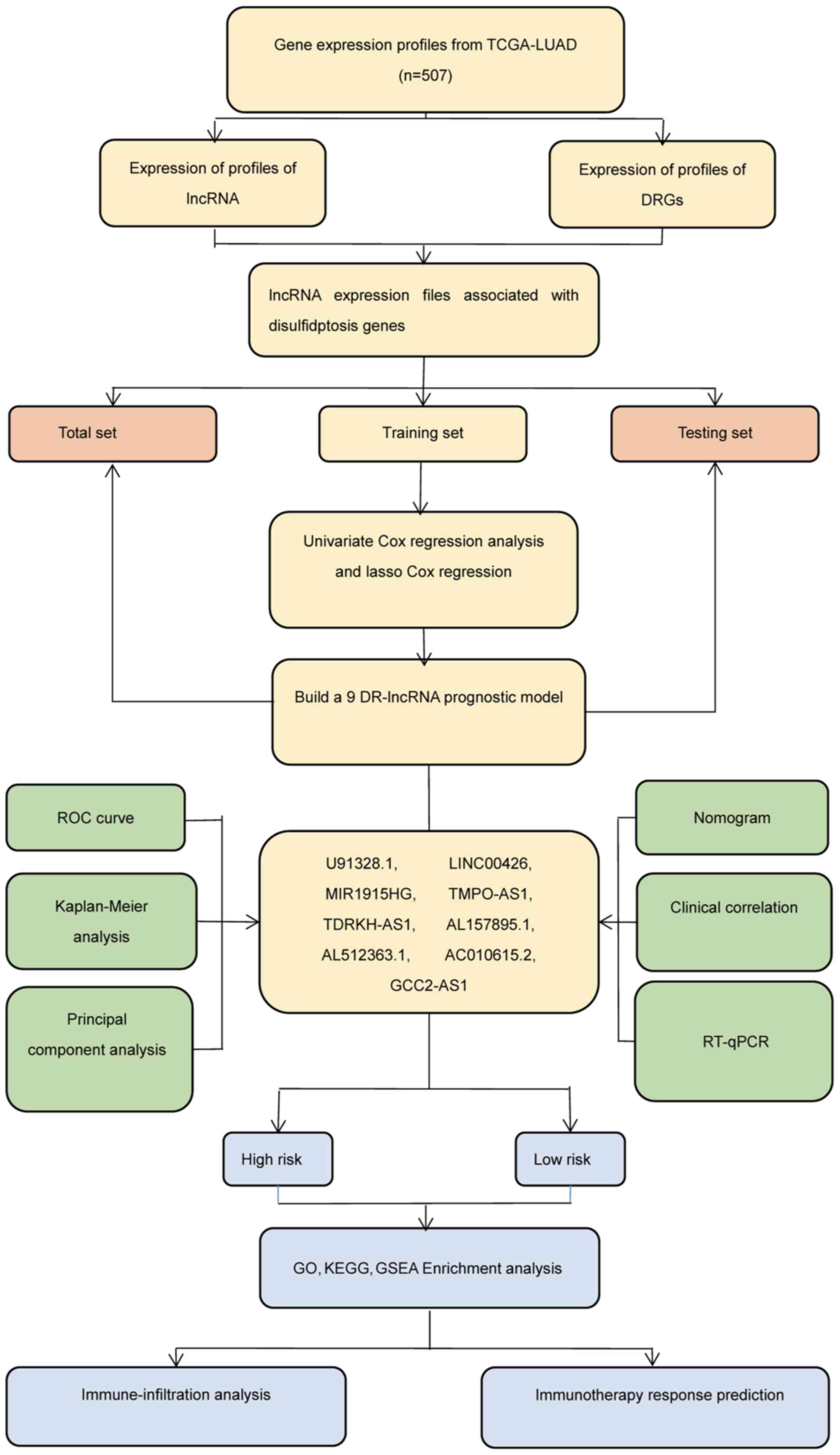
Introduction
According to the 2020 Global Cancer Statistics
report (1), lung cancer is the
leading cause of cancer-associated fatalities worldwide. Although
lung cancer ranks second in terms of the incidence rate, its
mortality rate remains the highest (2). Non-small cell lung cancer (NSCLC)
comprises ~85% of all lung cancer cases, with almost 60% of
patients experiencing local or distant metastasis. Among subtypes
of NSCLC, lung adenocarcinoma (LUAD) is the most prevalent
(3). Despite the availability of
various treatment modalities, including surgery, chemotherapy,
targeted therapy and immunotherapy, for the management of LUAD, the
overall 5-year survival rate remains <20% (4). Therefore, gaining a comprehensive
understanding of the underlying molecular mechanisms that drive the
development of LUAD and the identification of novel therapeutic
targets or biomarkers, are crucial for the effective improvement of
patient prognosis.
In the field of cancer metabolic therapy, programmed
cell death (PCD) fulfills a crucial role. A new form of PCD termed
disulfidptosis has recently been reported, disulfidptosis is
distinct from other forms of PCD such as necrosis, apoptosis,
autophagy and ferroptosis (5).
Disulfidptosis has been reported to be triggered by an increased
uptake of cysteine, coupled with an insufficient availability of
NADPH. The NADPH deficiency leads to the formation of disulfide
bonds within actin cytoskeletal proteins, causing dysfunction and
disruption of the actin network, ultimately resulting in cell death
(5,6). Actin, a versatile cytoskeletal
protein, is involved in numerous cellular processes, including
transcription, translation, cell morphogenesis, cellular mechanics,
intracellular transport and disulfide formation (7,8).
Aberrations in the cytoskeleton have been reported to be associated
with promoting and regulating PCD in both animal and plant systems
(9,10); therefore, understanding the
mechanisms underlying tumor-associated disulfidptosis is of
importance in developing understanding of the induction of tumor
cell death and the prevention of tumorigenesis.
Long non-coding RNAs (lncRNAs) are a class of RNA
molecules exceeding 200 nucleotides in length (10), which, unlike coding RNAs, do not
participate in protein translation. However, lncRNAs do exert a
critical role in gene regulation. Increasing evidence has
highlighted the importance of lncRNAs as key regulators, involved
in various physiological and pathological processes through
influencing of gene expression (11,12).
In recent years, high-throughput sequencing technologies have
enabled the identification of numerous non-coding genes that
significantly impact both tumorigenesis and tumor progression.
These lncRNAs have the ability to modulate not only the
proliferation, differentiation, invasion and metastasis of cancer
cells, but to also influence their metabolic reprogramming
(13,14). The significant involvement of
lncRNAs in tumorigenesis underscores their potential as promising
targets for precision cancer treatment; however, the precise role
of lncRNAs in relation to disulfidptosis in LUAD remains
incompletely understood.
Therefore, the present study aimed to identify and
validate a novel lncRNA-based prognostic marker associated with
disulfidptosis, with the intent to enhance prognostic prediction
for patients with LUAD. Furthermore, the disparities in cellular
processes, signaling pathways and immune status between high-risk
and low-risk groups was also performed. Ultimately, the goal was to
construct a prognostic nomogram to assist in clinical
decision-making and personalized management through the provision
of an estimation of the survival probability for patients with
LUAD.
Materials and methods
Clinical data collection for patients
with LUAD
Clinical data for patients with LUAD, was obtained
from The Cancer Genome Atlas (TCGA) database (https://gdc-portal.nci.nih.gov). RNA sequencing
(RNA-seq) data, clinical patient information and somatic mutation
data were obtained from the database. The dataset consisted of a
total of 600 samples, including 59 normal samples and 541 tumor
samples. To ensure the reliability of the prognostic analysis,
samples that had missing or non-informative prognostic data and
normal tissue were excluded from the present study. This resulted
in a final cohort of 507 tumor samples that were used for further
investigations. To assess the differences in characteristics among
the selected sample group, a χ2 test was performed
(Table I). The selection of genes
associated with disulfidptosis was based on a recently published
relevant study (5).
 | Table I.Clinical characteristics of 3 sets of
data randomly generated using data from The Cancer Genome Atlas
database. |
Table I.
Clinical characteristics of 3 sets of
data randomly generated using data from The Cancer Genome Atlas
database.
| Clinical
features | Total set, n
(%) | Train set, n
(%) | Test set, n
(%) | P-value |
|---|
| Age, years |
|
|
| 0.8184 |
|
≤65 | 239 (47.14) | 122 (48.03) | 117 (46.25) |
|
|
>65 | 258 (50.89) | 128 (50.39) | 130 (51.38) |
|
| Sex |
|
|
| 0.451 |
|
Female | 272 (53.65) | 141 (55.51) | 131 (51.78) |
|
|
Male | 235 (46.35) | 113 (44.49) | 122 (48.22) |
|
| Stage |
|
|
| 0.668 |
| I | 272 (53.65) | 131 (51.57) | 141 (55.73) |
|
| II | 120 (23.67) | 66 (25.98) | 54 (21.34) |
|
|
III | 81 (15.98) | 41 (16.14) | 40 (15.81) |
|
| IV | 26 (5.13) | 13 (5.12) | 13 (5.14) |
|
| T stage |
|
|
| 0.6613 |
| 1 | 169 (33.33) | 87 (34.25) | 82 (32.41) |
|
| 2 | 271 (53.45) | 130 (51.18) | 141 (55.73) |
|
| 3 | 45 (8.88) | 23 (9.06) | 22 (8.70) |
|
| 4 | 19 (3.75) | 12 (4.72) | 7 (2.77) |
|
| N stage |
|
|
| 0.177 |
| 0 | 327 (64.5) | 162 (63.78) | 165 (65.22) |
|
| 1 | 95 (18.74) | 54 (21.26) | 41 (16.21) |
|
| 2 | 71 (14) | 31 (12.20) | 40 (15.81) |
|
| 3 | 2 (0.39) | 2 (0.79) | 0 (0.00) |
|
| M |
|
|
| >0.999 |
| 0 | 338 (66.67) | 165 (64.96) | 173 (68.38) |
|
| 1 | 25 (4.93) | 12 (4.72) | 13 (5.14) |
|
Construction of a risk model
associated with disulfidptosis
To identify lncRNAs that exhibited co-expression
patterns with disulfidptosis-associated genes, Pearson's
correlation analysis was performed. Screening criteria of |R|≥0.4
and P<0.001 were applied, resulting in the identification of 91
lncRNAs using the limma (version 3.54.2) R package (15). Subsequently, dimensionality
reduction techniques were used to obtain disulfidptosis-related
(DR)-lncRNAs with prognostic significance. This process involved
two steps: First, univariate Cox regression analysis identified 17
DR-lncRNAs that showed significant associations with overall
survival (OS) in patients with LUAD. Subsequently, to further
reduce dimensionality and refine the selection of relevant
DR-lncRNAs, the lasso algorithm was used to further screen these 17
DR-lncRNAs using glmnet (version 4.1.7) in R (16). Finally, nine DR-lncRNAs that
exhibited significant associations with OS in patients with LUAD
were identified. Using the glmnet (version 4.1.7) and survival
(version 3.5.5) packages in R (16,17), a
lasso logistic regression model was constructed, which was
calculated as follows: Risk Score=∑ni=1
(LNCRNAExpi Coefi). Based on
the median risk score, patients were then categorized into low and
high-risk groups.
Risk model accuracy, independence
evaluation and generation of the nomogram
To evaluate the predictive ability of the risk model
for patients with LUAD, several analyses and visualizations were
performed. Kaplan-Meier (K-M) curve analysis was performed using
survival (version 3.5.5) and survminer (version 0.4.9) R packages
to assess the survival outcomes of different risk groups based on
the risk scores obtained from the model (18). K-M curves were used to determine
whether the risk model could effectively stratify patients into
distinct survival groups. The log rank test was used to calculate
the P-value of K-M survival curves and the ‘two stage’ function in
the TSHRC package (version 0.1.6), was used when survival curves
crossed over (19). Receiver
operating characteristic (ROC) curve analysis was performed using
the survival (version 3.5.5), survminer (version 0.4.9) and time
ROC (version 0.4) R packages to assess the predictive ability of
the risk model over specific time intervals, such as 1, 3 and 5
years (18). Principal component
analysis (PCA), using the limma (version 3.54.2) and scatterplot3d
(version 0.3.44) R packages (20),
was used for dimensionality reduction to visualize different
subgroups of patients based on their risk scores. Univariate and
multivariate Cox regression analysis performed using the survival
(version 3.5.5) R package were used to assess the associations
among the risk model, clinical factors and patient outcomes.
Univariate Cox regression analysis was used to assess the
individual impact of each variable, whereas multivariate Cox
regression analysis was used to determine whether the risk model
remained a reliable prognostic predictor after adjusting for other
clinical variables.
Through combining the risk model and various
clinical variables including sex, age, T, N and stage, a nomogram
was generated to predict the survival probability of patients with
LUAD using the rms (version 6.7.0), survival (version 3.5.5),
survcomp (version 1.48.0) and regplot (version 1.1) R packages
(21). A calibration curve was
generated using the rms (version 6.7.0), survival (version 3.5.5),
survcomp (version 1.48.0) and regplot (version 1.1) R packages to
evaluate the accuracy of the column chart by comparing the
predicted probabilities with the observed probabilities.
Functional enrichment analysis
To identify differentially expressed genes (DEGs)
based on different risk groups by using the limma (version 3.54.2),
criteria of |Log2FC|>1.0 and P<0.05 were applied (15). Once the DEGs had been identified,
gene ontology (GO) analysis was performed to explore the biological
functions associated with these genes using the clusterProfiler
(version 4.6.2) and ggpubr (version 0.6.0) packages in R (22). GO analysis comprised three
categories: Biological processes (BP), cellular components (CC) and
molecular functions (MF). Additionally Kyoto Encyclopedia of Genes
and Genomes (KEGG) analysis was performed using the clusterProfiler
(version 4.6.2) R package to assess the signaling pathways
associated with the DEGs. Finally, gene set enrichment analysis
(GSEA) using the clusterProfiler (version 4.6.2) and limma (version
3.54.2) R packages was performed to investigate the differential
signaling pathways involved in the low and high-risk groups.
The ESTIMATE algorithm was used to assess
differences in the tumor microenvironment (TME) between the
different risk groups using the limma (version 3.54.2) and estimate
(version 1.10.13) R packages. The CIBERSORT algorithm using the
limma (version 3.54.2) R package was employed to calculate the
relative expression levels of 22 immune cells within different risk
groups. Additionally, the single sample (ss)GSEA algorithm in the
GSVA (version 1.46.0) and limma (version 3.54.2) R package was used
to identify variations in immune cells and immune function across
different risk populations (20).
To evaluate the immune escape of tumor cells and their response to
immune checkpoint inhibitors (ICIs) within different risk groups,
the tumor immune dysfunction exclusion (TIDE) score was assessed
using the limma (version 3.54.2) and ggpubr (version 0.6.0) R
packages. Mutation frequencies and cancer maps for patients with
LUAD were generated using the maftools (version 2.14.0) R package
(23). Furthermore, tumor mutation
burden (TMB) of patients in the different risk groups was analyzed
using TCGA somatic mutation data. Based on the median TMB score,
patients with LUAD were categorized into low and high-risk TMB
groups. Differences in TMB and survival between the two risk sets
were analyzed using the survival (version3.5.5) and survminer
(version0.4.9) R packages. To predict drug responses in the
different risk groups of patients with LUAD, the oncoPredict
(version 0.2) and limma (version 3.54.2) R packages were employed
(23). This package was used to
calculate the half-maximal inhibitory concentration (IC50) values
of commonly used anti-tumor drugs and to predict drug reactions in
the various risk groups.
Cell culture and reverse
transcription-quantitative (RT-q)PCR analysis
The human LUAD A549 cell line and the human normal
lung epithelial BEAS-2B cell line were purchased from Procell Life
Science &Technology Co., Ltd. Cells were cultured in RPMI-1640
medium (Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc). The cell
cultures were maintained in a humidified environment at 37°C with
5% CO2. The cell density for RNA extraction was
1×106. Total RNA was extracted using the
TRIzol® reagent (Thermo, Fisher Scientific, Inc.), and
cDNA was obtained by reverse transcription using a reverse
transcriptase kit (Takara Biotechnology Co., Ltd.) . The cDNA was
amplified using the SYBR™ Green (Hunan Accurate Bio-Medical
Technology Co., Ltd.). RNA extraction, cDNA synthesis and qPCR were
all performed according to the manufacturer's protocols. The
reaction volume was 20 µl, including 10.0 µl SYBR ™ Green, 0.5 µl
each primer, 2 µl cDNA and 7.0 µl nucleic acid-free water. The
thermocycling conditions were as follows: Initial denaturation at
95°C for 30 sec, followed by 40 cycles of 95°C for 5 sec, 55°C for
30 sec and 72°C for 30 sec. β-actin was used as an internal
control. The mRNA expression levels were quantified using the
2−ΔΔCq method and normalized against expression levels
of β-actin (24). The primer
sequences used are presented in Table
II.
| Table II.lncRNA primer sequences |
Table II.
lncRNA primer sequences
| lncRNA | Sequence
(3′-5′) |
|---|
| Homo-β-actin | F:
CCTTCCTGGGCATGGAGTC |
|
| R:
TGATCTTCATTGTGCTGGGTG |
| Homo-LINC00426 | F:
CACTCCCTACACGTTCTAACCA |
|
| R:
ATCCCCCATTTGCTGTGTC |
| Homo-TDRKH-AS1 | F:
CACCTCTAGGCCAATTACCG |
|
| R:
GGTGCCACTCATGATTCAACG |
|
Homo-AC010615.2 | F:
CGGTGAACTGATGGTGCTGG |
|
| R:
GGCTCATGGTTGGGCTATCTTC |
| Homo-MIR1915HG | F:
CCGCGCTCACGATTGCTTT |
|
| R:
GCAAGCAGCATATAGCCTCGG |
| Homo-TMPO-AS1 | F:
CAAAAGACCCCAGAGCCGAACT |
|
| R:
TTGGGGCGTGGGCGAAG |
| Homo-GCC2-AS1 | F:
TTTCAGCACCCCAGGCTAGT |
|
| R:
TCACCGCCATCCTTGTTGTAG |
|
Homo-AL157895.1 | F:
AGTTCTCTGAGGGATAAGGAACAT |
|
| R:
TTCTAGGCATTCACAGGGTGC |
| Homo-U91328.1 | F:
ATGGGTCTCCGCTGATGCTT |
|
| R:
CTCAGACCTGTAGTCTTCCACCAG |
|
Homo-AL512363.1 | F:
CCTCTGTCTCACTTCAGCTGTT |
|
| R:
TGATTGGAAAACAAGACGCTGG |
Statistical analysis
All statistical analyses were performed using R
software (version 4.2.2) and GraphPad Prism (version 8.0.2;
Dotmatics). Differences between the two groups were assessed using
Student's t-test. The log-rank test and two stage hazard rate
comparison were used to assess differences between the K-M curves.
Univariate and multivariate Cox regression analyses were performed
to identify prognostic risk factors associated with LUAD. P<0.05
was considered to indicate a statistically significant
difference.
Results
Clinical data of patients with
LUAD
The present study included a total of 507 patients
with LUAD, who were divided into training and testing sets in a 1:1
ratio. The training set comprised 253 patients with LUAD and was
used for constructing the prognostic model and identifying
prognostic DR-lncRNAs. The remaining 254 patients with LUAD formed
the testing set, which was used to evaluate the accuracy of the
risk model developed from the training set. A comprehensive
comparison of key clinical characteristics, including age, sex,
grading and staging, as well as tumor (T), metastasis (M) and node
(N) status, among the training set, the testing set and the total
set of patients was performed (Table
I). Statistical analysis revealed no statistically significant
differences in these clinical characteristics among the three sets.
These findings suggested that the training, testing and all sets
were adequately matched in terms of important clinical variables,
minimizing potential confounding effects when evaluating the
accuracy of the prognostic model.
Construction of the DR-lncRNA risk
model for LUAD
The experimental plan of the present study was
summarized in a flow diagram (Fig.
1). Pearson's correlation analysis using the limma (version
3.54.2) R package (15) was
performed on expression levels of ten disulfidptosis-associated
genes using RNA-seq data from patients with LUAD, sourced from the
TCGA database. This analysis identified 91 differentially expressed
lncRNAs (Table SI). To visualize
the connections between these ten disulfidptosis genes and the 91
lncRNAs, a co-expression network was constructed using a Sankey
diagram (Fig. 2A). Subsequently,
univariate Cox regression analysis was performed to identify
DR-lncRNAs that were associated with OS, leading to the
identification of 17 lncRNAs significantly correlated with survival
(Fig. 2B). To further reduce
dimensionality and refine the selection of relevant DR-lncRNAs, the
lasso algorithm was used to further screen these 17 DR-lncRNAs.
Finally, nine DR-lncRNAs were identified that exhibited significant
associations with OS in patients with LUAD. (Fig. 2C and D). These DR-lncRNAs were
identified as U91328.1, LINC00426, MIR1915HG, TMPO-AS1, TDRKH-AS1,
AL157895.1, AL512363.1, AC010615.2 and GCC2-AS1. Using the
expression levels of these nine DR-lncRNAs and the coefficients
derived from the Cox regression model, a risk score was
subsequently calculated. The risk score formula yielded the
following calculation: Risk score=(−0.408862554222557 ×
U91328.1)+(−0.412743922052189 × LINC00426)+(0.23703345398073 ×
MIR1915HG)+ (0.225351354331566 × TMPO-AS1)+(−0.329373779699515 ×
TDRKH-AS1)+(−0.552378924475953 × AL157895.1)+(0.377529261105432 ×
AL512363.1)+(−0.225534318729328 × AC010615.2)+(0.565660789000357 ×
GCC2-AS1). Additionally, correlation testing was performed to
examine the association between the ten disulfidptosis genes and
the nine DR-lncRNAs (Fig. 2E). This
analysis provided valuable insights into the potential associations
and interactions among these genes and lncRNAs in the context of
disulfidptosis and LUAD.
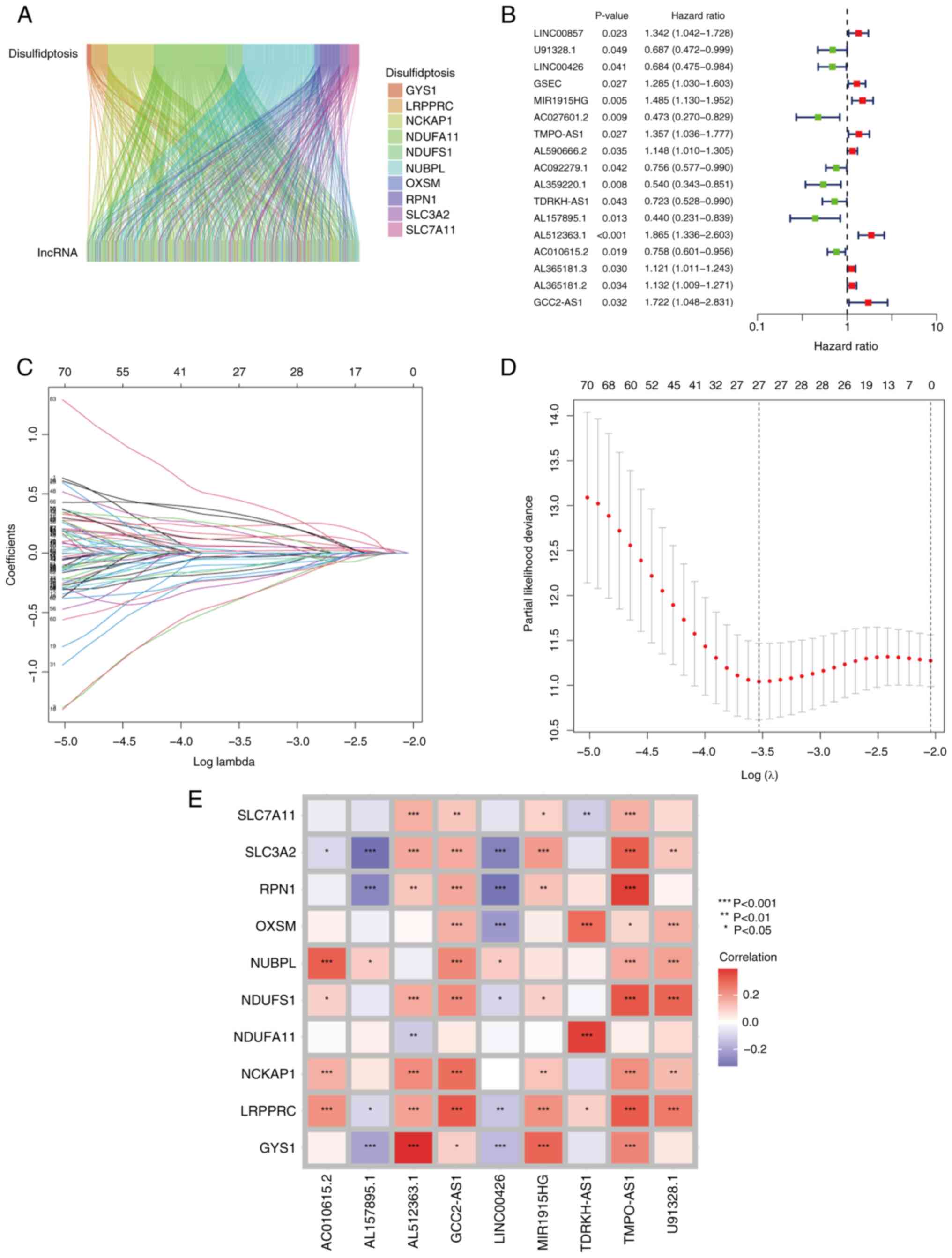 | Figure 2.Construction of DR-lncRNA risk model.
(A) Sankey diagram of the DR-lncRNAs associated with the ten
disulfidptosis-associated genes (GYS1, LRPPRC, NCKAP1, NDUFA11,
NDUFS1, NUBPL, OXSM, RPN1, SLC3A2 and SLC7A11). (B) Univariate Cox
regression analysis indicated that the 17 lncRNAs were
significantly associated with DR genes. (C and D) Lasso regression
analysis identified nine DR-lncRNAs. (E) Correlation heatmap
analysis of the ten DR genes, with nine independent prognosis
lncRNAs. *P<0.05, **P<0.01 and ***P<0.001. DR-,
disulfidptosis-related. DR, disulfidptosis-related. |
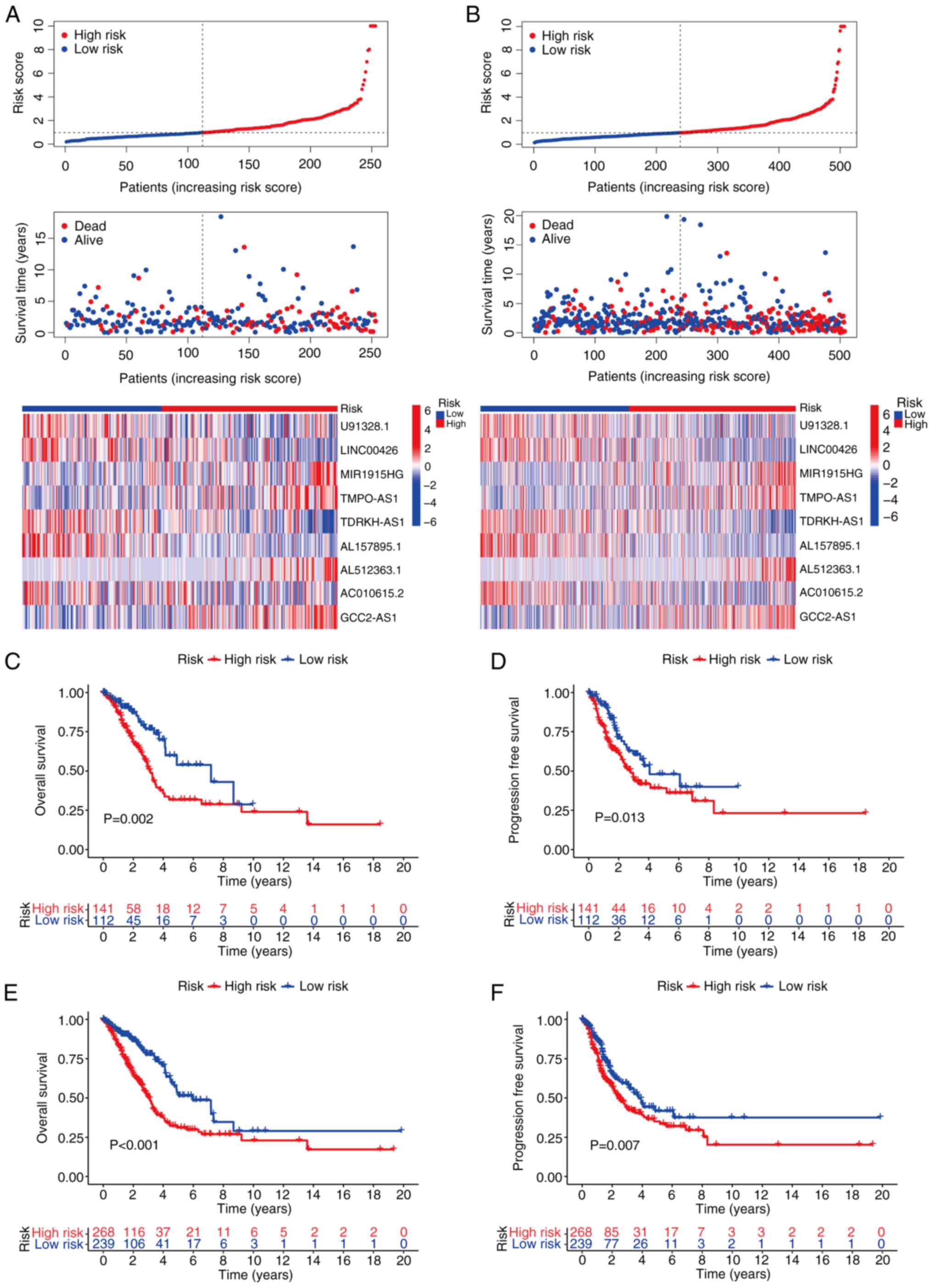
Validation of the model of the
disulfidptosis-related lncRNA signature (DRlncSig)
The testing set and the total set of patient samples
were used to assess the reliability of the established risk model.
The risk curves and scatter plots of the test groups were generated
to evaluate the performance of the risk model. The results
demonstrated that, compared with the high-risk group, the low-risk
group of patients with LUAD exhibited lower risk scores, longer
survival times and an improved prognosis. A heatmap analysis was
performed to investigate the potential role of the specific
DRlncSig in LUAD. The heatmap indicated that MIR1915HG, TMPO-AS1,
AL512363.1 and GCC2-AS1 may act as risk factors for LUAD (Fig. 3A). Furthermore, K-M curves were used
to examine the differences in OS and progression-free survival
(PFS) between the different risk groups. The results revealed that
patients classified as low risk had significantly better OS and PFS
values compared with those classified as high risk (Fig. 3C and D). These significant
differences in the testing set were consistent with those observed
in the total set of patients (Fig. 3B,
E and F). Taken together, these findings suggested that the
established risk model based on the identified DRlncSig was
reliable and was capable of stratifying patients with LUAD into
distinct risk groups with significantly different clinical outcomes
in terms of OS and PFS.
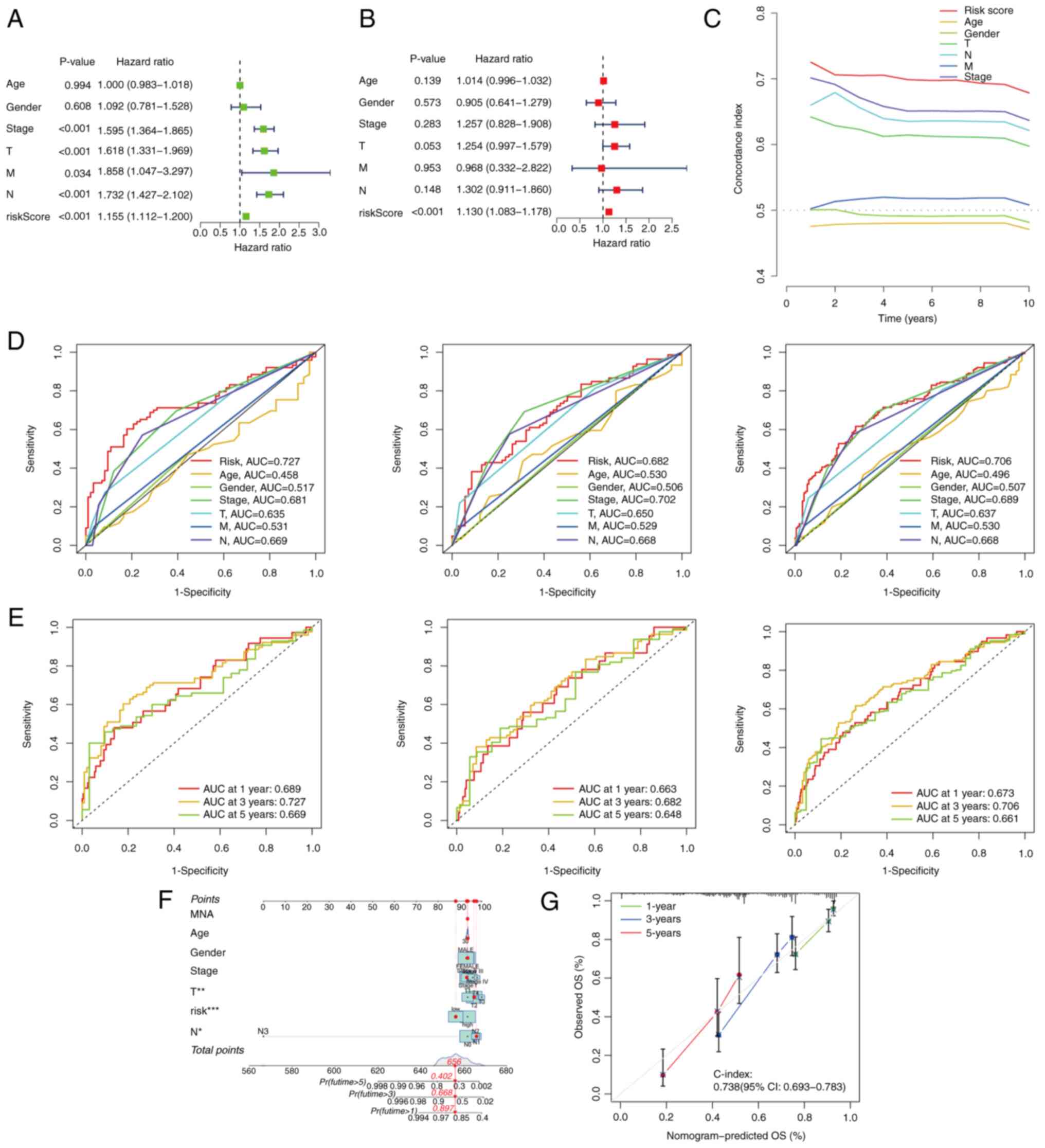
Effectiveness evaluation of the
DRlncSig
To evaluate the independent prognostic value of the
DR-lncRNAs, both univariate and multivariate Cox regression
analyses were performed. The results of the univariate Cox
regression analysis showed that variables such as T, N, M, stage
and risk score were significantly associated with OS in patients
with LUAD (Fig. 4A). Subsequently,
the multivariate Cox regression analysis demonstrated that the risk
score could serve as an independent prognostic factor for
predicting OS in patients with LUAD (Fig. 4B). Collectively, these findings
suggested that the risk model constructed using the nine DR-lncRNAs
had potential as an independent prognostic factor for patients with
LUAD. Furthermore, ROC analysis (Fig.
4D) from the training set, testing set and total set, and
C-index assessment from the total set (Fig. 4C) were performed to validate the
prognostic capabilities of the lncRNA signature. The results
obtained indicated that the risk score exhibited the highest area
under the curve (AUC) value (AUC=0.706), and higher C-index,
compared with other clinical risk indicators in the total set. In
the training set, the AUC values at 1, 3 and 5 years were 0.689,
0.727 and 0.669, respectively. Similarly, in the testing set, the
ROC curve showed AUC values of 0.663, 0.682 and 0.648 at 1, 3 and 5
years, respectively, providing strong evidence for the reliability
of the model. In the total set, the AUC values at 1, 3 and 5 years
were 0.673, 0.706 and 0.661 respectively (Fig. 4E). Additionally, a nomogram was
generated, incorporating multiple clinical factors, including T, N,
sex, age, stage and risk scores, to enhance the predictive ability
of the prognosis of LUAD patients for 1-, 3- and 5-year survival
(Fig. 4F). The calibration curve
was used to assess accuracy of the nomogram, confirming its
reliability (Fig. 4G).
K-M survival curve analysis of OS in high- and
low-risk groups of TCGA database with different clinic-pathological
characteristics. Further analysis was performed to examine the
predictive ability of the constructed signature in subgroups with
different clinical characteristics. The results illustrated that
the risk model successfully differentiated between high- and
low-risk groups, regardless of age, sex, TNM and the pathological
stage (Fig. 5). These findings
suggested that the established signature maintained its predictive
ability across diverse subgroups with varying clinical
characteristics. Therefore, the risk model based on the identified
DR-lncRNAs was demonstrated to be robust and applicable for risk
stratification in patients with LUAD, irrespective of demographic
factors and disease stage.
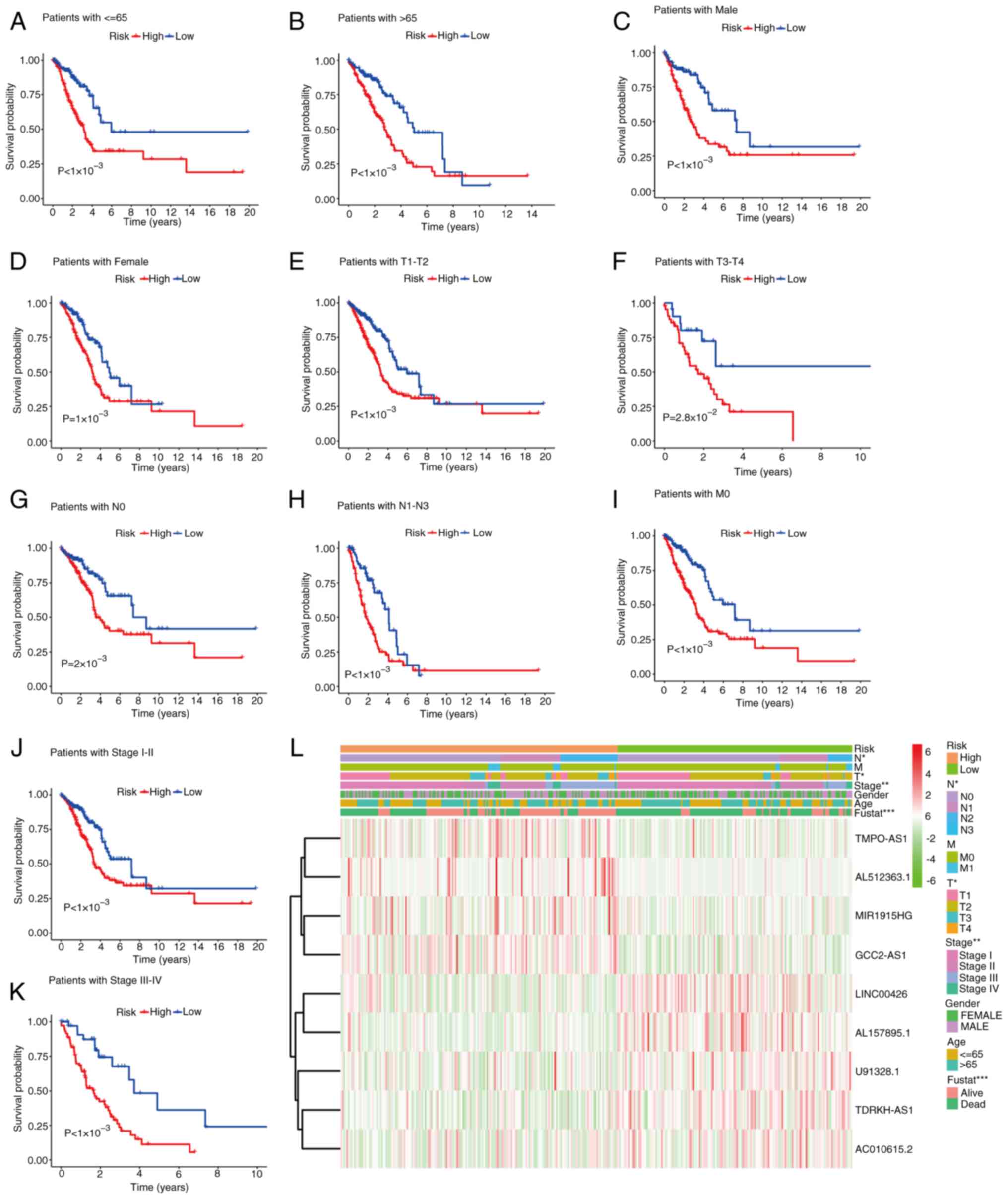 | Figure 5.Kaplan-Meier survival curve analysis
of the OS rate in the high- and low-risk groups of TCGA database
with different clinical pathological characteristics: (A) age ≤65
years, (B) age >65 years, (C) males, (D) females, (E) T1-T2, (F)
T3-T4, (G) N0, (H) N1-N3, (I) M0, (J) stage I–II and (K) stage
III–IV. (L) Heatmap of the clinical pathological characteristics.
*P<0.05, **P<0.01 and ***P<0.001. OS, overall survival;
TCGA, The Cancer Genome Atlas; T, tumor; N, node; M,
metastasis. |
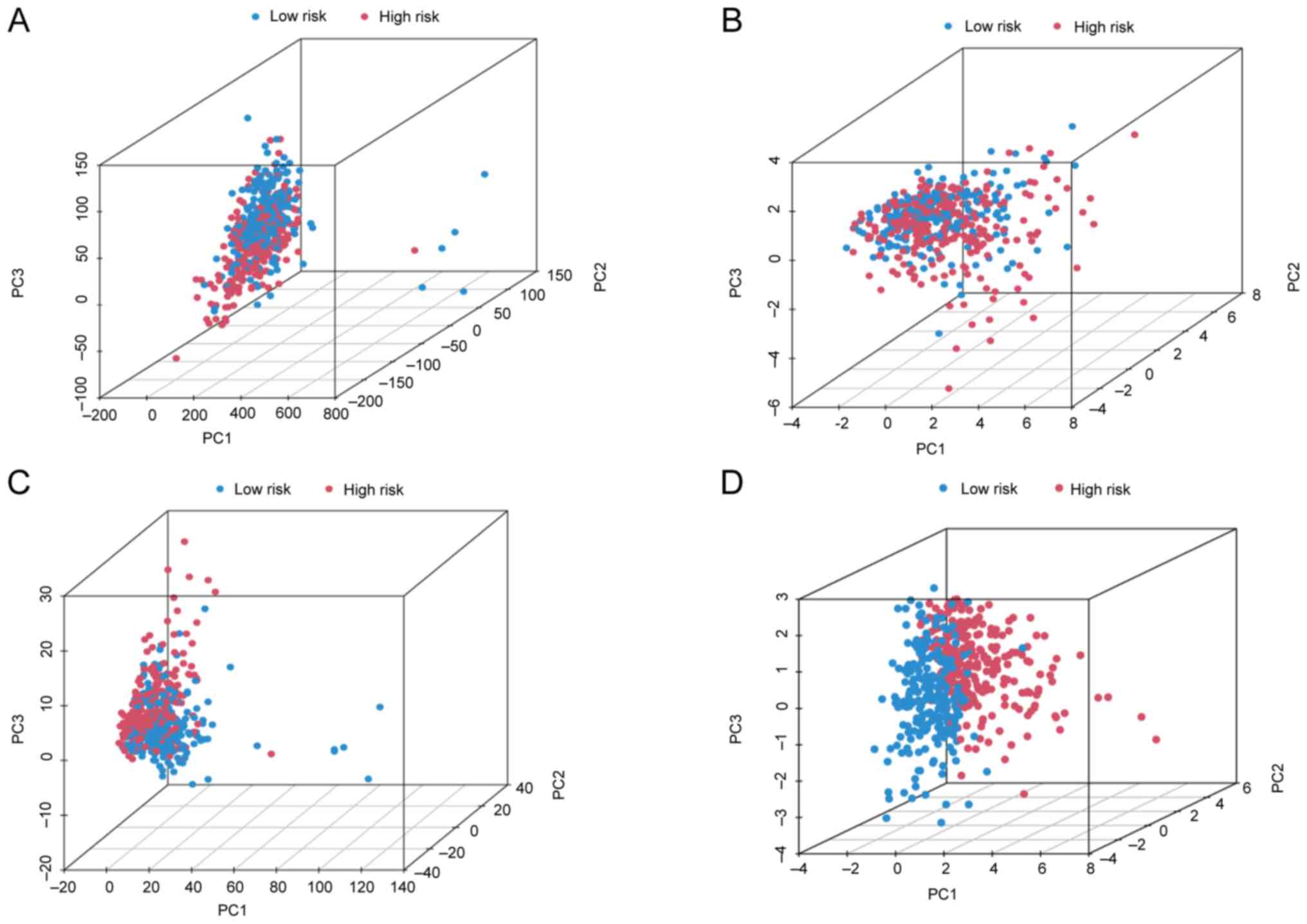
Investigation of the grouping ability
of the proposed model via PCA
PCA is a commonly used technique for dimensionality
reduction, which transforms a large number of variables into a
smaller set of principal components. These components retain most
of the original dataset's information while reducing its
dimensionality. In the present study, PCA was used to investigate
whether the DRlncSig could effectively discriminate between the
high- and low-risk patient groups. The results of the PCA analysis,
presented in Fig. 6, clearly
demonstrate a distinct separation between patients in the high- and
low-risk groups on the basis of their DRlncSig profiles. Taken
together, these findings suggested that the expression patterns of
the identified DRlncSig had the potential to accurately
differentiate between the high- and low-risk groups, suggesting
that the DRlncSig may serve as a promising biomarker for either
risk stratification or prognosis prediction in the studied disease
context.
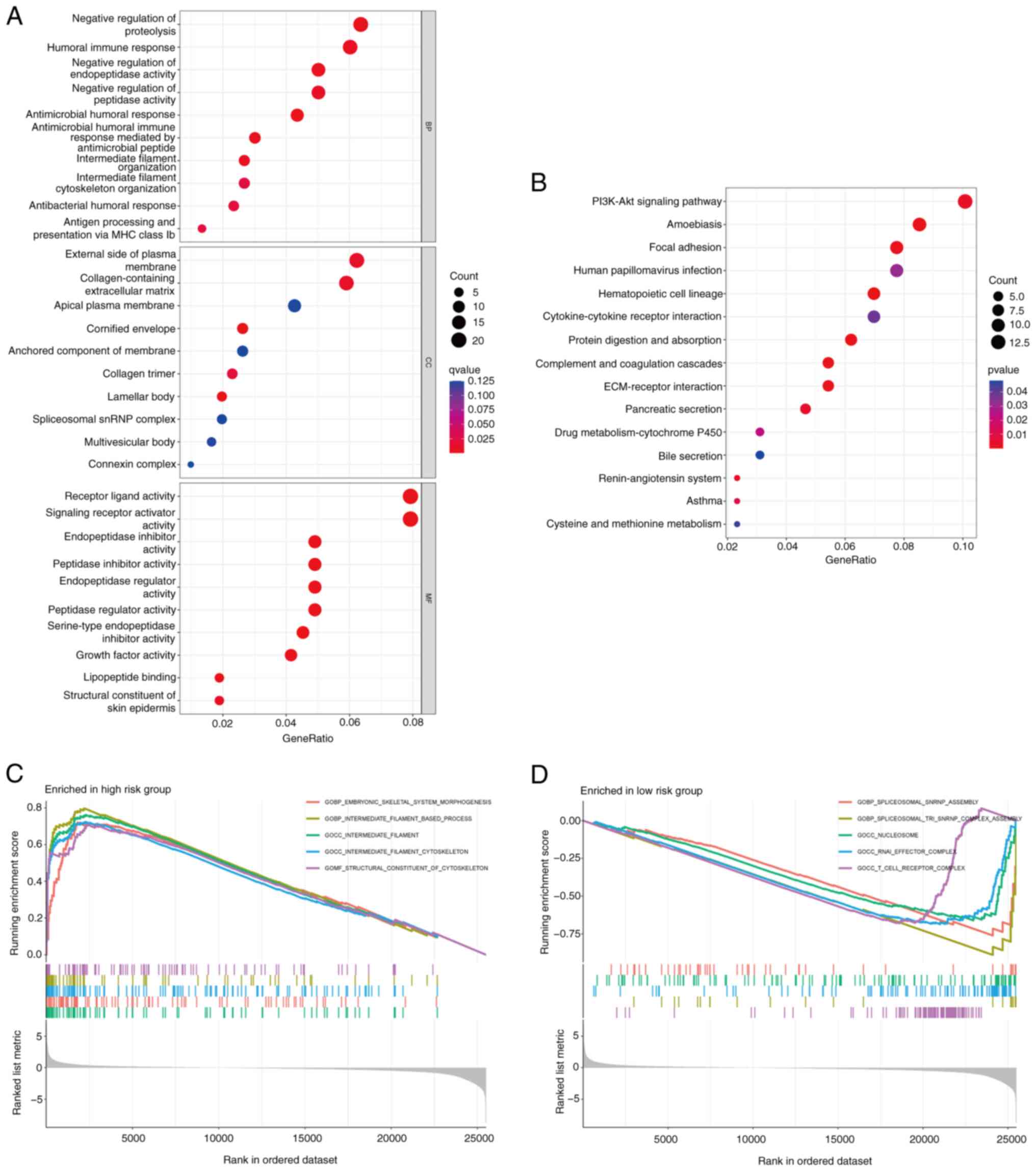
Functional enrichment analysis
To understand the underlying mechanisms contributing
to the significant differences between the different risk groups,
GO and KEGG enrichment analyses were performed, based on the nine
identified DR-lncRNAs. The GO analysis revealed that the DEGs were
significantly enriched in immune-related BP, including ‘negative
regulation of protein hydrolysis’, ‘humoral immune response’,
‘negative regulation of endopeptidase activity’ and ‘negative
regulation of peptidase activity’. In terms of CC, these DEGs were
notably enriched in the ‘external side of plasma membrane’ and the
‘collagen-containing extracellular matrix’. Furthermore, in terms
of MF, the DEGs were involved in ‘receptor ligand activity’ and
‘signaling receptor activator activity’ (Fig. 7A). Moreover, the KEGG enrichment
analysis demonstrated that these DEGs were primarily enriched in
pathways such as the ‘PI3K-AKT signaling’, ‘amoebiasis’ and ‘focal
adhesion’ pathways (Fig. 7B). In
addition, in the high-risk group, GSEA revealed significant
enrichment of pathways associated with ‘embryonic skeletal system
morphogenesis’, ‘intermediate filament-based process’ and the
‘intermediate filament cytoskeleton’ (Fig. 7C). However, the low-risk group
exhibited significant enrichment of pathways associated with
‘spliceosomal snRNP assembly’ and the ‘nucleosome’ (Fig. 7D).
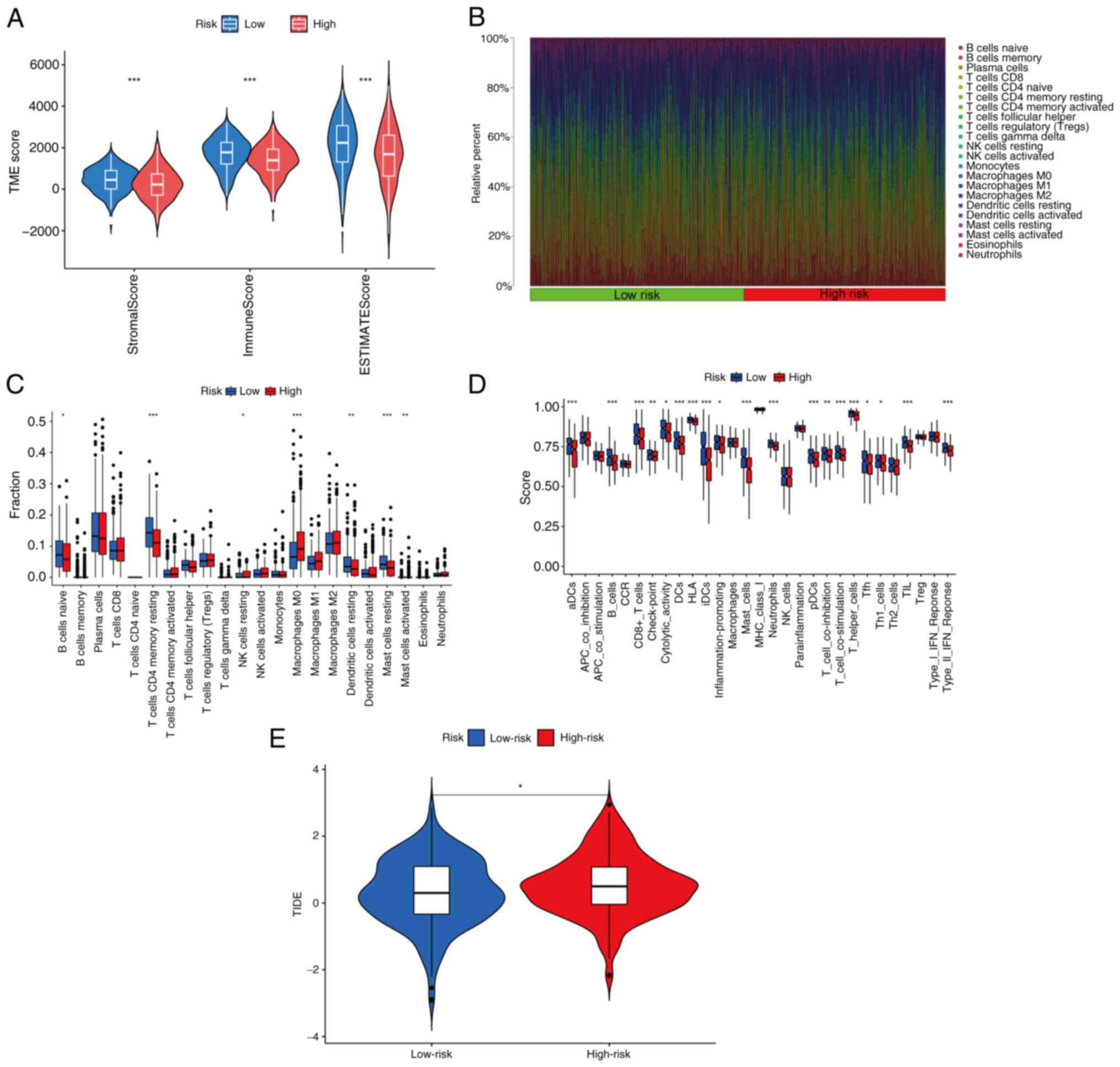
Landscape analysis of immune
infiltration and immunotherapy
The TME fulfills a crucial role in the progression
and treatment of LUAD tumors (25,26).
In the present study, the TME in the different risk groups was
investigated using multiple immune evaluation algorithms. First,
the ESTIMATE algorithm was used to assess TME scores. The immune
score, ESTIMATE score and stromal score of patients with LUAD in
the low-risk group were significantly higher compared with those in
the high-risk group (Fig. 8A).
These findings suggested that the TME characteristics differed
between the two risk groups, potentially influencing both the
prognosis and treatment response in patients with LUAD.
Subsequently, the CIBERSORT algorithm was used to analyze the
distribution of 22 immune cell types based on the risk model. The
results obtained indicated differences in the distribution of
immune cells between the risk groups (Fig. 8B). Specifically, in the low-risk
group, there was a lower proportion of M0 macrophages, activated
mast cells and natural killer (NK) cells in the resting state
compared with the high-risk group. However, resting memory CD4+ T
cells, naive B cells, resting dendritic cells and resting mast
cells accounted for a larger proportion in the low-risk group
(Fig. 8C). Furthermore, the ssGSEA
algorithm was used to analyze immune cell infiltration and immune
function in the different risk groups. The results demonstrated a
significant increase in activated DCs), B cells, CD8+ T cells, DCs,
mast cells, neutrophils, plasmacytoid DCs, T helper cells, T
follicular helper, Th1 cells and tumor-infiltrating lymphocytes in
the low-risk group. Regarding the immunological function, cytolytic
activity, human leukocyte antigens, IDCS, T cell co-inhibition, T
cell co-stimulation and type II IFN response were found to be
significantly activated in the low-risk group (Fig. 8D). These findings suggested that the
low-risk group may exhibit a more robust and active immune response
against the tumor. Finally, the TIDE algorithm was used to estimate
the relationship between the risk groups and immune therapy
response. The results obtained suggested that tumors in the
high-risk group were significantly more likely to experience immune
escape, and to exhibit worse treatment effects (Fig. 8E).
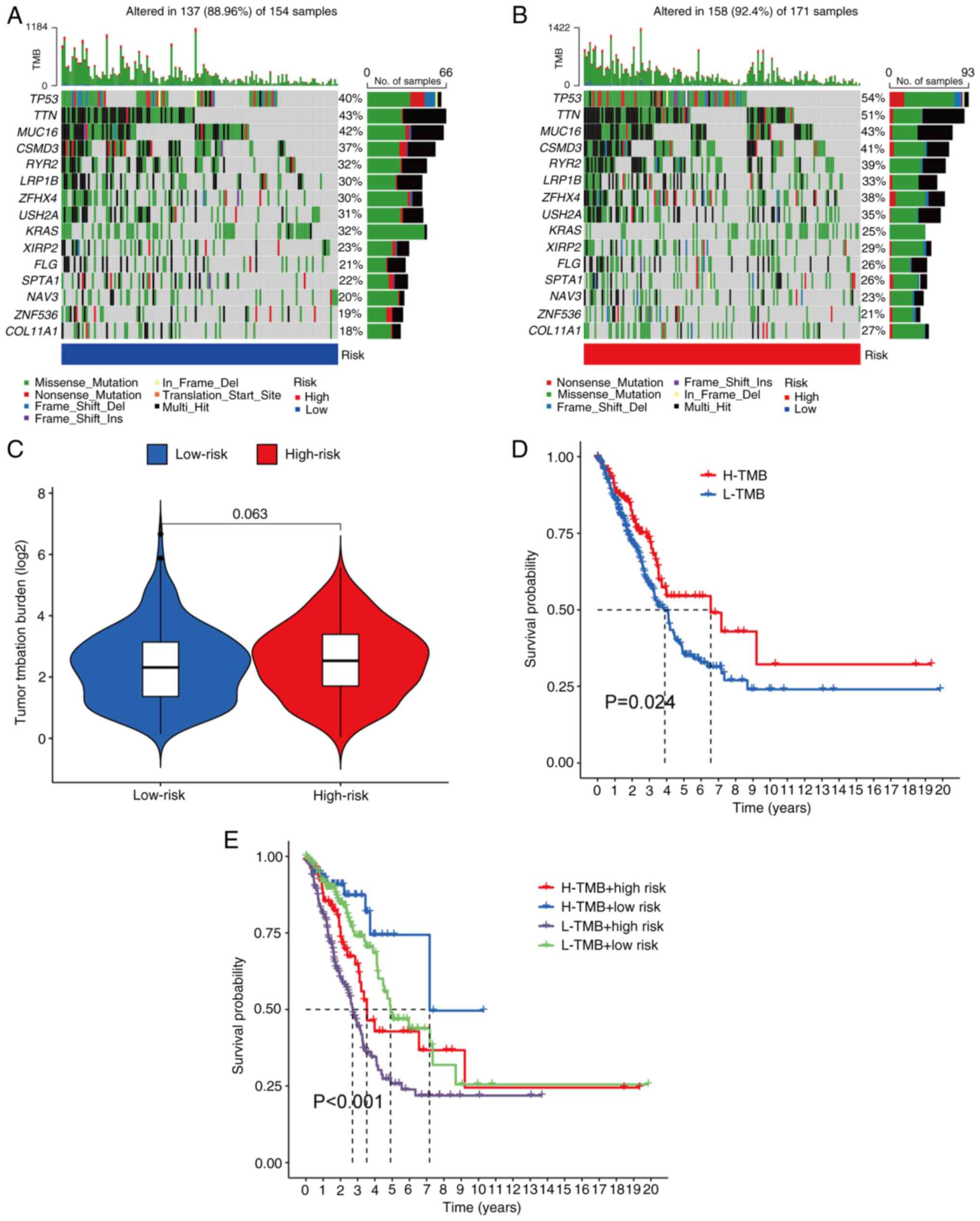
Somatic mutation landscape
analysis
A comparison of somatic mutations between the
different risk groups was performed in the patients with LUAD.
Among the 171 samples in the high-risk group, 158 samples (92.4%)
had mutations, whereas in the low-risk group, 137 out of 154
samples (88.96%) had mutations. The top 15 genes driving these
mutations were presented (Fig. 9A and
B). Furthermore, the TMB was analyzed in the different risk
groups; however, the difference was statistically significant
(Fig. 9C). Subsequently, based on
the median TMB score, patients with LUAD were divided into a low
TMB and high TMB group. K-M analysis revealed that the high TMB
group exhibited significantly improved OS rates compared with the
low TMB group (Fig. 9D). To predict
the prognosis of patients with LUAD and to determine which score
had the greater predictive value, the TMB and risk scores were
integrated. According to K-M analysis, patients with high TMB and
low risk scores had the highest OS rate, whereas patients with low
TMB and high-risk scores had the lowest OS (Fig. 9E).
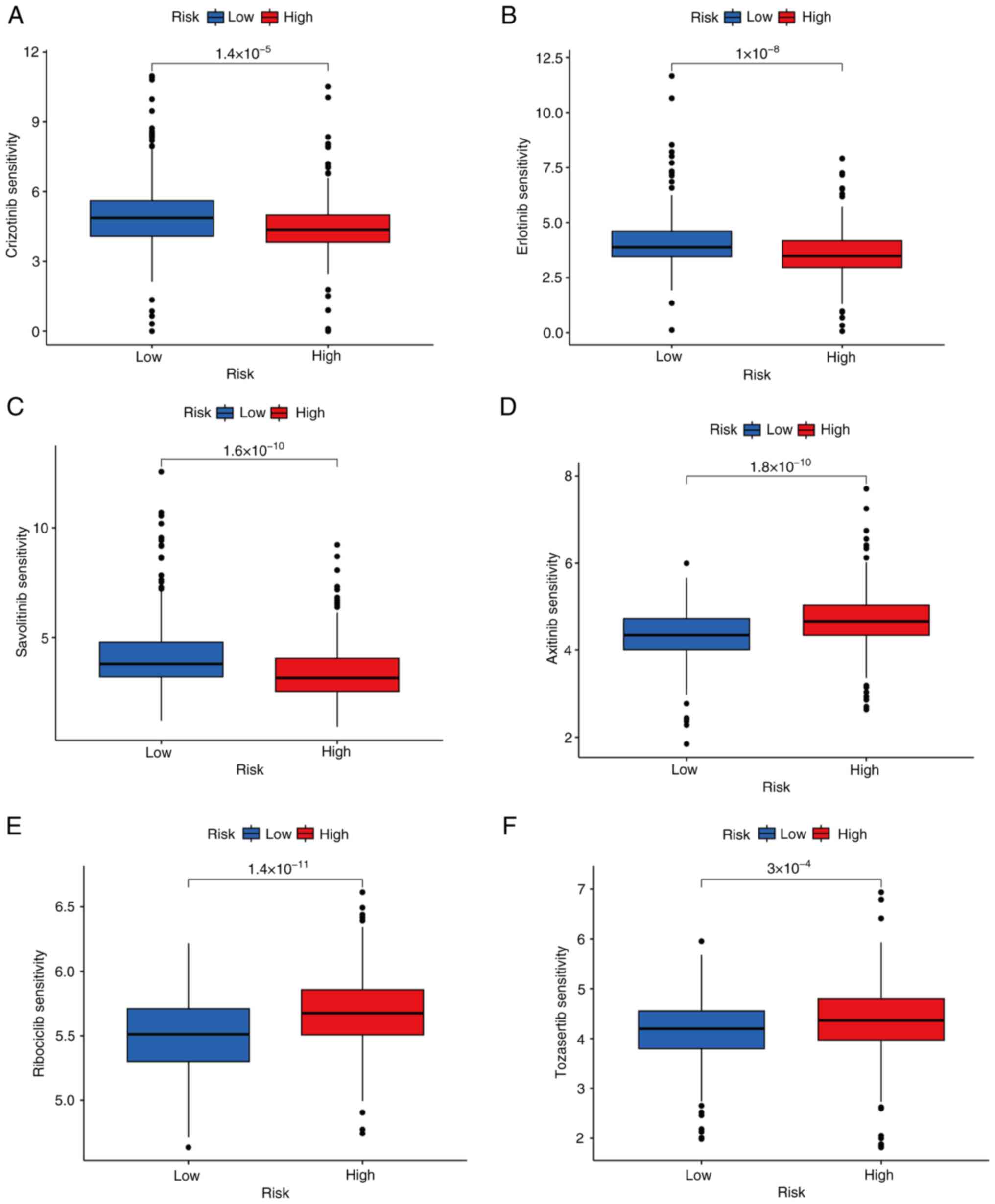
Drug-sensitivity prediction
To improve the effectiveness and targeting of
treatment for LUAD, the IC50 values of different anti-tumor drugs
in the high- and low-risk groups were evaluated. The
drug-sensitivity analysis revealed that samples in the high-risk
group exhibited a significantly higher level of drug-sensitivity to
crizotinib, erlotinib and savolitinib compared with samples in the
low-risk group (Fig. 10A-C).
However, high-risk group samples exhibited a significantly lower
level of drug-sensitivity to axitinib, ribociclib and tozasertib
compared with the low-risk group (Fig.
10D-F). These results suggested that patients in the low-risk
group may have a more favorable response towards axitinib,
ribociclib and tozasertib, whereas those in the high-risk group may
respond better to treatment with crizotinib, erlotinib and
savolitinib.
Cell-based in vitro experimental
validation
To further validate the reliability of the risk
model constructed using the DR-lncRNAs, RT-qPCR analysis was
performed to assess the mRNA expression levels of DR-lncRNAs in
LUAD. Subsequently, RT-qPCR analysis was performed on the A549 and
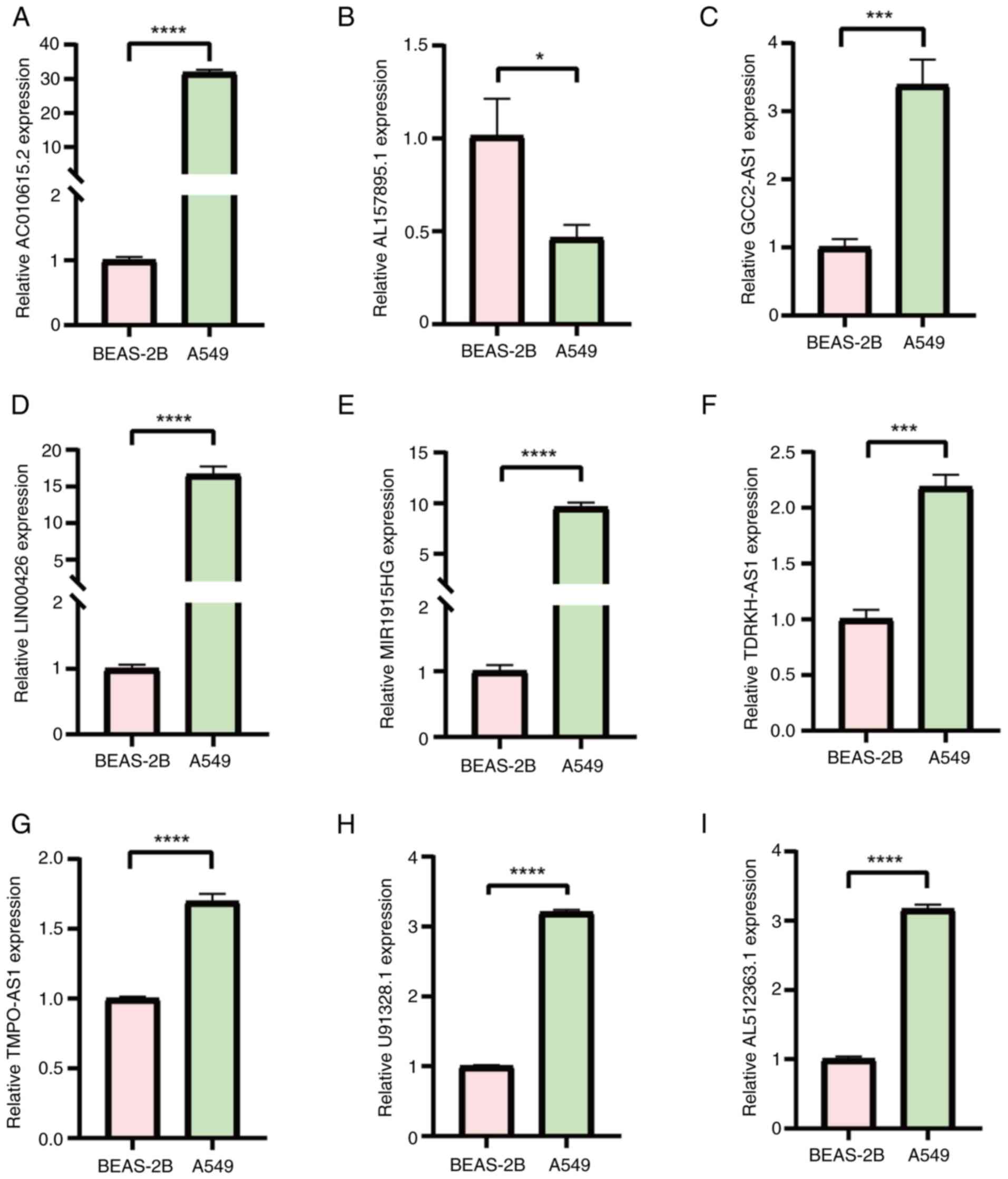
BEAS-2B cells. The results obtained revealed significant
differences in the expression levels of the eight tested DR-lncRNAs
when compared between the normal and LUAD cell lines. Specifically,
the mRNA expression levels of U91328.1, AC010615.2, MIR1915HG,
TMPO-AS1, LINC00426, TDRH-AS1, AL512363.1 and GCC2-AS1 were
significantly higher in the A549 cells, compared with the BEAS-2B
cells. Conversely, the mRNA expression level of AL157895.1 was
significantly lower in the A549 cells (Fig. 11).
lncRNA GCC2-AS1 is overexpressed in
LUAD
The validity of the model was demonstrated using
lncRNA GCC2-AS1, which had the highest risk score coefficient.
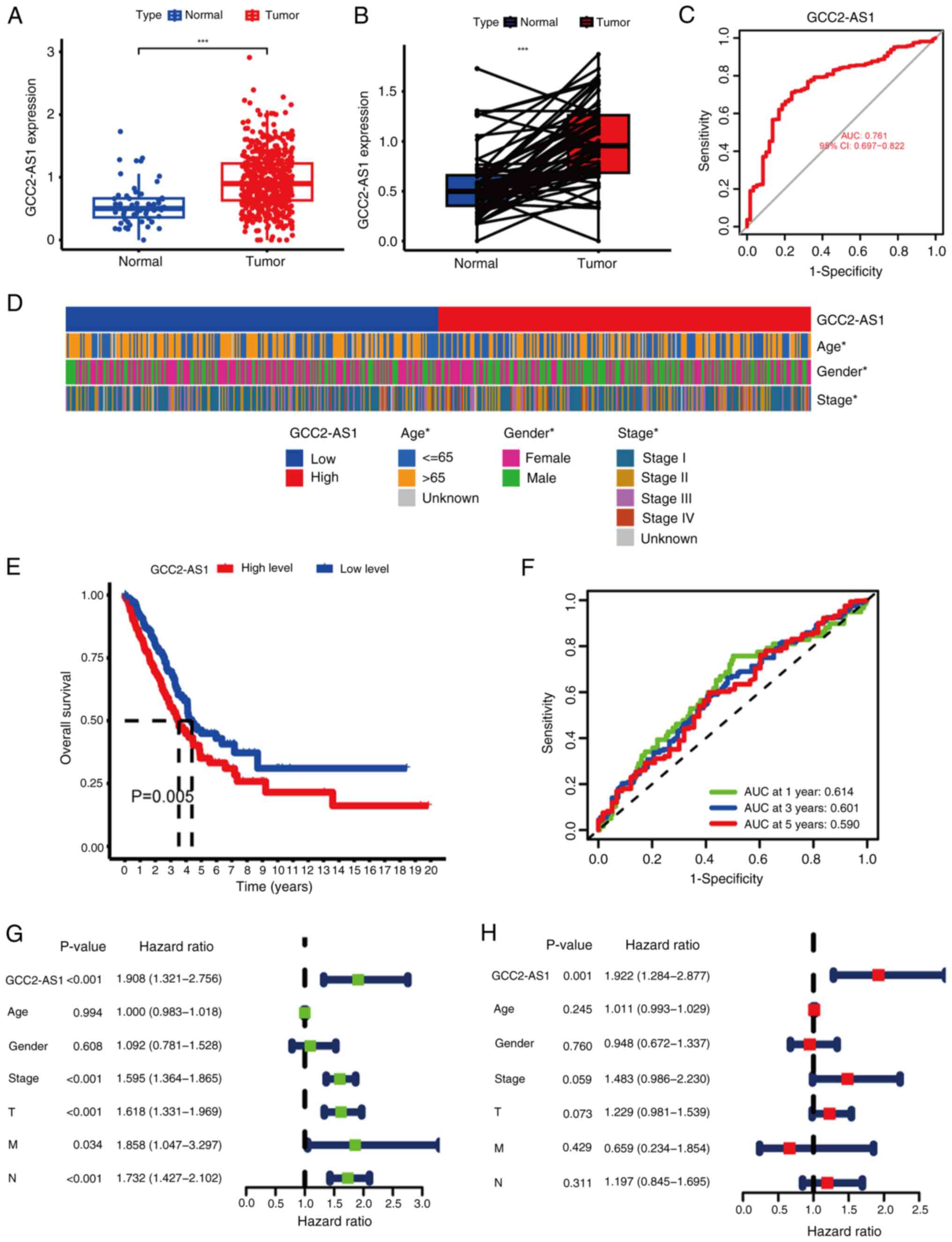
First, to compare the expression of GCC2-AS1 between the LUAD and
normal tissues, its expression in tumor tissues and normal lung
tissues was analyzed using data from TCGA database. GCC2-AS1 was
demonstrated to be significantly upregulated in LUAD tissues
(Fig. 12A). Additionally, in
paired LUAD cancer samples and adjacent normal samples from TCGA,
the expression level of GCC2-AS1 was significantly higher in LUAD
samples compared with their matched adjacent normal samples
(Fig. 12B). The aforementioned
results were consistent with the aforementioned RT-qPCR results for
GCC2-AS1 in the present study. An ROC analysis showed that GCC2-AS1
had a notable ability to discriminate between LUAD patients and
healthy individuals, with an AUC of 0.761 (Fig. 12C), which suggested that GCC2-AS1
could be used to help identify people with LUAD. Subsequently, to
examine the clinical relevance of GCC2-AS1 in LUAD, the patients
with LUAD were divided into high and low-expression groups, based
on the median expression level of GCC2-AS1. The correlations
between GCC2-AS1 expression and clinical parameters, including
stage, age and gender, were then evaluated. The results obtained
showed significant statistical differences in the expression levels
with respect to stage, gender and age (Fig. 12D). K-M analysis showed that
patients with LUAD with a high-expression of GCC2-AS1 had
significantly worse OS rates (Fig.
12E). Furthermore, ROC curve analysis revealed AUC values of
0.614, 0.601 and 0.590 at 1, 3 and 5 years, respectively, which
indicated the reliability of GCC2-AS1 as a prognostic factor
(Fig. 12F). To evaluate the
independent prognostic value of GCC2-AS1, both univariate and
multivariate Cox regression analyses were performed. The results of
the univariate Cox regression analysis showed that variables such
as T, N, stage and the expression of GCC2-AS1 were significantly
associated with OS in patients with LUAD (Fig. 12G). Subsequently, multivariate Cox
regression analysis indicated that GCC2-AS1 could serve as an
independent prognostic factor for predicting OS in patients with
LUAD (Fig. 12H). There have been
few previous studies on the association between GCC2-AS1 and
LUAD.
Discussion
PCD is a fundamental process in multicellular
organisms that regulates cell proliferation, maintains tissue
homeostasis and eliminates harmful or unnecessary cells from the
organism (27). Recently, a novel
form of PCD termed disulfidptosis has been proposed (5). Disulfidptosis involves the
accumulation of irreducible disulfides, leading to disulfide
stress, and ultimately resulting in disulfide-driven apoptosis.
Extensive research on lncRNAs has revealed their significant role
in promoting or inhibiting various types of tumors, including LUAD,
via the regulation of gene signals (28). LncRNAs are also potential biomarkers
for diagnostic and prognostic purposes (29,30).
However, the specific role of DR-lncRNAs in LUAD is very limited.
For example, Zhang et al (31) identified 127 DRlncRNAs and
established a prognostic model that consisted eight of them
(KTN1-AS1, AL365181.3, MANCR, LINC01352, AC090559.1, AC093673.1,
AP001094.3 and MHENCR) was verified. Song et al (32) developed a prognostic model based on
a different set of five DRlncRNAs (AL365181.2, GSEC, AC093673.1,
AC012615.1 and AL606834.1). The present study contributes to the
growing body of research on the prognostic potential of DRlncRNAs
in LUAD.
In the present study, univariate and lasso analyses
were conducted, which lead to the identification of nine DR-lncRNAs
associated with the prognosis of LUAD. Based on these findings, a
prognostic model was constructed. Through the incorporation of
clinical features and risk scores associated with prognosis, a
nomogram was developed that could lead to an improvement in the
prediction of OS in LUAD for 1, 3 and 5 years. To gain further
insights into the differences between the high and low-risk groups,
functional enrichment analysis was performed, and immune-associated
data were analyzed. It was observed that patients with poor
immunotherapy outcomes had higher risk scores. This suggested that
the identified DR-lncRNAs may have a role in modulating the
response to immunotherapy in LUAD. Integrating TMB with the risk
score indicated potential for the provision of a more comprehensive
prognostic assessment and in helping to identify patients with
distinct OS outcomes. Furthermore, the present study also
demonstrated that samples from the high-risk group of patients
exhibited greater drug-sensitivity to crizotinib, erlotinib and
savolitinib. This finding suggested that these targeted therapies
may be of use in the treatment of LUAD, particularly in patients
classified as high risk on the basis of the prognostic model. The
research methodology of the present study is similar to those
previously reported by Zhang et al (31) and Song et al
(32), as lasso and Cox methods were used to construct a model
of DRlncRNAs in LUAD. The risk model constructed using the DRlncRNA
profiles identified in the present study, was similar to those
previously reported by Zhang et al (31) and Song et al
(32), and could also predict the prognosis of LUAD patients and
their responses to immunotherapy and targeted therapy.
Nevertheless, the lncRNA names identified in risk model of the
present study differ from those reported in previous studies
(31,32), which highlighted the importance of
further identification of novel potential prognostic markers
related to disulfidptosis in the present risk model. Variations in
constructing risk models and identifying DRlncRNAs may arise from
multiple factors, such as selecting different sets of
disulfidptosis genes for the screening of DRlncRNAs. Specifically,
as research related to disulfidptosis is in its early stages, the
specific set of disulfidptosis genes has not yet been clearly
defined. Guided by the principles of reliability and credibility,
the 10 disulfidptosis genes selected in the present study were
based on the initial study be Liu et al (5) which proposed disulfidptosis,
representing the core genes initially identified for
disulfidptosis, whereas the genes selected in the other two studies
may have referenced additional literature (33,34),
comprising 16 and 25 disulfidptosis genes, respectively. The risk
model produced by the present study had a higher AUC value
(AUC=0.727, compared to 0.673 (31)
and 0.681 (32), which indicated
that the present model may demonstrate greater clinical
applicability, in terms of prognosis prediction. In summary, the
present study adds to understanding of the prognostic significance
of DRlncRNAs in LUAD, offering improved predictive models and
highlighting potential therapeutic targets.
The signature used in the present study included
nine lncRNAs, namely U91328.1, LINC00426, MIR1915HG, TMPO-AS1,
TDRKH-AS1, AL157895.1, AL512363.1 and AC010615.2. Several of these
lncRNAs have been previously reported in different cancer types.
For example, U91328.1 has been reported to be associated with poor
prognosis in colon cancer (35).
TMPO-AS1 has previously been reported to be linked to ferroptosis
and iron metabolism in LUAD (36).
TDRKH-AS1 has been reported to promote the proliferation and
invasion of colorectal cancer cells through the Wnt signaling
pathway (37). LINC00426 has been
reported to accelerate LUAD progression via the regulation of
miR-455-5p (38). MIR1915HG has
been reported to be associated with hypoxia in gastric
adenocarcinoma (39). AL157895.1
has been reported to be associated with the occurrence and
development of bladder cancer (40). However, to the best of our
knowledge, no previously published studies have reported on roles
for the other two lncRNAs, AL512363.1 and AC010615.2. The present
study has shown that these nine lncRNAs may exert a role in the
occurrence and development of LUAD and are closely associated with
the underlying mechanism of disulfidptosis.
Changes which occur in the TME fulfill a crucial
role in the progression of LUAD (25). The quantity and quality of
tumor-infiltrating lymphocytes are key factors that forecast
prognostic and therapeutic benefits in many types of cancer, such
as lung adenocarcinoma, oral squamous cell carcinoma and epithelial
ovarian cancer (41–44). The results of the present study
suggested that the risk groups identified based on the prognostic
model displayed distinct characteristics in terms of TME
composition, immune cell distribution and immune function.
Specifically, it was observed that the low-risk group exhibited a
higher TME score. Furthermore, the present study revealed that the
low-risk group exhibited increased immune cell infiltration and
activation of immune function. Previous studies have found that an
increased number of M0 macrophages is associated with poor
prognosis in LUAD (45), while
higher levels of CD8+ T lymphocyte and B lymphocyte infiltration
are associated with better overall survival in LUAD (46), which is consistent with our research
findings. This suggested that the immune system in the low-risk
group is more responsive and active against tumor cells. By
contrast, the high-risk group may have a more immunosuppressive
TME, which could contribute to a reduced immune response and
potential resistance to immune therapy. Considered together, these
findings suggested that the immune profiles and TME characteristics
differed between the high and low-risk groups. The low-risk group,
with a more favorable immune profile, may have a better potential
for positive treatment outcomes, including immunotherapy. These
insights into the associations between risk groups, TME and immune
function have provided information for understanding the
immunological aspects of LUAD and may aid in the development of
personalized treatment strategies.
ICIs have been reported to show promise as effective
immunotherapies for numerous types of cancer. These inhibitors
target specific molecules, such as programmed cell death protein 1
(PD-1)/programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte
associated protein 4, to enhance anti-tumor responses and to
prevent tumor cells from evading immune surveillance (47,48).
TMB has emerged as an important predictive marker for the response
to ICIs (49–51). High TMB has been reported to be
associated with a greater therapeutic efficacy of PD-1 or PD-L1
inhibitors (52). A study has
reported the correlation between high TMB and improved treatment
outcomes with ICIs (53). In the
present study, the potential of the proposed markers to serve as
reliable immune biomarkers for tumor treatment was assessed by
analyzing the TMB and TIDE scores in the different risk groups.
This analysis indicated the importance of somatic mutations and TMB
in LUAD. Through the integration of TMB with the risk score derived
from the prognostic model, the aim was to provide a more
comprehensive prognostic assessment, and to identify patients with
distinct OS outcomes. Overall, the findings of the present study
indicated the importance of considering TMB and its integration
with the risk score in evaluating immune responses and predicting
treatment outcomes in LUAD. This information could be valuable for
guiding personalized treatment decisions, and in of the improvement
of patient outcomes in the context of immunotherapy.
As targeted therapy are conventional therapeutic
strategies for intermediate and advanced LUAD (54), the sensitivity of anti-cancer drugs
among different risk groups of patients with LUAD was also
assessed. This analysis provided insights into individualized
treatment strategies. Our results showed the IC50 values of
targeted therapeutic agents such as crizotinib (55), erlotinib (56) and savolitinib (57) exhibited a notable decrease in
patients with high risk scores, implying that these patients may
display a more favorable treatment response to these drugs. With
this approach, it will be possible to better match patients with
the most appropriate anti-tumor drugs, leading to more effective
and targeted therapies.
The present study did, however, have certain
limitations. The disulfidptosis gene set selected was based on the
study by Liu et al (5), and
included GYS1, LRPPRC, NCKAP1, NDUFA11, NDUFS1, NUBPL, OXSM, RPN1,
SLC3A2 and SLC7A11. It has subsequently been demonstrated that ACTB
is also associated with disulfidptosis (58). In future studies, ACTB should be
included in the gene set for disulfidptosis and in-depth research
specifically targeting ACTB is required. Furthermore, the present
data were derived from the TCGA database and lacked external
dataset validation. Although the differential expression of these
lncRNAs was validated in a LUAD cell line and a human normal cell
line using RT-qPCR analysis, and bioinformatics analysis was
performed to examine the expression of the lncRNA GCC2-AS1 and its
relationship with lung adenocarcinoma, further in vivo and
in vitro experiments are needed to confirm the impact of
DR-lncRNAs on the occurrence and progression of LUAD.
In conclusion, in the present study a novel DRlncSig
was constructed, which provided a novel index to predict the
efficacy of therapeutic interventions and the prognosis of patients
with LUAD, which could be used to guide personalized
treatments.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was financially supported by the Shandong Province
Medical and Health Development Plan (grant no. 202304020860).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LZ, XS, JL and HLiu designed the study. JL performed
the acquisition and analysis of bioinformatic data. XG, YH, ZP, LL
and HLi performed cell culture and RT-qPCR analysis. XS wrote the
manuscript. LZ edited the manuscript. All authors read and approved
the final manuscript. XS and JL confirm the authenticity of all the
raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, AI tools were
used to improve the readability and language of the manuscript or
to generate images, and subsequently, the authors revised and
edited the content produced by the AI tools as necessary, taking
full responsibility for the ultimate content of the present
manuscript.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global Cancer Statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barta JA, Powell CA and Wisnivesky JP:
Global epidemiology of lung cancer. Ann Glob Health. 85:82019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zappa C and Mousa SA: Non-small cell lung
cancer: Current treatment and future advances. Transl Lung Cancer
Res. 5:288–300. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Spella M and Stathopoulos GT: Immune
resistance in lung adenocarcinoma. Cancers (Basel). 13:3842021.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu X, Nie L, Zhang Y, Yan Y, Wang C,
Colic M, Olszewski K, Horbath A, Chen X, Lei G, et al: Actin
cytoskeleton vulnerability to disulfide stress mediates
disulfidptosis. Nat Cell Biol. 25:404–414. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Machesky LM: Deadly actin collapse by
disulfidptosis. Nat Cell Biol. 25:375–376. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ren W, Zhao W, Cao L and Huang J:
Involvement of the actin machinery in programmed cell death. Front
Cell Dev Biol. 8:6348492021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y and Chang SKC: Color and texture
of surimi-like gels made of protein isolate extracted from catfish
byproducts are improved by washing and adding soy whey. J Food Sci.
87:3057–3070. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Franklin-Tong VE and Gourlay CW: A role
for actin in regulating apoptosis/programmed cell death: Evidence
spanning yeast, plants and animals. Biochem J. 413:389–404. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smertenko A and Franklin-Tong VE:
Organisation and regulation of the cytoskeleton in plant programmed
cell death. Cell Death Differ. 18:1263–1270. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li Y, Jiang T, Zhou W, Li J, Li X, Wang Q,
Jin X, Yin J, Chen L, Zhang Y, et al: Pan-cancer characterization
of immune-related lncRNAs identifies potential oncogenic
biomarkers. Nat Commun. 11:10002020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Y, Zhang J, Huo C, Ding N, Li J, Xiao
J, Lin X, Cai B, Zhang Y and Xu J: Dynamic organization of lncRNA
and circular RNA regulators collectively controlled cardiac
differentiation in humans. EBioMedicine. 24:137–146. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huarte M: The emerging role of lncRNAs in
cancer. Nat Med. 21:1253–1261. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luo J, Langer LF and Liu J: A novel role
of LncRNA in regulating tumor metabolism and angiogenesis under
hypoxia. Cancer Commun (Lond). 39:22019.PubMed/NCBI
|
|
15
|
Xu M, Zhou H, Hu P, Pan Y, Wang S, Liu L
and Liu X: Identification and validation of immune and oxidative
stress-related diagnostic markers for diabetic nephropathy by WGCNA
and machine learning. Front Immunol. 14:10845312023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Friedman J, Hastie T and Tibshirani R:
Regularization paths for generalized linear models via coordinate
descent. J Stat Softw. 33:1–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang M, Zheng H, Xu K, Yuan Q, Aihaiti Y,
Cai Y and Xu P: A novel signature to guide osteosarcoma prognosis
and immune microenvironment: Cuproptosis-related lncRNA. Front
Immunol. 13:9192312022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Q, Sun T, Wang G, Zhang M, Zhu Y, Shi
X and Ding Z: Cuproptosis-related LncRNA signature for predicting
prognosis of hepatocellular carcinoma: A comprehensive analysis.
Dis Markers. 2022:32652122022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li H, Han D, Hou Y, Chen H and Chen Z:
Statistical inference methods for two crossing survival curves: A
comparison of methods. PLoS One. 10:e01167742015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang F, Lin H, Su Q and Li C:
Cuproptosis-related lncRNA predict prognosis and immune response of
lung adenocarcinoma. World J Surg Oncol. 20:2752022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iasonos A, Schrag D, Raj GV and Panageas
KS: How to build and interpret a nomogram for cancer prognosis. J
Clin Oncol. 26:1364–1370. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu N, Hou J, Si J, Yang N, Chen B, Wei X
and Zhu L: SIRT1 and ZNF350 as novel biomarkers for osteoporosis: A
bioinformatics analysis and experimental validation. Mol Biol Rep.
51:5302024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mayakonda A, Lin DC, Assenov Y, Plass C
and Koeffler HP: Maftools: Efficient and comprehensive analysis of
somatic variants in cancer. Genome Res. 28:1747–1756. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu J, Li L, Zhang H, Zhao Y, Zhang H, Wu S
and Xu B: A risk model developed based on tumor microenvironment
predicts overall survival and associates with tumor immunity of
patients with lung adenocarcinoma. Oncogene. 40:4413–4424. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang L, He YT, Dong S, Wei XW, Chen ZH,
Zhang B, Chen WD, Yang XR, Wang F, Shang XM, et al: Single-cell
transcriptome analysis revealed a suppressive tumor immune
microenvironment in EGFR mutant lung adenocarcinoma. J Immunother
Cancer. 10:e0035342022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Christgen S, Tweedell RE and Kanneganti
TD: Programming inflammatory cell death for therapy. Pharmacol
Ther. 232:1080102022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tan C, Du H, Wang Y, Zhao J, Cheng X and
Lan H: LncRNA GABPB1-IT1 inhibits the tumorigenesis of renal cancer
via the miR-21/PTEN axis. J Biochem Mol Toxicol. 37:e232882023.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng Z, Lu C, Wang H, Wang N, Cui S, Yu
C, Wang C, Zuo Q, Wang S, Lv Y, et al: Long noncoding RNA
LHFPL3-AS2 suppresses metastasis of non-small cell lung cancer by
interacting with SFPQ to regulate TXNIP expression. Cancer Lett.
531:1–13. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cui Y, Zhang C, Ma S and Guan F:
TFAP2A-induced SLC2A1-AS1 promotes cancer cell proliferation. Biol
Chem. 402:717–727. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang HB, Pan JY and Zhu T: A
disulfidptosis-related lncRNA prognostic model to predict survival
and response to immunotherapy in lung adenocarcinoma. Front
Pharmacol. 14:12541192023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Song Z, Cao X, Wang X, Li Y, Zhang W, Wang
Y and Chen L: A disulfidptosis-related lncRNA signature for
predicting prognosis and evaluating the tumor immune
microenvironment of lung adenocarcinoma. Sci Rep. 14:46212024.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang L, Liu J, Li S, Liu X, Zheng F, Xu S,
Fu B and Xiong J: Based on disulfidptosis, revealing the prognostic
and immunological characteristics of renal cell carcinoma with
tumor thrombus of vena cava and identifying potential therapeutic
target AJAP1. J Cancer Res Clin Oncol. 149:9787–9804. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao S, Wang L, Ding W, Ye B, Cheng C,
Shao J, Liu J and Zhou H: Crosstalk of disulfidptosis-related
subtypes, establishment of a prognostic signature and immune
infiltration characteristics in bladder cancer based on a machine
learning survival framework. Front Endocrinol (Lausanne).
14:11804042023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou SZ, Pan YL, Deng QC, Yin CJ, Zhou DJ,
Liu ML, Zhou J and Wu XJ: A prognostic signature for colon
adenocarcinoma patients based on m6A-related lncRNAs. J Oncol.
2023:77977102023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yao J, Chen X, Liu X, Li R, Zhou X and Qu
Y: Characterization of a ferroptosis and iron-metabolism related
lncRNA signature in lung adenocarcinoma. Cancer Cell Int.
21:3402021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiao Y, Zhou J, Jin Y, Yang Y, Song M,
Zhang L, Zhou J and Zhang J: Long non-coding RNA TDRKH-AS1
promotes colorectal cancer cell proliferation and invasion through
the β-Catenin activated Wnt signaling pathway. Front Oncol.
10:6392020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li H, Mu Q, Zhang G, Shen Z, Zhang Y, Bai
J, Zhang L, Zhou D, Zheng Q, Shi L, et al: Linc00426 accelerates
lung adenocarcinoma progression by regulating miR-455-5p as a
molecular sponge. Cell Death Dis. 11:10512020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fan Z, Wang Y and Niu R: Identification of
the three subtypes and the prognostic characteristics of stomach
adenocarcinoma: Analysis of the hypoxia-related long non-coding
RNAs. Funct Integr Genomics. 22:919–936. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang C, Bai X, Peng X, Shi W, Li Y, Chen
G, Yu H, Feng Z and Deng Y: Starvation-induced long non-coding RNAs
are significant for prognosis evaluation of bladder cancer. Aging
(Albany NY). 14:10067–10080. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hwang C, Lee SJ, Lee JH, Kim KH, Suh DS,
Kwon BS and Choi KU: Stromal tumor-infiltrating lymphocytes
evaluated on H&E-stained slides are an independent prognostic
factor in epithelial ovarian cancer and ovarian serous carcinoma.
Oncol Lett. 17:4557–4565. 2019.PubMed/NCBI
|
|
42
|
Paijens ST, Vledder A, de Bruyn M and
Nijman HW: Tumor-infiltrating lymphocytes in the immunotherapy era.
Cell Mol Immunol. 18:842–859. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shaban M, Khurram SA, Fraz MM, Alsubaie N,
Masood I, Mushtaq S, Hassan M, Loya A and Rajpoot NM: A novel
digital score for abundance of tumour infiltrating lymphocytes
predicts disease free survival in oral squamous cell carcinoma. Sci
Rep. 9:133412019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pan X, Lin H, Han C, Feng Z, Wang Y, Lin
J, Qiu B, Yan L, Li B, Xu Z, et al: Computerized tumor-infiltrating
lymphocytes density score predicts survival of patients with
resectable lung adenocarcinoma. iScience. 25:1056052022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xue Q, Wang Y, Zheng Q, Chen L, Lin Y, Jin
Y, Shen X and Li Y: Prognostic value of tumor immune
microenvironment factors in patients with stage I lung
adenocarcinoma. Am J Cancer Res. 13:950–963. 2023.PubMed/NCBI
|
|
46
|
Liu X, Wu S, Yang Y, Zhao M, Zhu G and Hou
Z: The prognostic landscape of tumor-infiltrating immune cell and
immunomodulators in lung cancer. Biomed Pharmacother. 95:55–61.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu C, Zheng S, Wang Z, Wang S, Wang X,
Yang L, Xu H, Cao Z, Feng X, Xue Q, et al: KRAS-G12D mutation
drives immune suppression and the primary resistance of
anti-PD-1/PD-L1 immunotherapy in non-small cell lung cancer. Cancer
Commun (Lond). 42:828–847. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wen Y, Tang F, Tu C, Hornicek F, Duan Z
and Min L: Immune checkpoints in osteosarcoma: Recent advances and
therapeutic potential. Cancer Lett. 547:2158872022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jardim DL, Goodman A, de Melo Gagliato D
and Kurzrock R: The challenges of tumor mutational burden as an
immunotherapy biomarker. Cancer Cell. 39:154–173. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu L, Bai X, Wang J, Tang XR, Wu DH, Du
SS, Du XJ, Zhang YW, Zhu HB, Fang Y, et al: Combination of TMB and
CNA stratifies prognostic and predictive responses to immunotherapy
across metastatic cancer. Clin Cancer Res. 25:7413–7423. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Samstein RM, Lee CH, Shoushtari AN,
Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ,
Omuro A, et al: Tumor mutational load predicts survival after
immunotherapy across multiple cancer types. Nat Genet. 51:202–206.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ricciuti B, Wang X, Alessi JV, Rizvi H,
Mahadevan NR, Li YY, Polio A, Lindsay J, Umeton R, Sinha R, et al:
Association of high tumor mutation burden in non-small cell lung
cancers with increased immune infiltration and improved clinical
outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA
Oncol. 8:1160–1168. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cao D, Xu H, Xu X, Guo T and Ge W: High
tumor mutation burden predicts better efficacy of immunotherapy: A
pooled analysis of 103078 cancer patients. Oncoimmunology.
8:e16292582019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Imyanitov EN, Iyevleva AG and Levchenko
EV: Molecular testing and targeted therapy for non-small cell lung
cancer: Current status and perspectives. Crit Rev Oncol Hematol.
157:1031942021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shaw AT, Bauer TM, de Marinis F, Felip E,
Goto Y, Liu G, Mazieres J, Kim DW, Mok T, Polli A, et al:
First-line lorlatinib or crizotinib in advanced ALK-positive lung
cancer. N Engl J Med. 383:2018–2029. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Abdelgalil AA, Al-Kahtani HM and
Al-Jenoobi FI: Erlotinib. Profiles Drug Subst Excip Relat Methodol.
45:93–117. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lee TS, Kim JY, Lee MH, Cho IR, Paik WH,
Ryu JK, Kim YT and Lee SH: Savolitinib: A promising targeting agent
for cancer. Cancers (Basel). 15:47082023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ni L, Yang H, Wu X, Zhou K and Wang S: The
expression and prognostic value of disulfidptosis progress in lung
adenocarcinoma. Aging (Albany NY). 15:7741–7759. 2023.PubMed/NCBI
|