Introduction
Oral cancer is one of the most common cancers
worldwide, and accounts for 4% of all cancer cases (1). Oral squamous cell carcinoma (OSCC) is
the most common tumor that occurs in the oral and maxillofacial
regions, causing marked mortality (2). At present, the main clinical
treatments for OSCC are surgery, radiotherapy and chemotherapy
(3). In 2020, 377,713 cases of OSCC
were reported worldwide (4). The
Global Cancer Observatory predicts that by 2040, the incidence of
OSCC will increase by ~40%, with a concomitant increase in
mortality, the overall 5-year survival rate of OSCC remains <50%
(3,5,6).
Considering the special physiological characteristics of the oral
cavity, the treatment of OSCC not only emphasizes the improvement
in the survival rate of patients, but also aims at improving their
quality of life (7). However, the
unique structural characteristics of the oral cavity make it
complicated and challenging to complete postoperative
reconstruction, which usually results in poor quality of life for
patients (8). Therefore, it is
evident that novel effective drugs for the treatment of OSCC are
required.
Procaine (PCA) is a common local anesthetic drug
mainly used in oral surgeries. Recent studies have shown that in
addition to anesthesia, PCA has been proven to be an inhibitor of
DNA methylation (9,10). Villar-Garea et al (11) found that PCA not only inhibited
MCF-7breast cancer cell proliferation, but was also tightly bound
to DNA rich in CpG islands, causing DNA demethylation events. In
addition, a study indicated that PCA could inhibit the growth of
liver cancer cells both in vitro and in vivo, and has
a demethylation effect (12).
Despite the fact that the antitumor effect of PCA has been
partially studied, these studies have focused on the epigenetic
effects of PCA, and have not explored the mechanism of cell
proliferation inhibition by PCA. In particular, the mechanism of
action of PCA in human oral tongue squamous cell carcinoma CAL27
cells is still unclear. Therefore, in the present study, the aim
was to elucidate the effect of PCA on tongue squamous cell
carcinoma. The potential mechanism of PCA as an anticancer drug in
CAL27 cells was also studied. The results could provide novel ideas
for the future applications of PCA as an antitumor drug in clinical
treatment.
Materials and methods
Reagents
DMEM, fetal bovine serum (FBS) and
penicillin/streptomycin were purchased from Gibco; Thermo Fisher
Scientific, Inc. PCA (cat. no. 51-05-8) was purchased from Thermo
Fisher Scientific, Inc. The anti-GAPDH (cat. no. 5174), anti-p21
(cat. no. 2947), anti-Bcl-2 (cat. no. 2870), anti-Bax (cat. no.
5023), anti-survivin (cat. no. 2808), anti-LC3B (cat. no. 3868),
anti-p62 (cat. no. 8025), anti-ERK (cat. no. 9102),
anti-phosphorylated (p)-ERK (cat. no. 4370), anti-AKT (cat. no.
9272), anti-p-AKT (cat. no. 4060) and anti-PI3K (cat. no. 4249)
polyclonal antibodies were purchased from Cell Signaling
Technology, Inc. IRDye 800CW goat anti-rabbit IgG (cat. no.
925-32211) was purchased from LI-COR Biosciences. Cell Counting Kit
(CCK-8) was purchased from Dojindo Laboratories, Inc. Rhodamine
123, crystal violet dye and RIPA cell lysate were purchased from
Beyotime Institute of Biotechnology. Western blotting protein
prestaining marker, primers, β-actin, CDK2 and cyclin E were
obtained from Shanghai Sangong Pharmaceutical Co., Ltd.
Hydroxychloroquine sulfate (CQ) was purchased from Selleck
Chemicals. The cell apoptosis detective kit [Annexin V-propidium
iodide (PI)] was purchased from Jiangsu Kaiji Biotechnology Co.,
Ltd.
Cell culture
The human oral tongue squamous cell carcinoma CAL27
and SCC-15 cell lines were obtained from the American Type Culture
Collection. Cells were cultured in DMEM with 10% FBS and 1%
penicillin/streptomycin, and maintained in a humidified chamber of
95% air, 5% CO2 and at 37°C.
Cell viability assay
The cells were seeded into 96-well plates with
8×103 cells/well and cultured overnight. After treated
with different doses of PCA (0.5, 1.0 and 2.0 mg/ml) for 24 h, the
cells were incubated with 10 µl CCK-8. After 2 h, a microplate
reader was used to measure absorbance at 450 nm.
Colony formation assay
Aggregations of cells comprising a minimum of 50
cells were recognized as colonies. The cells were seeded into
6-well plates (2×103 cells/well) and treated with
different concentrations (0, 0.0675, 0.125, 0.25, 0.5 and 1.0
mg/ml) of PCA at 37°C for 2 weeks. Cell colonies were fixed with
cold 100% methanol for 15 min at room temperature and stained with
0.1% crystal violet for 20 min at room temperature. Formed colonies
were observed and captured by light microscope (IX51; Olympus
Corporation). Colonies were quantified using Image J v1.8.0
(National Institutes of Health).
Intracellular reactive oxygen species
(ROS) assay
In order to clarify whether the up-regulation of ROS
levels in cells is the main reason for the inhibition of cell
proliferation by PCA, ROS levels were measured in CAL27 cells and
SCC-15 cells after co-administration of the ROS scavenger NAC and
different concentrations of PCA in each group. The cells were
washed with PBS and re-suspended in 10 µM fluorescent dye
2′-7′dichlorofluorescin diacetate (DCFH-DA). Next, cells were
incubated in the dark for 30 min at 37°C. The experimental
procedure was completed according to the experimental step-by-step
instructions of the ROS detection kit (Beyotime Institute of
Biotechnology; cat. no. S0033S). After washing twice, the cells
were observed and photographed under a fluorescence microscope, and
intracellular ROS levels were semi-quantitatively detected in a
flow cytometer (BD Biosciences). The results of cell flow analysis
were statistically analyzed to obtain peak plots using FlowJo
software v10.6.2 (BD Biosciences).
Wound healing assay
The cells were scratched using micro-pipette tips,
and imaged using a microscope. The width of the scratch was
measured and referred to as Wbefore. Then, the cells were treated
with different concentrations (0, 0.5, 1.0 and 2.0 mg/ml) of PCA
and starved with 2% serum DMEM. Cell migration was monitored and
images were captures using a light microscope at 0, 6, 12, 24 and
48 h. In the present experiment, the distance of statistical cell
migration was used as a quantitative method. By measuring the width
of the same scratch, which is called Wafter, the migration distance
is calculated by subtracting Wafter from Wbefore. The migration of
the control was set as 100. The resultant scale on the image is 100
µm.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.) reagent. PrimeScript™ RT Master Mix
(Takara Bio, Inc.; cat. no. RR037A) according to the manufacturer's
instructions. Relative mRNA levels were quantified with SYBR Green
PCR Kit (Roche Diagnostics) and normalized to GAPDH using the
following primers: GAPDH-F, 5′-AGAAGGCTGGGGCTCATTTG-3′, and
GAPDH-R, 5′-AGGGGCCATCCACAGTCTTC-3′; P21-F,
5′-GGAGACTCTCAGGGTCGAAA-3′, P21-R, 5′-GGATTAGGGCTTCCTCTTGG-3′. The
thermocycling parameters included: 40 cycles at 95°C for 15 sec,
60°C for 15 sec and 72°C for 30 sec. Then another 10 min at 95°C,
15 sec at 95°C and 30 sec at 60°C for 40 cycles. The mRNA levels of
target genes were normalized to GAPDH and data of relative
expression was quantified using the 2−ΔΔcq method
(13). Data were obtained from
triplicate experiments.
Mitochondrial membrane potential (ΔΨm)
assay
ΔΨm was assessed using Rhodamine 123. Cells were
incubated with 2 µM Rhodamine 123 in the dark for 30 min, and ΔΨm
levels were detected in a flow cytometer (BD Biosciences).
Mitochondrial fluorescence intensity-determined ΔΨm was assessed
using High Content Screening (HCS) (14).
Cell cycle analysis
The cells were seeded into 6-well plates with
2×104 cells/well and cultured overnight. The cells were
harvested and fixed with 70% ethanol at −20°C for 30 min. After
washing with PBS, cells were stained (20 µg/ml PI, 200 µg/ml
RNase). The cell cycle profiling was analyzed using a FACScan™ flow
cytometer (BD Biosciences) using 488 nm excitation. The results of
cell cycle analysis were statistically analyzed to obtain peak
plots using FlowJo software v10.6.2 (BD Biosciences). Doublets were
excluded from analysis, and 10,000 events were collected in the
single-cell gate per sample.
Apoptosis analysis
The cells were seeded into 6-well plates with
2×104 cells/well and cultured overnight. The cells were
washed twice with cold PBS and added trypsin at 37°C for 2 min for
digestion. After exposure, the cells were harvested and stained
with Annexin V-FITC and PI as recommended by the manufacturer.
Samples were analyzed by FACScan™ flow cytometer (BD Biosciences).
The results of apoptosis analysis were statistically analyzed by
using FlowJo software v10.6.2 (BD Biosciences). The signals of
early and late apoptotic cells were located in the lower and upper
right quadrant of the resulting dot plot, respectively.
Western blotting
Total protein was extracted using RIPA buffer and a
protease inhibitor cocktail (Beyotime Institute of Biotechnology)
to prevent protein degradation. Cells were disrupted through
sonication, ensuring complete lysis. A BCA protein assay kit (cat.
no. P0012) was obtained from Beyotime Institute of Biotechnology
and used following manufacturer's instructions for protein
quantification. Cell lysates (30–50 µg) were separated by 12.5%
SDS-PAGE and transferred onto PVDF membranes (Thermo Fisher
Scientific, Inc.). After blocking with 5% non-fat milk in PBST
buffer with 0.1% Tween for 1 h at room temperature, the blots were
incubated with primary antibodies [anti-GAPDH (1:1,000), anti-p21
(1:1,000), anti-Bcl-2 (1:1,000), anti-Bax (1:1,000), anti-survivin
(1:1,000), anti-LC3B (1:1,000), anti-p62 (1:1,000), anti-ERK
(1:1,000), anti-p-ERK (1:1,000), anti-AKT (1:1,000.), anti-pAKT
(1:1,000) and anti-PI3K (1:1,000) polyclonal antibodies] at 4°C
overnight. Membranes were then incubated with secondary antibodies
(IRDye 800CW goat anti-rabbit IgG; 1:20,000] at room temperature
for 1 h, and then detected using Odyssey infrared imaging system
(LI-COR Biosciences). Densitometry were semi-quantified using Image
J v1.8.0 (National Institutes of Health).
Immunofluorescence
The cells were seeded into 96-well plates with
8×103 cells/well and cultured overnight. The cells were
treated with different concentrations of PCA (0, 0.5, 1.0 and 2.0
mg/ml) for 24 h. Then, the cells were fixed with 4%
paraformaldehyde for 15 min at room temperature, washed with PBS
before permeabilization using 0.1% Triton X-100 for 10 min at room
temperature. After washing with PBS, cells were blocked for 30 min
in 5% BSA containment solution at 37°C and then, incubated with
primary antibody LC3B (1:400; Cell Signaling Technology, Inc.; cat.
no. 3868) for 1 h at 37°C, washed with PBS and incubated with
fluorescently-labelled secondary antibody [FITC-conjugated
Affinipure Goat Anti-Mouse IgG(H+L); 1:200; cat. no. A00003-1;
Proteintech Group, Inc.] for 1 h at 37°C. Finally, the nuclei were
visualized by Hoechst33342 counterstaining for 5 min at room
temperature. After washing with PBS, immunofluorescent cells were
visualized using HSC and analyzed with the integrated Columbus
(version 6.0) image data storage and analysis system (PerkinElmer,
Inc.). The resultant scale on the image is 100 µm.
Statistical analysis
All data from three times experiments are presented
as mean ± standard deviation. Statistical analyses were carried out
using SPSS (version 16.0; IBM Corp.). Differences between two
groups were assessed using a paired Student's t-test, and
differences between ≥3 groups were assessed using one-way or
two-way ANOVA followed by Bonferroni's post hoc test. P<0.05 and
P<0.01 were considered to indicate statistically significant
differences.
Results
Effects of PCA on cell viability and
colony formation activity
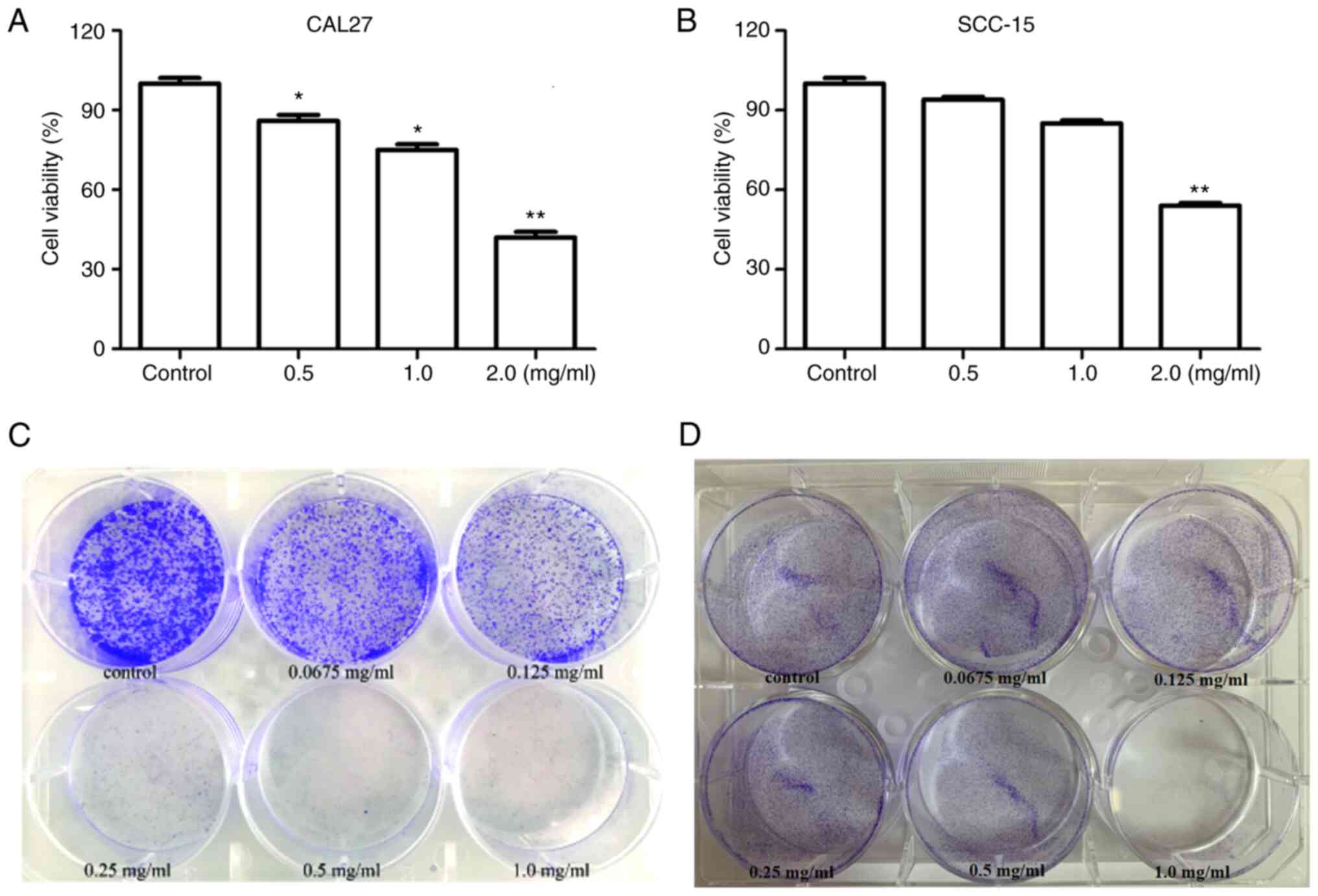
To determine the effect of PCA on cell viability,
CAL27 and SCC-15 cells were treated with different concentrations
of PCA, and then a CCK-8 assay was carried out. PCA exhibited
significant concentration-dependent anti-proliferative effects in
CAL27 cells (Fig. 1A). The results
showed that cell viability was significantly reduced at PCA
concentration of 0.5 and 1.0 mg/ml (P<0.05). Cell activity was
only 40% at the highest PCA concentration tested (2.0 mg/ml),
compared with that in the control group (P<0.01). Similar
results were observed in SCC-15 cells (Fig. 1B). PCA exhibited a significant
inhibitory effect at 2 mg/ml on SCC-15 cells. These results
indicate that CAL27 cells are more sensitive to PCA than SCC-15
cells.
Consequently, a colony formation assay was conducted
to detect changes in the self-renewal ability of the two cell
lines. Cell viability significantly decreased in a dose-dependent
manner (Fig. 1C). Even at the
lowest concentration (0.0675 mg/ml), PCA significantly inhibited
CAL27 cell colony formation compared with the control. PCA had a
similar inhibitory effect in SCC-15 cells (Fig. 1D). The anti-proliferative effects of
PCA in SCC-15 cells were observed at the lowest dose (0.0675
mg/ml). These results are consistent those of the cell
proliferation assays for both CAL27 and SCC-15 cells.
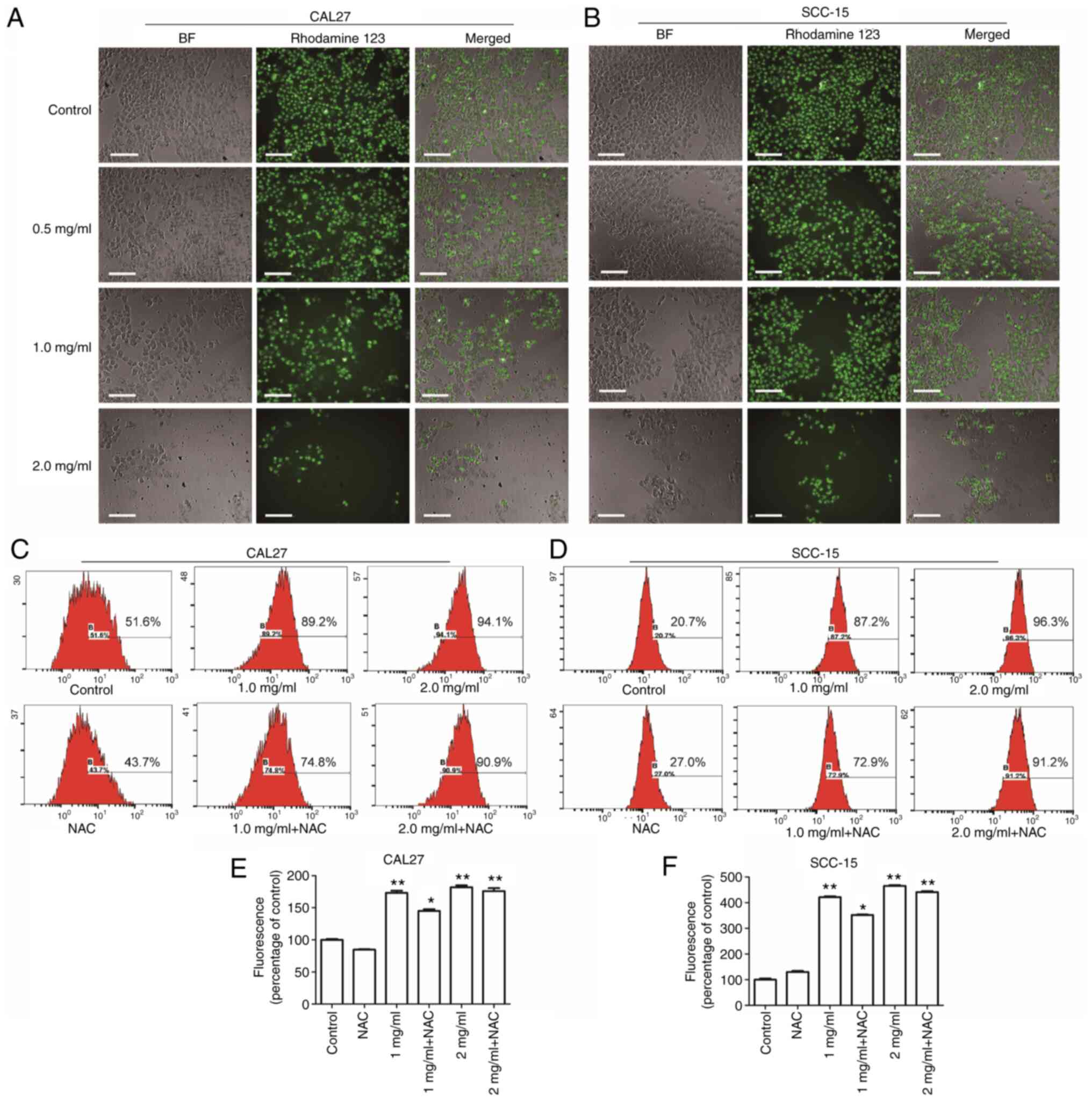
Effects of PCA on ΔΨm and
intracellular ROS generation
Mitochondria are the center of the cellular
oxidative respiratory chain as well as the center of apoptosis
regulation. Decreased ΔΨm is known to be closely related to
mitochondrial dysfunction (15).
Therefore, the effects of PCA on mitochondrial function in human
tongue squamous carcinoma cells was examined by HCS. CAL27 cells
(Fig. 2A; left panels) and SCC-15
cells (Fig. 2B; left panels)
treated with PCA were imaged under a light microscope; the
mitochondrial membranes were stained with Rhodamine 123 (middle
panels). Compared with the control group, the cells treated with
PCA induced a reduction in the ΔΨm, suggesting that PCA induced a
loss of ΔΨm in both CAL27 cells and SCC-15 cells. Moreover, it was
observed that as the PCA concentration increased, the green
fluorescence and number of cells gradually decreased. The results
are consistent with those of the CCK-8 assay results.
DCFH-DA was used to assess ROS production to
investigate whether ROS would be generated in CAL27 cells after PCA
treatment. The intracellular ROS levels in CAL27 cells increased
significantly after PCA treatment (Fig.
2C and E). These effects were dose dependent. However, this
increase in DCFH-DA-based fluorescence was not inhibited by
pretreatment with the ROS scavenger N-acetyl-L-cysteine (NAC),
which did not reduce the PCA-induced increase in fluorescence.
These results indicate that ROS production was not the main cause
of the inhibited proliferation of CAL27 cells exposed to PCA.
Similar results were found in SCC-15 cells (Fig. 2D and F). A significant
dose-dependent loss of ΔΨm was observed in SCC-15 cells after PCA
treatment (Fig. 2B). Furthermore,
NAC partially reversed PCA-induced ROS production, which is
consistent with the results obtained in CAL27 cells (NAC + 1 mg/ml
PCA; *P<0.05; Fig. 2E).
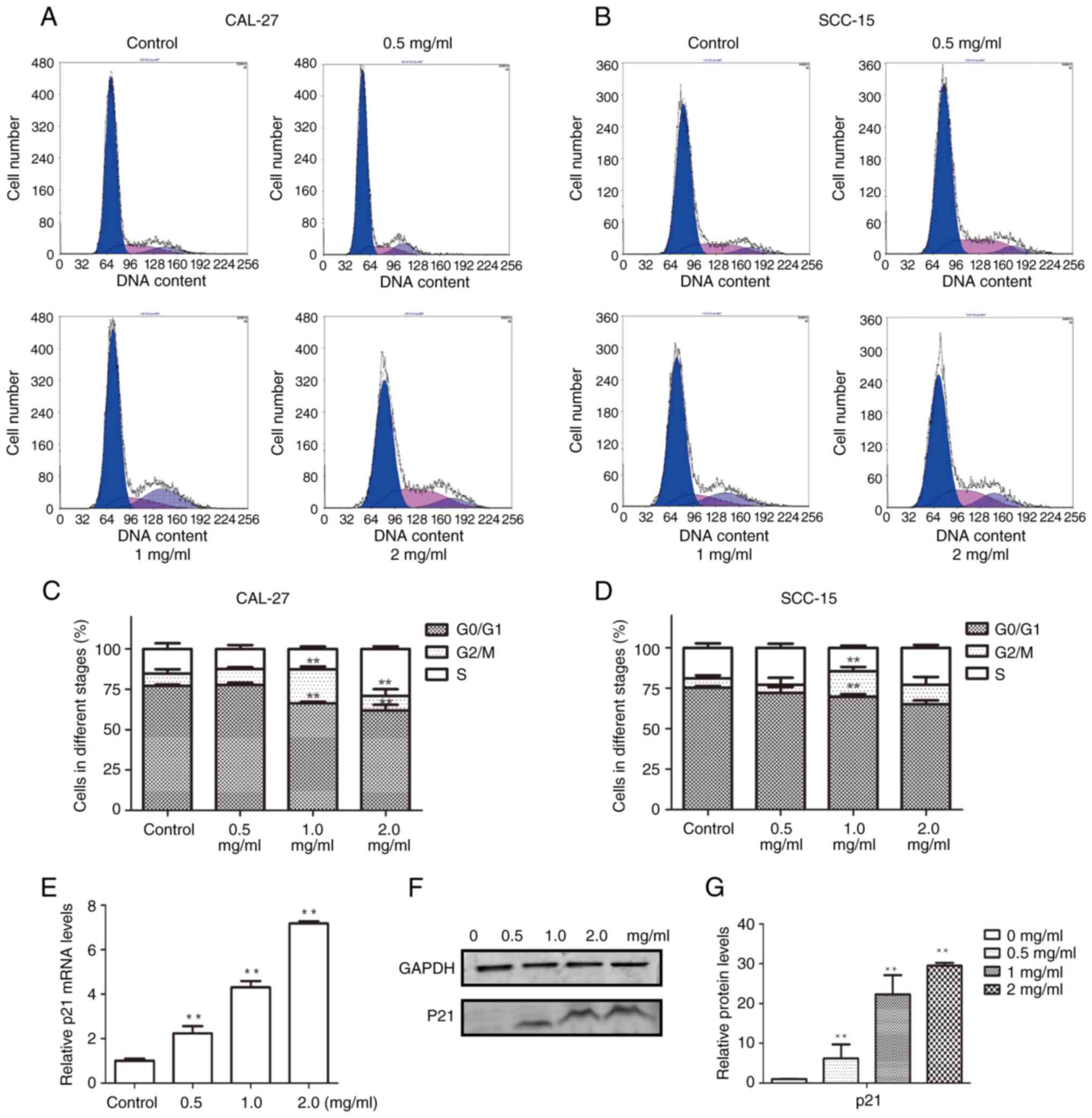
Effects of PCA on cell cycle
To elucidate the mechanism by which PCA inhibits
cell proliferation, the cell cycle distribution was examined using
flow cytometry. The G0/G1 phase population was reduced in CAL27
cells after PCA treatment, whereas the G2/M phase cell population
was significantly increased, suggesting that PCA caused G2/M phase
arrest in CAL27 cells (Fig. 3A and
C). Notably, no remarkable G2/M arrest was observed at the
highest concentration of PCA (2 mg/ml), although an increase in the
S phase cell population was observed. This indicated that the cells
were inhibited in the S phase at a high PCA dose.
Similar results were observed in SCC-15 cells
(Fig. 3B and D) as PCA induced G2/M
phase arrest. The potential mechanisms by which PCA induced G2/M
phase arrest in CAL27 cells were further explored.
RT-qPCR was performed to analyze the expression of
the cyclin-dependent kinase inhibitor p21, which is related to S
phase arrest. p21 mRNA expression levels significantly increased
with increasing PCA concentrations (*P<0.05, **P<0.01;
Fig. 3E). Moreover, p21 protein
expression increased in a PCA dose-dependent manner, which is
consistent with the RT-qPCR results (Fig. 3F and G).
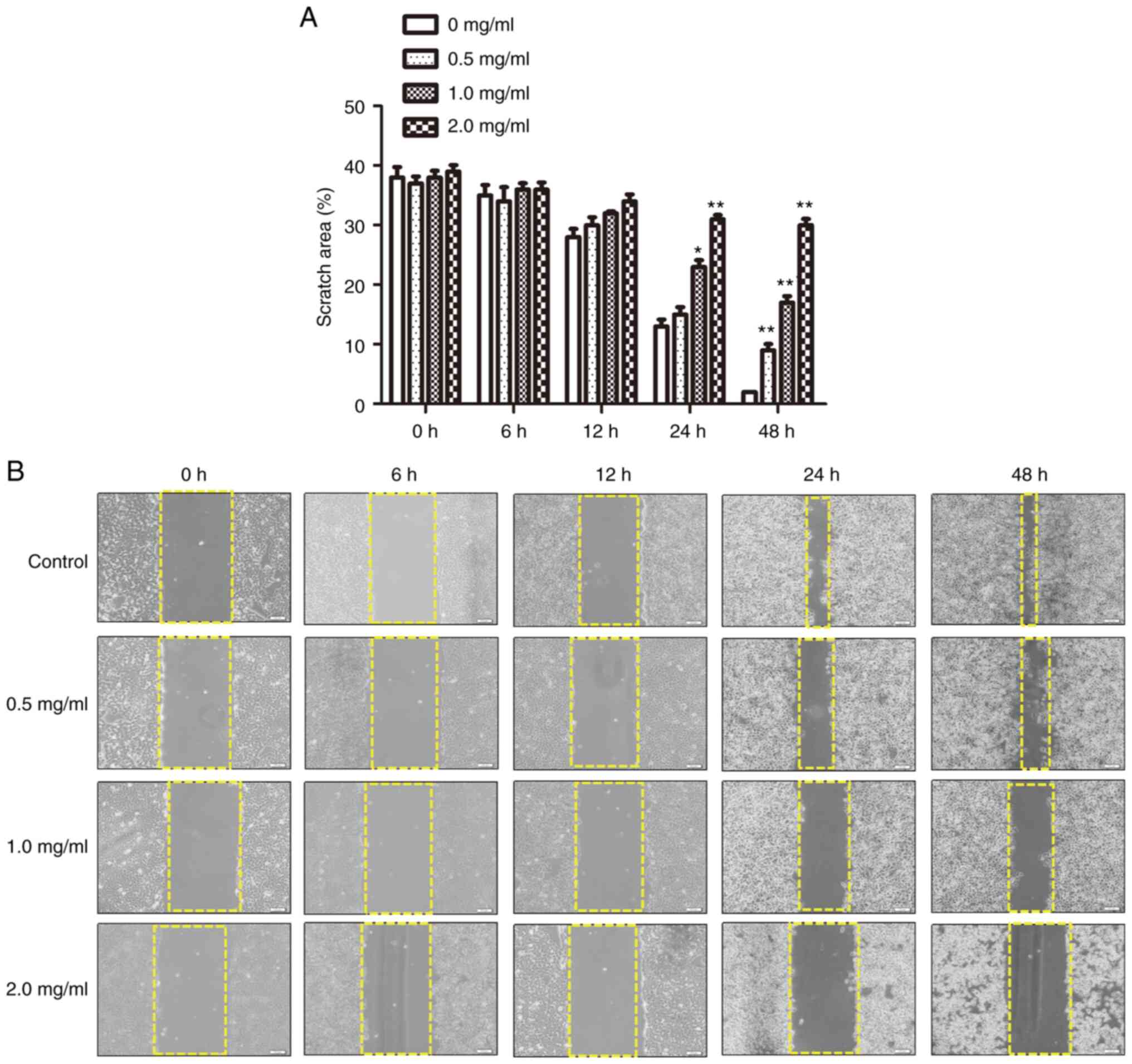
Effects of PCA on cell migration
The effects of PCA on SCC-15 and CAL27 cells were
compared using CCK8, immunofluorescence and cell proliferation
assays. The results were found to be similar for both cell lines.
In addition, the results of the CCK8 assay, colony formation assay,
intracellular ROS assay, ΔΨm assay, cell cycle analysis and
immunofluorescence analysis all revealed that CAL27 cells are more
sensitive to PCA than SCC-15 cells. Therefore, for subsequent
in-depth mechanistic studies, CAL27 cells were focused on. In the
present experiment, the effect of PCA on CAL27 cell migration was
investigated in greater depth. A number of drugs affect both cell
morphology and migration. To determine the inhibitory effects of
PCA on cell migration, a wound healing assay using CAL27 cells was
performed (Fig. 4A and B). PCA
inhibited cell migration in a concentration- and time-dependent
manner. The scratch area increased with increasing PCA
concentration, especially at 24 and 48 h. Compared with the control
group, the scratching area healed with 2.0 mg/ml PCA (*P<0.05,
**P<0.01), which showed that PCA significantly inhibited the
cell healing rate.
Effects of PCA on apoptosis
PCA blocked cell proliferation and induced growth
arrest in CAL27 cells. To further clarify the underlying
mechanisms, CAL27 cells were treated with different concentrations
of PCA, dual-stained with Annexin-V-FITC and PI and analyzed using
flow cytometry. The early and late apoptotic rates of PCA-treated
cells were observed in a dose-dependent manner (Fig. 5A). At the highest concentration
tested (2 mg/ml), ~11.0% of the cells underwent late apoptosis
compared with 1.6% of the control cells. These results suggest that
PCA treatment increased apoptosis in a dose -and time-dependent
manner in CAL27 cells.
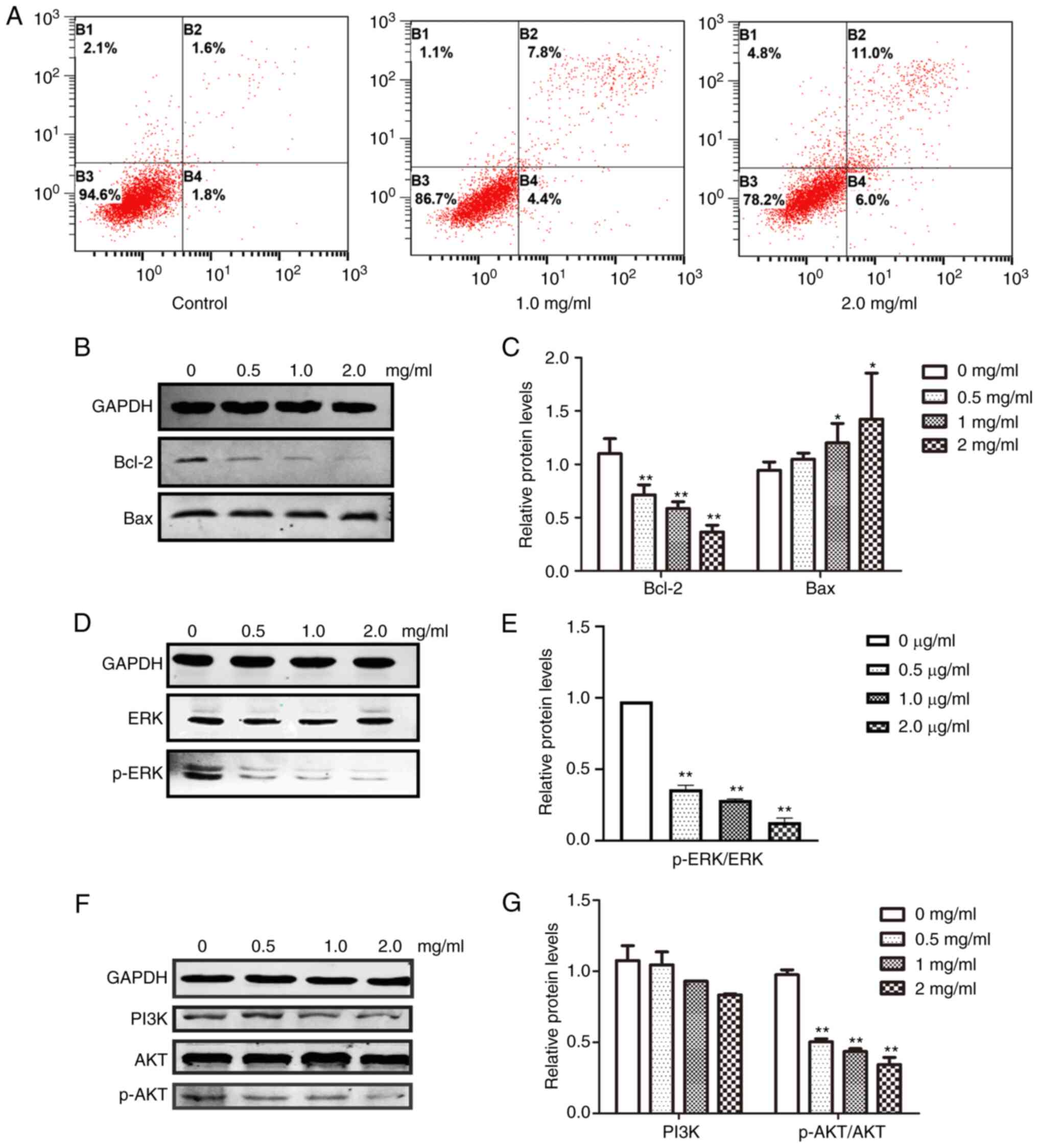
The effect of PCA on anti-apoptotic (Bcl-2) and
apoptotic (Bax) protein expression was then analyzed. As it was
anticipated, Bcl-2 expression was significantly suppressed
(**P<0.01), whereas Bax expression was slightly increased
(*P<0.05), suggesting that PCA-mediated inhibition of
proliferation occurred through apoptosis induction (Fig. 5B and C).
The effects of PCA on the ERK and PI3K/AKT signaling
pathways were examined to determine which signaling pathways play
important roles in PCA-induced apoptosis. Both ERK and p-ERK
protein expression levels were significantly reduced in a
dose-dependent manner in PCA-treated CAL27 cells compared with the
control group (Fig. 5C and D).
These results suggest that PCA inhibits the ERK signaling pathway.
Moreover, with increasing concentrations of PCA, both p-AKT and
PI3K expression levels were decreased, indicating that PCA also
inhibits the PI3K/AKT signaling pathway.
Effects of PCA on cell autophagy
The LC3 protein expression level, a marker of
autophagy, was determined to investigate whether autophagy
participates in the effect of PCA on CAL27 cell proliferation. The
cell nucleus was stained with Hoechst33342, whereas autophagosomes
were visualized as FITC-labeled LC3B (Fig. 6A). With an increase in PCA
concentration, the number of live cells gradually decreased, and
the intensity of green fluorescence gradually increased. Moreover,
green fluorescence was mainly distributed in the cytoplasm and was
not co-localized with the nucleus. These results indicated that PCA
induced autophagy. Western blotting showed similar results. The
LC3II/I expression levels increased in a concentration-dependent
manner in PCA-pretreated CAL27 cells compared with the control
group, suggesting that PCA induces autophagy in CAL27 cells
(Fig. 6B and C). To determine
whether PCA could induce complete autophagy, the levels of the
autophagy substrate p62 were examined. The p62 protein expression
levels also showed a significant concentration-dependent increase,
suggesting that autophagy flux might be inhibited by PCA.
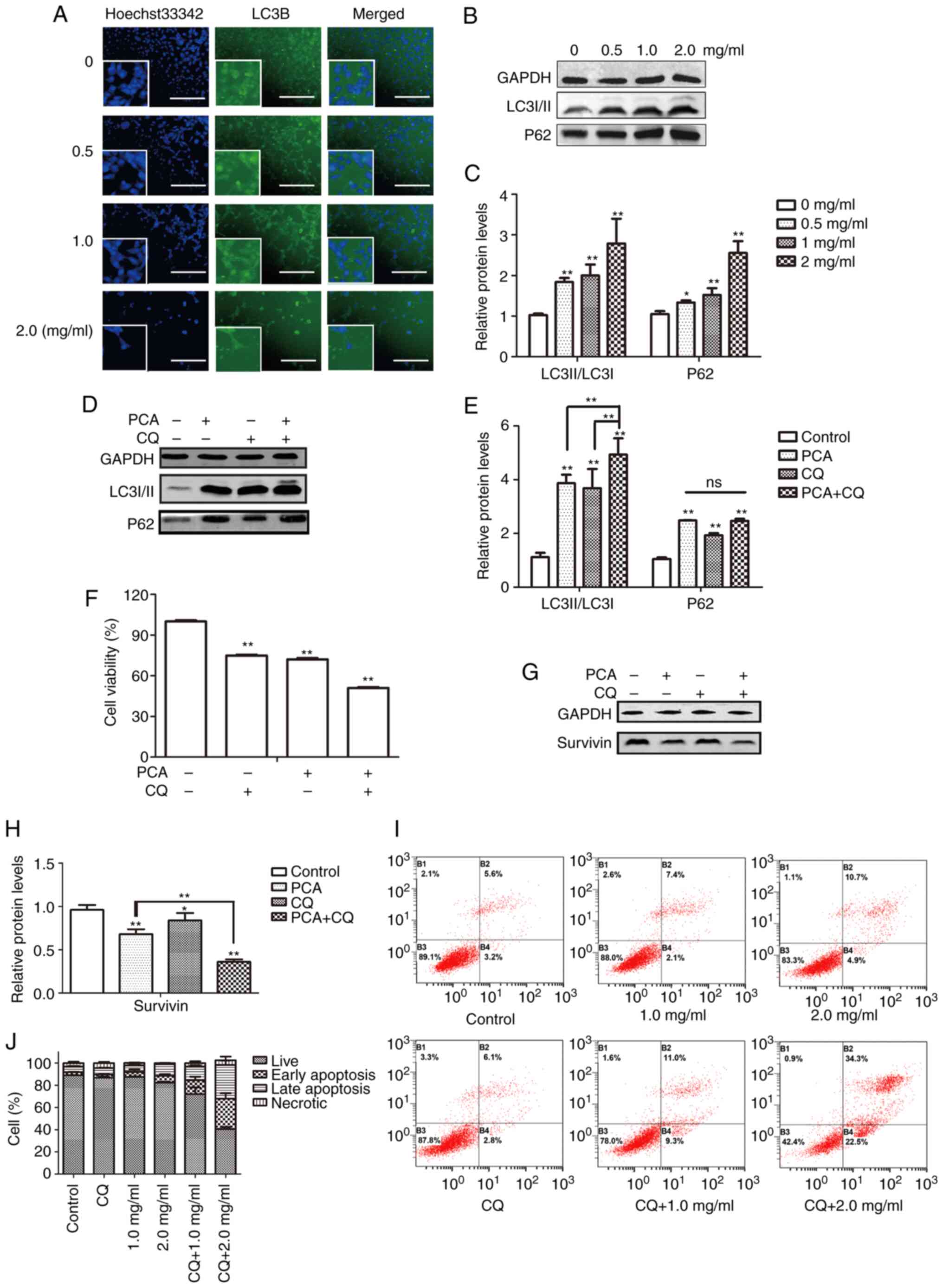 | Figure 6.Effects of PCA on cell autophagy. (A)
Cells were treated with PCA for 24 h. Cells expressing LC3B were
stained by FITC. Cells were stained with Hoechst33342, and
fluorescence was visualized (scale bar, 100 µm) Original
magnifications, ×200. Higher magnification displayed in the lower
left corner (×400). Cells were preincubated with or without CQ
before exposure to PCA for 24 h. (B) Expression levels of p62 and
LC3 in absence of CQ determined by western blotting. (C)
Quantification of p62 and LC3 expression levels in absence of CQ.
(D) Expression levels of p62 and LC3 in presence of CQ determined
by western blotting. (E) Quantification of p62 and LC3 expression
levels in presence of CQ. (F) Effects of PCA on cell viability in
presence of CQ were determined using a Cell Counting Kit-8 assay.
(G) Expression levels of cell apoptosis-regulated proteins
determined by western blotting. (H) Quantification of survivin
expression. (I) Effects of PCA on cell apoptosis in presence of CQ
were determined by flow cytometry. (J) Summary of the apoptosis
assay results displayed as percentages of cells in different cell
death stages: Live cells, early apoptosis, late apoptosis and
necrosis. In Fig. 6B, D and G,
GAPDH was used as the loading control (*P<0.05, **P<0.01).
PCA, procaine; CQ, chloroquine; ns, not significant. All
experiments were assessed using one-way or two-way ANOVA for
analysis. |
Effects of autophagy on apoptosis
induced by PCA
Autophagy is regarded as a double-edged sword in
tumor development and treatment because it kills tumor cells and
protects them from injury (16,17).
To clarify the role of autophagy in PCA-induced apoptosis, changes
in autophagic flux were examined by treating cells with the
lysosomal inhibitor CQ alone or in combination with PCA and
measuring LC3II/I and p62 expression levels using western blotting.
PCA combined with CQ significantly enhanced the transformation of
LC3I to LC3II in CAL27 cells (Fig. 6D
and E). It was also observed that the addition of CQ had no
significant effect on the change in p62 expression.
Next, it was observed that compared with PCA-treated
cells, PCA combined with CQ significantly reduced cell activity for
24 h (Fig. 6F). The apoptotic
effects of PCA combined with CQ were then investigated. A total of
~34.3% of CAL27 cells demonstrated late apoptosis at the highest
dose (2 mg/ml) of PCA combined with CQ compared with 10.7% of the
cells treated with PCA alone, indicating that the apoptosis rate in
cells treated with PCA combined with CQ was higher than that in
cells treated with PCA alone (Fig.
6I).
Similarly, protein expression levels were detected
in the surviving cells. Compared with the PCA-treated group,
survivin protein expression was significantly reduced in the PCA +
CQ group (Fig. 6H). These results
suggest that the inhibition of autophagy could enhance the
sensitivity of PCA.
Discussion
Novel drugs often take decades to transition from
laboratories to clinical application. Therefore, an increasing
number of researchers are turning their attention to the ‘new use
of old drugs’. ‘Old drugs’ are drugs that have been marketed or are
undergoing clinical trials, and ‘new use’ refers to the discovery
of new indications and their use in disease treatment (18). For instance, aspirin has been used
as an antipyretic and analgesic for >100 years. Owing to more
in-depth clinical research, researchers have discovered that
aspirin plays a role in the prevention and treatment of
cardiovascular diseases and also in cancer prevention and treatment
(19,20). Another example is the metformin,
which reduces blood sugar, promotes antitumor activity and also
treats abnormal metabolic diseases (21,22). A
previous study demonstrated that metformin can also regulate the
metabolic flux of stem cells in the stomach and promote their
differentiation into gastric parietal cells that produce gastric
acid, protecting the stomach (23).
Several recent studies have reported that anesthetics may have
antitumor effects, which has aroused great interest. For example,
Chen et al (24) found that
propofol exerts an antitumor effect by inhibiting autophagic flux
in HeLa cells (24). Another study
showed that 400 mM lidocaine inhibited the proliferation of CAL27
cells with high EGFR expression without cytotoxicity (25). As a representative anesthetic, PCA
has also shown antitumor effects in different types of cancer,
including lung (26), colon
(13) and breast (27) cancer. In the aforementioned studies,
the range of antitumor PCA doses was micromolar to millimolar; 2 µM
(26), 2 mM (12) and 10 mM (26). In the present study, the PCA dose
associated with significant antitumor properties was 1–2 mg/ml
in vitro, which is equivalent to 8.5 mM and is roughly
similar to the dose used to induce PCA antitumor activity in the
aforementioned published studies. Meanwhile, it was also observed
that the FDA-approved dose of PCA for injection is 1 or 2%, which
is ~10 mg/ml. This dose differs from the doses used in the present
study. Some ‘old drugs’, which have been approved for clinical use,
are used as ‘new efficacy drugs’ by altering the dose, such as with
aspirin and metformin. If PCA is to be used clinically as an
antitumor agent in the future, further animal and clinical trials
are required.
In the current study, it was shown that PCA had
different antiproliferative effects by investigating two OSCC cell
lines. CAL27 cells were more sensitive to PCA treatment than SCC-15
cells, indicating that the in vitro inhibitory effects of
PCA depend on the cancer cell line.
Tumorigenesis is a phenomenon of uncontrolled cell
proliferation caused by abnormal cell cycle regulation; therefore,
blocking the cell cycle is usually an important means of tumor
treatment (28). The results of the
present study showed that PCA induced G2/M arrest and upregulated
the expression of the cyclin 1 kinase inhibitor p21, which is known
to inhibit mammalian cell proliferation, induce cell cycle arrest
and inhibit the cyclin-cdk complex (29). These results indicate that p21 may
play a key regulatory role during PCA-induced G2/M arrest in CAL27
cells.
The mitochondrial pathway is a main signaling
pathway that induces apoptosis (30). Numerous anticancer drugs exert
antitumor effects by activating the mitochondrial apoptosis
pathway. The anti-apoptotic protein Bcl-2 and the pro-apoptotic
protein Bax control cell apoptosis by regulating mitochondrial
membrane permeability (31). The
present study found that PCA downregulated Bcl-2 and upregulated
Bax expression, caused ROS explosion in cells and decreased the
ΔΨm, suggesting that PCA may induce CAL27 cell apoptosis by
activating the mitochondrial pathway.
In addition to the classic antitumor mechanisms,
such as apoptosis induction and cell cycle arrest, it was observed
that PCA can induce autophagy. Autophagy is a double-edged sword
that promotes and inhibits tumorigenesis (31–34).
Therefore, the regulation of autophagy is expected to become a
novel method for tumor prevention and treatment. LC3 and p62 are
markers of autophagosome formation and degradation, respectively,
and are used to detect autophagic flux (35). The increase in LC3II levels
represents an increase in autophagosomes or the blockade of
autophagosomal maturation and completion of the autophagy pathway.
For example, cetuximab induces p27 accumulation and type II LC3B
induction in retinoblastoma cells (36). Recent studies have shown that
autophagy preferentially degrades p62 as a particular substrate
(37,38). The total p62 expression level was
negatively associated with autophagic activity. Autophagosomes are
intermediate structures involved in dynamic cellular autophagy. At
any point in time, the autophagosome production rate and the rate
at which autolysosomes are converted into autolysosomes are
balanced (39). It was shown that
PCA induced LC3II protein accumulation and increased p62 levels. To
demonstrate this, the well-known autophagy inhibitor CQ was used to
detect autophagy flux. CQ can inhibit late autophagy by increasing
lysosomal pH and inhibiting lysosomal enzyme chelation, thereby
preventing autophagic degradation (11). The results showed that LC3II levels,
after CQ combined with PCA were significantly higher than LC3II
levels induced by PCA or CQ alone. These data indicate that PCA can
cause the accumulation of autophagosomes, which cannot be ignored.
The accumulation of autophagosomes induced by PCA may be due to an
increase in autophagosome generation caused by early autophagy
activation, or the blocking of autophagosomal clearance caused by
late autophagy inhibition. This question needs to be answered in
the future (40).
Recently, the ERK pathway has become an attractive
therapeutic target, and several drugs targeting this pathway have
shown promise in clinical trials. PCA reportedly affects HCT 116
cell proliferation, migration and apoptosis and inactivates the ERK
pathway by regulating RhoA (13).
Moreover, Li et al (13)
revealed that PCA inhibited MG63 cells viability by upregulating
microRNA-133b, thereby inactivating the AKT/ERK pathway. The
current study also confirmed that PCA produces antitumor effects by
reducing p-ERK in CAL27 cells, suggesting that PCA may play an
inhibitory role in various malignant tumors by inhibiting the ERK
signaling pathway.
In apoptosis-related pathways, specific signals and
growth factors activate the PI3K/AKT pathway, which regulates cell
proliferation, differentiation, metabolism, migration and other
biological processes. For example, Yang et al (41) found that oridonin induced G2/M phase
arrest and apoptosis and inhibited OSCC cell proliferation by
inhibiting the PI3K/AKT signaling pathway. The results of the
current study further confirmed that PCA inhibited proliferation,
and induced cell cycle arrest and apoptosis in CAL27 cells by
inhibiting PI3K/AKT phosphorylation.
The current study has some limitations. In
vivo experiments are required to elucidate the antitumor
activity and mechanism of PCA. Therefore, besides OSCC cell lines,
nude mice xenografts or clinical samples are worthy for validation.
In addition, although no study has yet demonstrated the role of PCA
in the tumor microenvironment, considering the network effects in
the organism, hypothesizing that PCA may be involved in the
progression of OSCC by modulating immune infiltration through
interactions with immune cells, vasculature and nerves is
reasonable. These findings warrant further study in the future.
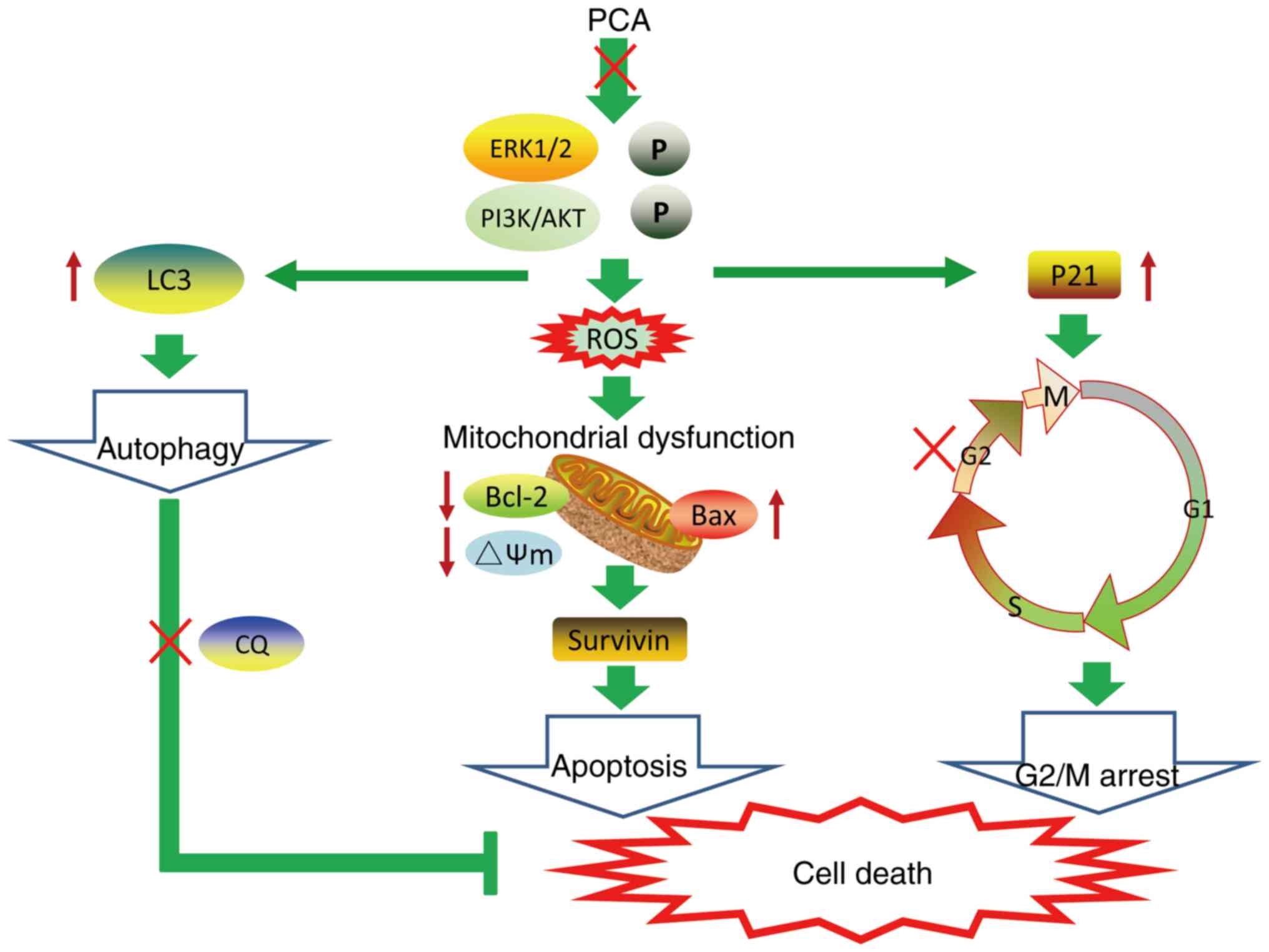
A schematic model of PCA-induced cell death is shown
in Fig. 7. The findings of the
present study demonstrate that PCA inhibits cell proliferation and
migration by impairing mitochondrial function and inducing G2/M
cycle arrest and apoptosis. The inhibitory effects of PCA on CAL27
cell growth may be dependent on PI3K/AKT and ERK pathway
inhibition. Meanwhile, it was shown that PCA elicited autophagy and
blocked autophagic flux. The combination of PCA and the autophagy
inhibitor CQ enhanced CAL27 cell sensitivity to PCA. The results of
the current study indicate the potential PCA as a novel drug
against tongue squamous cell carcinoma.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Jilin Provincial Department
of Finance (grant no. 2023SCZ64), the National Natural Science
Foundation of China (grant no. 81903881), and the Department of
Science and Technology of Jilin Province (grant nos. 20190103086JH
and 20200201398JC).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
CZ and MH performed the experiments. NS, LY and TZ
analyzed the data. CZ and MH wrote the manuscript. TZ and XW
designed and supervised the study. MH and XW confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Omura K: Current status of oral cancer
treatment strategies: Surgical treatments for oral squamous cell
carcinoma. Int J Clin Oncol. 19:423–430. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tampa M, Mitran MI, Mitran CI, Sarbu MI,
Matei C, Nicolae I, Caruntu A, Tocut SM, Popa MI, Caruntu C and
Georgescu SR: Mediators of inflammation-A potential source of
biomarkers in oral squamous cell carcinoma. J Immunol Res.
12:10617802018.PubMed/NCBI
|
|
3
|
Lien JC, Lin MW, Chang SJ, Lai KC, Huang
AC, Yu FS and Chung JG: Tetrandrine induces programmed cell death
in human oral cancer CAL 27 cells through the reactive oxygen
species production and caspase-dependent pathways and associated
with beclin-1-induced cell autophagy. Environ Toxicol. 32:329–343.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ng JH, Iyer NG, Tan MH and Edgren G:
Changing epidemiology of oral squamous cell carcinoma of the
tongue: A global study. Head Neck. 39:297–304. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chu H, Li Z, Gan Z, Yang Z, Wu Z and Rong
M: LncRNA ELF3-AS1 is involved in the regulation of oral squamous
cell carcinoma cell proliferation by reprogramming glucose
metabolism. Onco Targets Ther. 12:6857–6863. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tan Y, Wang Z, Xu M, Li B, Huang Z, Qin S,
Nice EC, Tang J and Huang C: Oral squamous cell carcinomas: State
of the field and emerging directions. Int J Oral Sci. 15:442023.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Adelstein D, Gillison ML, Pfister DG,
Spencer S, Adkins D, Brizel DM, Burtness B, Busse PM, Caudell JJ,
Cmelak AJ, et al: NCCN guidelines insights: Head and neck cancers,
version 2.2017. J Natl Compr Canc Netw. 15:761–770. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mouw KW, Haraf DJ, Stenson KM, Cohen EE,
Xi X, Witt ME, List M, Blair EA, Vokes EE and Salama JK: Factors
associated with long-term speech and swallowing outcomes after
chemoradiotherapy for locoregionally advanced head and neck cancer.
Arch Otolaryngol Head Neck Surg. 136:1226–1234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li YC, Wang Y, Li DD, Zhang Y, Zhao TC and
Li CF: Procaine is a specific DNA methylation inhibitor with
anti-tumor effect for human gastric cancer. J Cell Biochem.
119:2440–2449. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schumann NAB, Mendonça AS, Silveira MM,
Vargas LN, Leme LO, de Sousa RV and Franco MM: Procaine and
S-Adenosyl-l-Homocysteine affect the expression of genes related to
the epigenetic machinery and change the DNA methylation status of
in vitro cultured bovine skin fibroblasts. DNA Cell Biol. 39:37–49.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Villar-Garea A, Fraga MF, Espada J and
Esteller M: Procaine is a DNA-demethylating agent with
growth-inhibitory effects in human cancer cells. Cancer Res.
63:4984–4989. 2003.PubMed/NCBI
|
|
12
|
Tada M, Imazeki F, Fukai K, Sakamoto A,
Arai M, Mikata R, Tokuhisa T and Yokosuka O: Procaine inhibits the
proliferation and DNA methylation in human hepatoma cells. Hepatol
Int. 1:355–364. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li C, Gao S, Li X, Li C and Ma L: Procaine
inhibits the proliferation and migration of colon cancer cells
through inactivation of the ERK/MAPK/FAK pathways by regulation of
RhoA. Oncol Res. 26:209–217. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen X, Li LY, Jiang JL, Li K, Su ZB,
Zhang FQ, Zhang WJ and Zhao GQ: Propofol elicits autophagy via
endoplasmic reticulum stress and calcium exchange in C2C12 myoblast
cell line. PLoS One. 13:e01979342018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao X, Qi T, Kong C, Hao M, Wang Y, Li J,
Liu B, Gao Y and Jiang J: Photothermal exposure of
polydopamine-coated branched Au-Ag nanoparticles induces cell cycle
arrest, apoptosis, and autophagy in human bladder cancer cells. Int
J Nanomedicine. 13:6413–6428. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li X, He S and Ma B: Autophagy and
autophagy-related proteins in cancer. Mol Cancer. 19:122020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marsh T and Debnath J: Autophagy
suppresses breast cancer metastasis by degrading NBR1. Autophagy.
16:1164–1165. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nosengo N: Can you teach old drugs new
tricks? Nature. 534:314–316. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sperling CD, Verdoodt F, Aalborg GL,
Dehlendorff C, Friis S and Kjaer SK: Low-dose aspirin use and
endometrial cancer mortality-a Danish nationwide cohort study. Int
J Epidemiol. 49:330–337. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo CG, Cheung KS, Zhang F, Chan EW, Chen
L, Wong IC and Leung WK: Incidences, temporal trends and risks of
hospitalisation for gastrointestinal bleeding in new or chronic
low-dose aspirin users after treatment for Helicobacter pylori: A
territory-wide cohort study. Gut. 69:445–452. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guardado-Mendoza R, Salazar-López SS,
Álvarez-Canales M, Farfán-Vázquez D, Martínez-López YE,
Jiménez-Ceja LM, Suárez-Pérez EL, Angulo-Romero F, Evia-Viscarra
ML, Montes de Oca-Loyola ML, et al: The combination of linagliptin,
metformin and lifestyle modification to prevent type 2 diabetes
(PRELLIM). A randomized clinical trial. Metabolism. 104:1540542020.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dong TS, Chang HH, Hauer M, Lagishetty V,
Katzka W, Rozengurt E, Jacobs JP and Eibl G: Metformin alters the
duodenal microbiome and decreases the incidence of pancreatic
ductal adenocarcinoma promoted by diet-induced obesity. Am J
Physiol Gastrointest Liver Physiol. 317:G763–G772. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miao ZF, Adkins-Threats M, Burclaff JR,
Osaki LH, Sun JX, Kefalov Y, He Z, Wang ZN and Mills JC: A
Metformin-Responsive metabolic pathway controls distinct steps in
gastric progenitor fate decisions and maturation. Cell Stem Cell.
26:910–925.e6. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen X, Li K and Zhao G: Propofol Inhibits
HeLa cells by impairing autophagic flux via AMP-Activated Protein
Kinase (AMPK) activation and endoplasmic reticulum stress regulated
by calcium. Med Sci Monit. 24:2339–2349. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sakaguchi M, Kuroda Y and Hirose M: The
antiproliferative effect of lidocaine on human tongue cancer cells
with inhibition of the activity of epidermal growth factor
receptor. Anesth Analg. 102:1103–1107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma XW, Li Y, Han XC and Xin QZ: The effect
of low dosage of procaine on lung cancer cell proliferation. Eur
Rev Med Pharmacol Sci. 20:4791–4795. 2016.PubMed/NCBI
|
|
27
|
Chen C, Wang N, Huang T, Cheng G, Hu Y,
Wang B, Zhang Y and Wang C: Chloroprocaine antagonizes progression
of breast cancer by regulating LINC00494/miR-3619-5p/MED19 axis. J
Biochem Mol Toxicol. 38:e235242024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin AB, McNeely SC and Beckmann RP:
Achieving precision death with cell-cycle inhibitors that target
DNA replication and repair. Clin Cancer Res. 23:3232–3240. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Z, Sturgis EM, Zhang F, Lei D, Liu Z,
Xu L, Song X, Wei Q and Li G: Genetic variants of p27 and p21 as
predictors for risk of second primary malignancy in patients with
index squamous cell carcinoma of head and neck. Mol Cancer.
11:172012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pfeffer CM and Singh ATK: Apoptosis: A
target for anticancer therapy. Int J Mol Sci. 19:4482018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao W, Wang X, Zhou Y, Wang X and Yu Y:
Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor
immunotherapy. Signal Transduct Target Ther. 7:1962022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou J, Hu SE, Tan SH, Cao R, Chen Y, Xia
D, Zhu X, Yang XF, Ong CN and Shen HM: Andrographolide sensitizes
cisplatin-induced apoptosis via suppression of
autophagosome-lysosome fusion in human cancer cells. Autophagy.
8:338–349. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Okuyama K, Suzuki K, Naruse T, Tsuchihashi
H, Yanamoto S, Kaida A, Miura M, Umeda M and Yamashita S: Prolonged
cetuximab treatment promotes p27Kip1-mediated G1 arrest and
autophagy in head and neck squamous cell carcinoma. Sci Rep.
11:52592021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lamark T, Svenning S and Johansen T:
Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays
Biochem. 61:609–624. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Deng Z, Lim J, Wang Q, Purtell K, Wu S,
Palomo GM, Tan H, Manfredi G, Zhao Y, Peng J, et al:
ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy
and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy.
16:917–931. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mizushima N, Yamamoto A, Matsui M, Matsui
M, Yoshimori T, Yoshimori T and Ohsumi Y: In vivo analysis of
autophagy in response to nutrient starvation using transgenic mice
expressing a fluorescent autophagosome marker. Mol Biol Cell.
15:1101–1111. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang J, Ren X, Zhang L, Li Y, Cheng B and
Xia J: Oridonin inhibits oral cancer growth and PI3K/Akt signaling
pathway. Biomed Pharmacother. 100:226–232. 2018. View Article : Google Scholar : PubMed/NCBI
|