Introduction
Prostate cancer remains the most prevalent
malignancy diagnosed in men worldwide and the second most common
cause of cancer-related death in men (1,2). In
2023, 288,300 new cases of prostate cancer and 34,700 prostate
cancer-related deaths were expected in the United States (3). Due to the lack of apparent symptoms at
the initial stages, patients with prostate cancer are generally
diagnosed at an advanced stage with metastasis, which is associated
with a high mortality rate (4). The
androgen receptor is the main factor involved in the pathogenesis
of prostate cancer. Therefore, androgen deprivation therapy (ADT)
focusing on halting tumor growth has been the mainstay of prostate
cancer treatment; however, the majority of patients develop
castration resistance within 3 years following ADT failure, and
even progress to the incurable metastatic castration-resistant
prostate cancer stage, contributing to a poor 5-year survival rate
(5–7). Therefore, it is urgently required to
understand the detailed mechanisms underlying prostate cancer
development, and to identify potential biomarkers of prostate
cancer progression and novel therapeutic targets.
Epithelial cell transforming sequence 2 (ECT2), a
guanine nucleotide exchange factor of Rho GTPases, is encoded by
the human ECT2 gene and is located on chromosome 3q26, a
region prone to chromosome alterations in human tumors (8). Accumulating evidence has revealed that
ECT2 serves a role in normal cellular activities, including
cytokinesis and cell division, and participates in malignant
transformation, tumor initiation and metastasis (9,10).
ECT2 expression is upregulated in several types of human cancer,
such as breast cancer, colorectal cancer, gastric cancer and
esophageal squamous cell carcinoma, and the high ECT2 expression is
associated with poor outcomes of patients with malignant tumors
(11–14). ECT2 has been identified as an
oncogene for human tumors. For instance, aberrant expression of
ECT2 can drive colorectal cancer progression and growth (11), and ECT2 promotes the proliferation
of glioma through stabilizing E2F transcription factor 1 (13). A previous study revealed that ECT2
expression is enhanced in human prostate cancer tissues, and this
expression is positively associated with tumor invasion and
increased distant metastasis, suggesting that ECT2 is an
independent prognostic marker of poor survival (15). Nevertheless, to the best of our
knowledge, the involvement of ECT2 in the malignant progression of
prostate cancer has not yet been addressed.
Therefore, the present study aimed to investigate
the specific role of ECT2 in prostate cancer, and to explore the
potential molecular mechanism, which will help identify novel
targets and provide a theoretical basis for the treatment of
prostate cancer.
Materials and methods
Bioinformatics analysis
The expression profile of ECT2 in prostate cancer
was examined using the University of Alabama at Birmingham Cancer
data analysis (UALCAN) portal (http://ualcan.path.uab.edu) (16) database based on 52 adjacent normal
tissues and 497 tumor samples.
Cell culture
The RWPE-1 human normal prostate epithelial cell
line was purchased from American Type Culture Collection and
cultured in keratinocyte serum-free medium (Invitrogen; Thermo
Fisher Scientific, Inc.) with 0.05 mg/ml bovine pituitary extract
and 5 ng/ml human recombinant epidermal growth factor. The LNCaP,
DU145, PC-3 and 22RV1 human prostate cancer cell lines were
obtained from Procell Life Science & Technology Co., Ltd. LNCaP
and 22RV1 cells were incubated with RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.) and 1% penicillin/streptomycin mixture
(HyClone; Cytiva). DU145 cells were incubated with Minimum
Essential Medium (Procell Life Science & Technology Co., Ltd.)
with 10% FBS and 1% penicillin/streptomycin, while PC-3 cells were
incubated with Ham's F-12K medium (Procell Life Science &
Technology Co., Ltd.) in the presence of 10% FBS and 1%
penicillin/streptomycin. All cells were cultured at 37°C in a
humified incubator with 5% CO2.
Western blotting
Total protein was isolated from PC-3 cells using
RIPA lysis buffer (Beyotime Institute of Biotechnology). The
protein concentration was determined using a BCA kit (Beyotime
Institute of Biotechnology). The same amounts of proteins (30
µg/lane) were separated by electrophoresis using a 12% SDS-PAGE
gel, and then transferred to polyvinylidene fluoride membranes.
Following blocking with 5% nonfat milk at room temperature for 2 h,
the membranes were probed with primary antibodies against ECT2
(1:1,000; cat. no. ab86604; Abcam), MMP2 (1:1,000; cat. no.
10373-2-AP; Proteintech Group, Inc.), MMP9 (1:2,000, cat. no.
30592-1-AP; Proteintech Group, Inc.), E-cadherin (1:10,000; cat.
no. ab40772; Abcam), N-cadherin (1:10,000; cat. no. ab76011;
Abcam), Vimentin (1:1,000; cat. no. ab92547; Abcam), hexokinase 2
(HK2; 1:1,000; cat. no. ab209847; Abcam), pyruvate kinase M2 (PKM2;
1:1,000; cat. no. ab85555; Abcam), lactate dehydrogenase A (LDHA;
1:1,000; cat. no. ab5248; Abcam), ETS1 (1:1,000; cat. no. ab186844;
Abcam) and GAPDH (1:2,500; cat. no. ab9485; Abcam) at 4°C
overnight. The membranes were incubated with HRP-conjugated goat
anti-rabbit secondary antibody (1:5,000; cat. no. ab6721; Abcam)
for 2 h at room temperature. The blots were further developed using
an enhanced chemiluminescence kit (Amersham; Cytiva) and
semi-quantified using ImageJ software Version 1.52 (National
Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from PC-3 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). The RNA concentration and purity were determined using a
spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.). Subsequently, 1 µg RNA was reverse-transcribed into cDNA
using a First Strand cDNA Synthesis Kit (Sangon Biotech Co., Ltd.).
The following conditions were used for RT: 42°C for 2 min, 37°C for
15 min and 85°C for 5 sec. Thereafter, SYBR Green qPCR Master Mix
(Applied Biosystems; Thermo Fisher Scientific, Inc.) was used for
the qPCR assay according to the manufacturer's instructions. The
qPCR thermocycling conditions were as follows: Initial denaturation
at 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec, 55°C
for 5 sec and 72°C for 10 sec. The primer sequences used in the
present study were as follows: ECT2 forward,
5′-ACTACTGGGAGGACTAGCTTG-3′ and reverse,
5′-CACTCTTGTTTCAATCTGAGGCA-3′; ETS1 forward,
5′-CCCGTACGTCCCCCACTCCT-3′ and reverse, 5′-TGGGACATCTGCACATTCCA-3′;
and GAPDH forward, 5′-CAGGAGGCATTGCTGATGAT-3′ and reverse,
5′-GAAGGCTGGGGCTCATTT-3′. The mRNA levels of the genes were
calculated using the 2−∆∆Cq method (17). GAPDH was used as the internal
control.
Cell transfection
Short hairpin RNAs (shs) (pGPU6 vector) targeting
ECT2 (sh-ECT2-1 sense,
5′-CCGGGCTGAGCATTCCCTTTCCATACTCGAGTATGGAAAGGGAATGCTCAGCTTTTTG-3′
and antisense,
5′-AATTCAAAAAGCTGAGCATTCCCTTTCCATACTCGAGTATGGAAAGGGAATGCTCAGC-3′;
and sh-ECT2-2 sense,
5′-CCGGCGGAATGAACAGGATTTCTATCTCGAGATAGAAATCCTGTTCATTCCGTTTTTG-3′
and antisense,
5′-AATTCAAAAACGGAATGAACAGGATTTCTATCTCGAGATAGAAATCCTGTTCATTCCG-3′)
and ETS1 (sh-ETS1-1 sense,
5′-CCGGGTGCAGATGTCCCACTATTAACTCGAGTTAATAGTGGGACATCTGCACTTTTTG-3′
and antisense,
5′-AATTCAAAAAGTGCAGATGTCCCACTATTAACTCGAGTTAATAGTGGGACATCTGCAC-3′;
and sh-ETS1-2: sense,
5′-CCGGTGTGAAACCATATCAAGTTAACTCGAGTTAACTTGATATGGTTTCACATTTTTG-3′
and antisense,
5′-AATTCAAAAATGTGAAACCATATCAAGTTAACTCGAGTTAACTTGATATGGTTTCACA-3′)
were constructed by Shanghai GenePharma Co., Ltd., with scrambled
shRNA (pGPU6) as the negative control (sh-NC; sense,
5′-CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTTG-3′
and antisense,
5′-AATTCAAAAACAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTG-3′).
The Homo sapiens ETS1 full-length open reading frame was
amplified and inserted into a pcDNA3.1 vector to construct an ETS1
overexpression vector (oe-ETS1; Shanghai GenePharma Co., Ltd.).
Empty pcDNA3.1 vector served as the negative control (oe-NC;
Shanghai GenePharma Co., Ltd.). shs plasmids (50 nM) and/or
oe-ETS1/oe-NC (15 nM) were transfected into PC-3 cells using
Lipofectamine® 3000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) for an incubation at 37°C for 6 h according to
the manufacturer's instructions. PC-3 cells were not transfected
served as the control. Following 48 h of culture at 37°C, the cells
were harvested for subsequent experiments.
Cell proliferation assay
Cell proliferation was assessed using Cell Counting
Kit-8 (CCK-8) and colony formation assays. For the CCK-8 assay,
PC-3 cells were seeded into 96-well plates (1.0×104
cells/well) and incubated in a 5% CO2 incubator at 37°C
for the indicated durations (24, 48 and 72 h). CCK-8 solution (10
µl; Dojindo Molecular Technologies, Inc.) was added to each well
for an additional incubation at 37°C for 2 h. The absorbance at 450
nm was recorded using a microplate reader. The relative cell
viability (%) was calculated using the following formula:
(absorbance in treated group-absorbance blank)/(absorbance in
control group at 24 h-absorbance blank) ×100.
In addition, PC-3 cells were seeded in 6-well plates
(500 cells/well) and incubated in a 5% CO2 incubator at
37°C for 2 weeks. The colonies (>50 cells) were fixed with
methanol for 15 min at room temperature and stained with 0.1%
crystal violet for 10 min at room temperature. Images were captured
using light microscopy and colonies were observed by eye.
Assessment of cell migration and
invasion
Cell migration was assessed using a wound healing
assay. In brief, PC-3 cells were seeded in 6-well plates and
incubated in a 5% CO2 incubator at 37°C. When the cells
reached 100% confluency, a single scratch wound was created using a
200-µl pipette tip. The plates were washed with PBS and
subsequently cultured with serum-free medium for 24 h. Images were
captured at 0 and 24 h using light microscopy. The cell migration
rate (%) was calculated using the following formula: (0 h wound
width −24 h width)/0 h wound width ×100.
Cell invasion was detected using an 8-µm pore
Transwell chamber (Corning, Inc.) precoated with Matrigel (BD
Biosciences) for 30 min at 37°C. A total of 2.0×105 PC-3
cells were resuspended in serum-free medium and subsequently seeded
into the upper Transwell chamber. The complete medium containing
10% FBS was plated in the lower chamber. Following incubation in a
5% CO2 incubator at 37°C for 48 h, the non-invasive
cells in the upper chamber were removed using a cotton swab. The
invasive cells were fixed with methanol for 15 min at room
temperature and stained with crystal violet for 10 min at room
temperature. Finally, the images were observed using light
microscopy and the invasive cells were counted using ImageJ
software Version 1.52 (National Institutes of Health).
Measurement of lactate release,
glucose uptake, oxygen consumption rate (OCR) and extracellular
acidification rate (ECAR)
The cell culture medium was harvested and the
lactate release and glucose uptake were evaluated using lactate
assay (cat. no. MAK064) and glucose uptake assay kits (cat. no.
MAK542) (Sigma-Aldrich; Merck KGaA), respectively, according to the
manufacturer's guidelines.
In addition, cells (1.0×104 cells/well)
were seeded into XF96 cell culture microplates and incubated at
37°C overnight. The OCR and ECAR were detected using a Cell Mito
Stress Test (cat. no. 103015-100) and Glycolysis Stress Test Kit
(cat. no. 103020-100), respectively, on a Seahorse XFe96 Analyzer
(all Agilent Technologies, Inc.), according to the manufacturer's
guidelines.
Luciferase reporter assay
The promoter region of ECT2 (−2,000 to transcription
start site) or mutant promoter region of ECT2 without ETS1-binding
sites was cloned into the pGL3-Basic vector (Promega Corporation)
to construct the luciferase reporter vectors. PC-3 cells were
co-transfected with the luciferase reporter vectors and
oe-NC/oe-ETS1 using Lipofectamine 3000 reagent. Following cell
culture for 48 h post-transfection, the luciferase activity was
detected using the Dual-Luciferase Reporter Assay System (Promega
Corporation) according to the manufacturer's instructions, and
normalized to Renilla luciferase activity.
Chromatin immunoprecipitation (ChIP)
assay
The HumanTFDB (http://bioinfo.life.hust.edu.cn/HumanTFDB#!/) website
was adopted to predict presumptive binding sites between the
transcription factor ETS1 and the ECT2 promoter region, which was
then verified using a ChIP assay, carried out using the SimpleChIP
Enzymatic Chromatin IP Kit (Cell Signaling Technology, Inc.)
according to the manufacturer's guidelines. In brief, PC-3 cells
were fixed with 1% paraformaldehyde at 37°C for 10 min for
crosslinking, quenched with 125 mM glycine at room temperature for
5 min, resuspended in SDS Lysis Buffer (MilliporeSigma) and
subsequently sonicated into DNA fragments. After centrifugation at
12,000 × g for 5 min at 4°C, 100 µl of supernatant was incubated
with 5 µg anti-ETS1 (cat. no. 14069; Cell Signaling Technology,
Inc.) or anti-IgG (cat. no. 2729; Cell Signaling Technology, Inc.)
antibodies. Protein A/G agarose beads (40 µl) (Santa Cruz
Biotechnology, Inc.) were added to each IP reaction (100 µl) and
incubated for 60 min at 4°C. The chromatin fragments were
immunoprecipitated. After being washed with low-salt wash buffer,
high-salt wash buffer and LiCI wash buffer, and being rinsed with
TE buffer, the precipitated DNA was amplified and detected by qPCR
as aforementioned.
Statistical analysis
Continuous variables are presented as the mean ±
standard deviation from three repeats. Statistical analysis was
conducted using an unpaired Student's t-test for two groups and
one-way ANOVA with Tukey's post hoc test for more than two groups.
The analysis was performed using GraphPad Prism software (version
8.0; Dotmatics). P<0.05 was considered to indicate a
statistically significant difference.
Results
ECT2 expression is upregulated in
prostate cancer
Using the UALCAN database, it was determined that
the expression levels of ECT2 were significantly upregulated in
prostate cancer tumor samples compared with normal samples
(Fig. 1A). Subsequently, the
expression levels of ECT2 were examined in normal prostate
epithelial cells and prostate cancer cells to confirm the abnormal
ECT2 expression in prostate cancer. As shown in Fig. 1B and C, the mRNA and the protein
expression levels of ECT2 in prostate cancer cell lines (LNCaP,
DU145, PC-3 and 22RV1 cells) were considerably higher than those in
RWPE-1 cells. Among these cell lines, ECT2 expression was highest
in PC-3 cells. Therefore, PC-3 cells were used for subsequent
experiments.
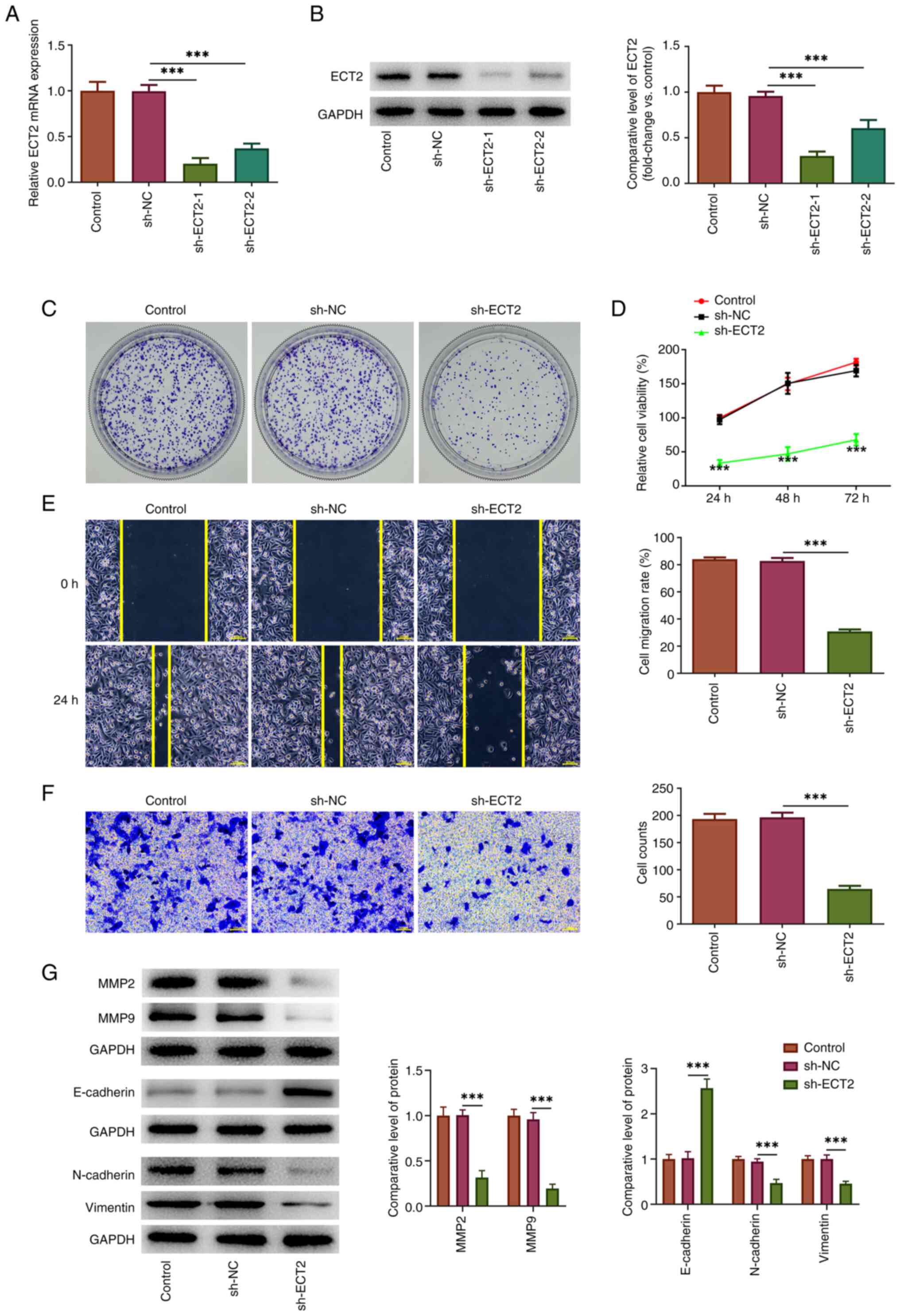
Knockdown of ECT2 expression reduces
the proliferation, migration and invasion of PC-3 cells
To explore the regulatory role of ECT2 in prostate
cancer, loss-of-function experiments were performed. As shown in
Fig. 2A and B, the expression
levels of ECT2 were significantly downregulated following
transfection with sh-ECT2-1/2. The sh-ECT2-1 vector (designated
sh-ECT2 hereafter) was used in subsequent experiments due to its
superior transfection efficacy. The subsequent cellular behavior
assays revealed that knockdown of ECT2 expression could effectively
inhibit the proliferation of prostate cancer cells, as shown by the
reduced colony formation and cell viability in the sh-ECT2 group
compared with the sh-NC group (Fig. 2C
and D). Furthermore, wound healing and Transwell assays
revealed that knockdown of ECT2 expression could reduce the wound
closure and number of invasive cells, indicating decreased
migration and invasion of PC-3 cells following ECT2 knockdown
(Fig. 2E and F). Additionally, the
inhibitory effect of knockdown of ECT2 expression on the expression
levels of MMP2 and MMP9 (invasion-related proteins) in PC-3 cells
further confirmed the anti-invasive effect of knockdown of ECT2
expression. In addition, the findings in Fig. 2G also revealed that knockdown of
ECT2 expression significantly increased the expression levels of
E-cadherin, a hallmark of epithelial cells, while significantly
reducing the protein expression levels of N-cadherin and Vimentin
(hallmarks of mesenchymal cells), suggesting that ECT2 knockdown in
prostate cancer cells might retard the epithelial-mesenchymal
transition which commonly occurs during cancer metastasis (18).
Knockdown of ECT2 expression restricts
aerobic glycolysis of PC-3 cells
Prostate cancer cells can alter their glucose
metabolism mode to aerobic glycolysis to meet the energy
requirements for cell proliferation, migration, invasion and
metastasis (19). Given that
knockdown of ECT2 expression significantly inhibited the
proliferation and invasion of PC-3 cells, additional experiments
were performed to assess whether this effect was associated with
changes in aerobic glycolysis. As shown in Fig. 3A and B, lactate release and glucose
uptake were significantly decreased in the sh-ECT2 group compared
with the sh-NC group. Accordingly, the decreased ECAR and increased
OCR following sh-ECT2 transfection revealed that knockdown of ECT2
expression enhanced the glycolytic capacity, while it decreased ATP
production and maximal respiration, suggesting that it impeded
aerobic glycolysis in PC-3 cells (Fig.
3C). This was further verified by western blot analysis, which
indicated that the expression levels of several critical enzymes in
the aerobic glycolysis process, including LDHA, HK2 and PKM2, were
significantly downregulated following knockdown of ECT2 expression
in PC-3 cells (Fig. 3D).
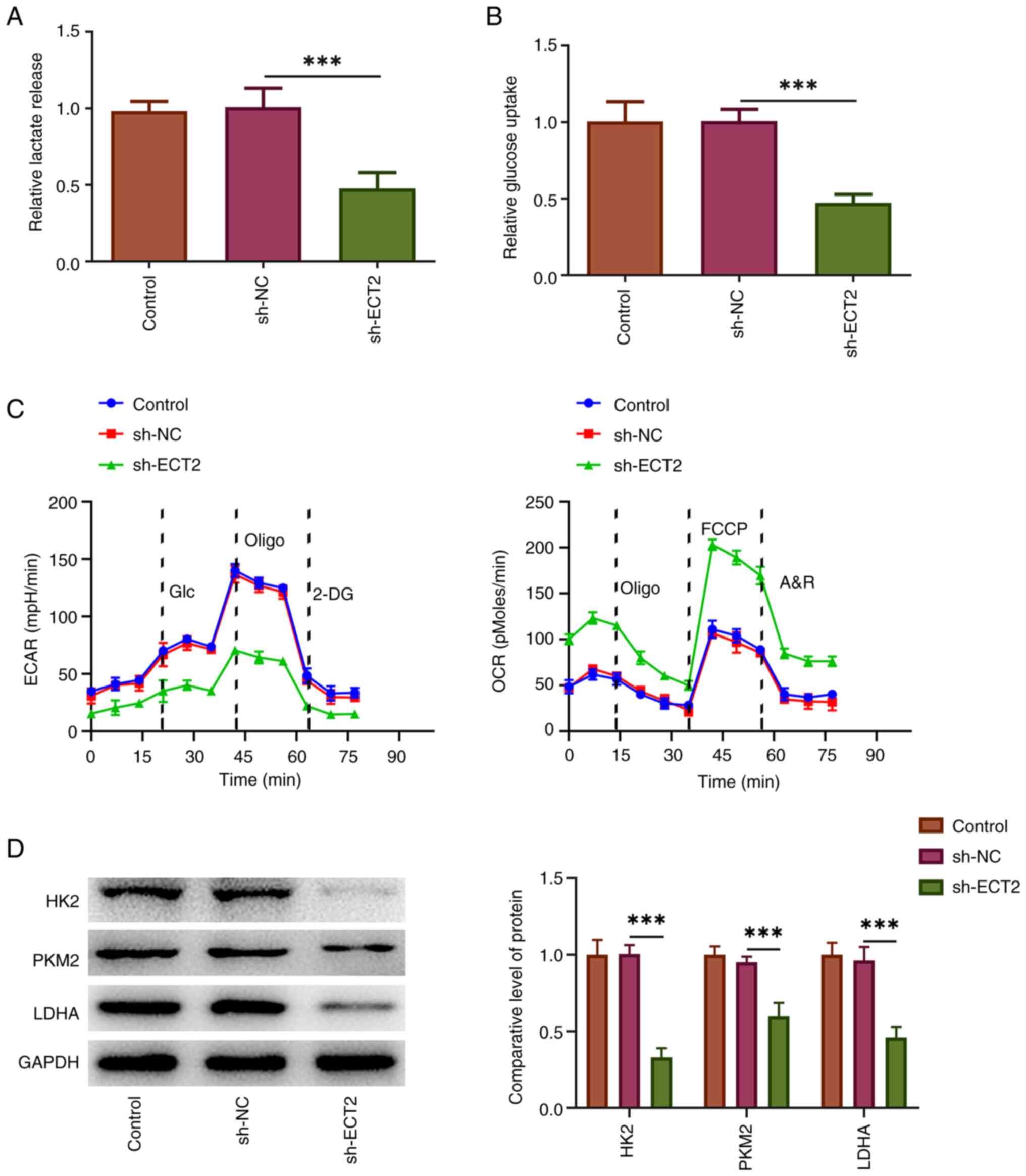 | Figure 3.Knockdown of ECT2 expression
restricts aerobic glycolysis in PC-3 cells. The cell culture medium
was harvested and the (A) lactate release and (B) glucose uptake
were examined using commercial kits. (C) The ECAR and OCR of PC-3
cells were assessed using a Seahorse XFe96 Analyzer to evaluate the
glycolytic capacity and maximal respiration. (D) Western blotting
was performed to examine the expression levels of several critical
enzymes in the aerobic glycolysis process. ***P<0.001. 2-DG,
2-deoxy-D-glucose; A&R, antimycin A/rotenone; ECAR,
extracellular acidification rate; ECT2, epithelial cell
transforming sequence 2; FCCP, carbonylcyanide-4-(trifluoromethoxy)
phenylhydrazone; Glc, glucose; HK2, hexokinase 2; LDHA, lactate
dehydrogenase A; NC, negative control; OCR, oxygen consumption
rate; Oligo, oligomycin; PKM2, pyruvate kinase M2; sh, short
hairpin RNA. |
Transcription factor ETS1 binds to the
ECT2 promoter and positively regulates ECT2 expression
Additional experiments were conducted to clarify the
molecular mechanism of ECT2 and its regulation in prostate cancer
cells. The HumanTFDB (http://bioinfo.life.hust.edu.cn/HumanTFDB#!/) website
predicted that presumptive binding sites may exist between the
transcription factor ETS1 and the ECT2 promoter region (Fig. 4A). It was observed that the
expression levels of ETS1 were significantly upregulated in PC-3
cells compared with RWPE-1 cells (Fig.
4B and C). To explore the interaction between ETS1 and ECT2,
PC-3 cells were transfected with oe-ETS1 plasmid to overexpress
ETS1 or sh-ETS1-1/2 to interfere with ETS1 expression. sh-ETS1-1
was used for subsequent experiments due to its superior
transfection efficacy (Fig. 4D and
E). ETS1 overexpression significantly upregulated ECT2
expression, while knockdown of ETS1 expression significantly
downregulated ECT2 expression (Fig. 4F
and G), demonstrating that ETS1 could positively regulate ECT2.
In addition, to confirm the binding site of ETS1 in the ECT2
promoter, luciferase reporter and ChIP assays were conducted. The
data revealed that the luciferase activity in cells co-transfected
with ECT2-wide-type (ECT2-WT) and oe-ETS1 was markedly increased in
comparison to that in cells co-transfected with ECT2-WT and oe-NC.
There was no difference of the luciferase activity in cells with
ECT2-mutant-type (Fig. 4H).
Furthermore, the enrichment of precipitated chromatin fragments
containing binding sites to the ECT2 promoter in the anti-ETS1
group was significantly higher than that in the IgG group (Fig. 4I). Therefore, these data confirmed
that ETS1 could directly bind to the ECT2 promoter and positively
regulate ECT2 expression at the transcriptional level in PC-3
cells.
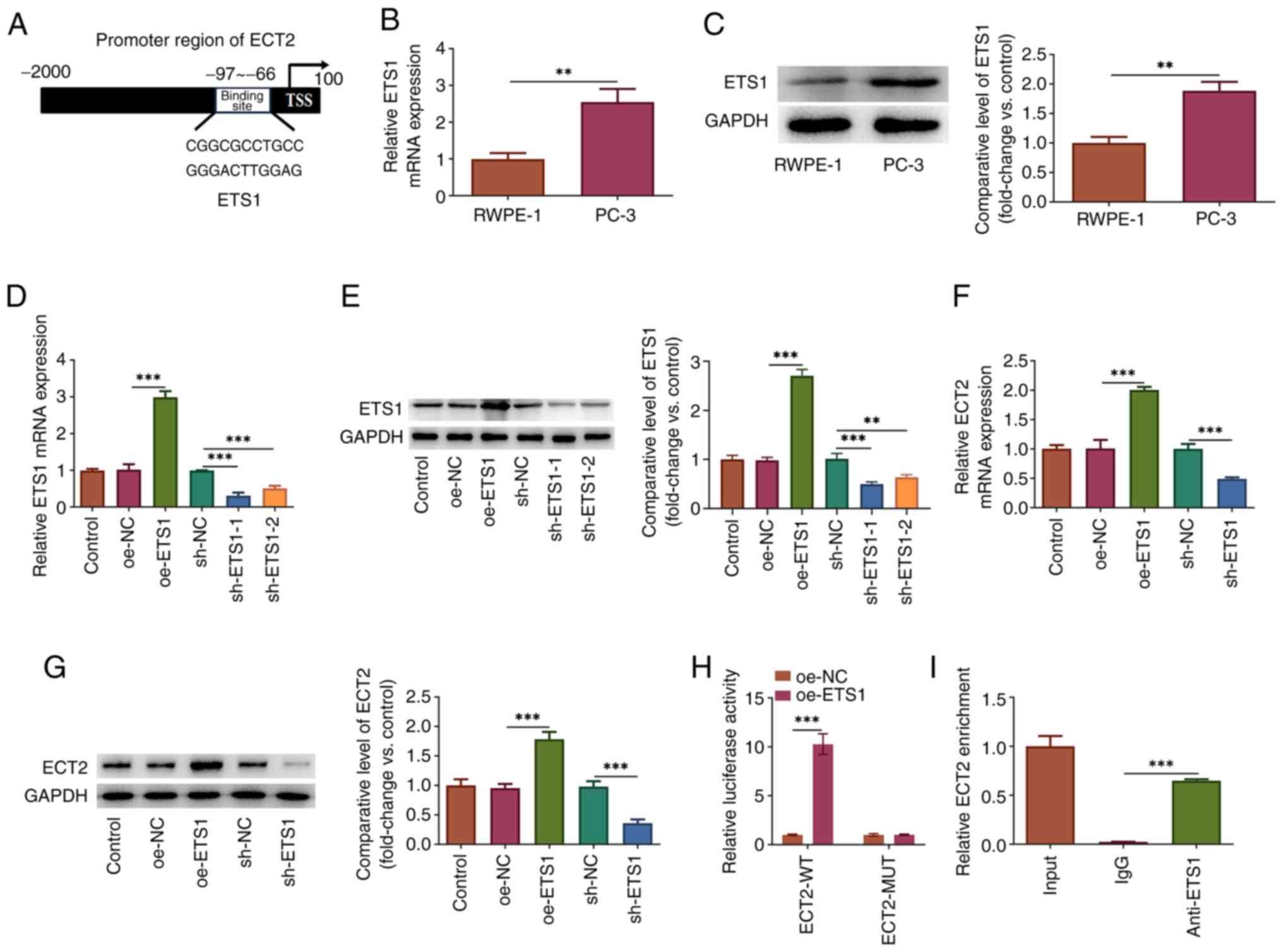 | Figure 4.ETS1 transcriptionally binds to the
ECT2 promoter and positively regulates ECT2 expression. (A)
Predicted binding site between the transcriptional factor ETS1 and
the ECT2 promoter. Expression levels of ETS1 in PC-3 and RWPE-1
cells were examined by (B) RT-qPCR and (C) western blot analyses.
(D and E) PC-3 cells were transfected with oe-ETS1 to overexpress
ETS1 or with sh-ETS1-1/2 to interfere with ETS1 expression. The
mRNA and protein expression levels of ETS1 were examined using (D)
RT-qPCR and (E) western blot analyses, respectively. The mRNA and
protein expression levels of ECT2 were examined using (F) RT-qPCR
and (G) western blot analyses, respectively. (H) The binding
relationship between ETS1 and the ECT2 promoter was verified using
a luciferase reporter assay. (I) A chromatin immunoprecipitation
assay was conducted and the precipitated chromatin fragments were
examined by qPCR. **P<0.01 and ***P<0.001. ECT2, epithelial
cell transforming sequence 2; MUT, mutant; NC, negative control;
oe, overexpression vector; RT-qPCR, reverse
transcription-quantitative PCR; sh, short hairpin RNA; TSS,
transcription start site; WT, wild-type. |
Impact of knockdown of ECT2 expression
on the malignant behavior and aerobic glycolysis of PC-3 cells is
weakened by ETS1 overexpression
To verify the involvement of ETS1 underlying
ECT2-mediated prostate cancer progression, gain- and
loss-of-function experiments were conducted in PC-3 cells. As shown
in Fig. 5A, PC-3 cells were
transfected with sh-ECT2/sh-NC alone or co-transfected with sh-ECT2
and oe-ETS1/oe-NC, and the CCK-8 assay revealed that knockdown of
ECT2 expression inhibited cell viability, which was partly reversed
by additional ETS1 overexpression. Knockdown of ECT2
expression-caused reduction in colonies was partly abolished
following ETS1 overexpression (Fig.
5B). Subsequently, wound healing and Transwell assays revealed
that the inhibitory effects of knockdown of ECT2 expression on cell
migration and invasion were partly reversed by ETS1 overexpression
(Fig. 5C and D). Furthermore,
simultaneous transfection with sh-ECT2 and oe-ETS1 significantly
increased the protein expression levels of MMP2, MMP9, N-cadherin
and Vimentin and decreased the protein expression levels of
E-cadherin compared with those following transfection with sh-ECT2
and oe-NC (Fig. 5E). In addition,
simultaneous transfection of the cells with sh-ECT2 and oe-ETS1
significantly increased lactate release and glucose uptake compared
with transfection with sh-ECT2 and oe-NC. This was accompanied by
upregulated ECAR and downregulated OCR, revealing that the
inhibitory effect of knockdown of ECT2 expression on aerobic
glycolysis was partly weakened by ETS1 overexpression (Fig. 6A-C). Furthermore, ECT2
knockdown-reduced protein expression levels of HK2, PKM2 and LDHA
were partly restored by additional ETS1 overexpression (Fig. 6D).
 | Figure 6.Impact of knockdown of ECT2
expression on aerobic glycolysis in PC-3 cells is weakened by ETS1
overexpression. The cell culture medium was harvested and the (A)
lactate release and (B) glucose uptake were examined using
commercial kits. (C) The ECAR and OCR of PC-3 cells were assessed
using a Seahorse XFe96 Analyzer to evaluate the glycolytic capacity
and maximal respiration. (D) Western blotting was performed to
examine the expression levels of several critical enzymes in the
aerobic glycolysis process. *P<0.05 and ***P<0.001. 2-DG,
2-deoxy-D-glucose; A&R, antimycin A/rotenone; ECAR,
extracellular acidification rate; ECT2, epithelial cell
transforming sequence 2; FCCP, carbonylcyanide-4-(trifluoromethoxy)
phenylhydrazone; Glc, glucose; HK2, hexokinase 2; LDHA, lactate
dehydrogenase A; NC, negative control; OCR, oxygen consumption
rate; oe, overexpression vector; Oligo, oligomycin; PKM2, pyruvate
kinase M2; sh, short hairpin RNA. |
Discussion
Prostate cancer is the most common cancer in men
worldwide (1,2). It is of great importance to identify
novel biomarkers and develop effective therapeutic targets for the
treatment of prostate cancer. The present study demonstrated that
ECT2 was highly expressed in prostate cancer. Knockdown of ECT2
expression could reduce aerobic glycolysis of prostate cancer, and
thus, inhibit cell proliferation, invasion and migration.
Furthermore, the transcription factor ETS1 could directly bind to
the ECT2 promoter and positively regulate ECT2. The regulatory role
of ECT2 in prostate cancer may be partly mediated by ETS1. Taken
together, the data demonstrated that ECT2 may be a promising
therapeutic target in human prostate cancer.
Numerous studies have shown that cancer cells
reprogram their metabolism to facilitate growth, survival and
metastasis (19,20). The alteration of aerobic glycolysis,
also known as the ‘Warburg effect’, is a well-recognized hallmark
of cancer cell metabolism (20).
Increased glycolysis, which is accompanied by increased glucose
intake and fermentation of glucose to lactate, is essential to
fulfill the demands of energy requirements and macromolecule
synthesis in cancer cells, and it also modulates the tumor stroma
to a pro-tumorigenic microenvironment, thereby promoting cancer
cell proliferation (21,22). Therefore, restriction of aerobic
glycolysis may provide possible therapeutic targets or drugs for
cancer therapy (22–24). At present, the role of ECT2 in
glucose metabolic reprogramming of cancer has not been fully
investigated. The limited findings have revealed that ECT2 could
enhance aerobic glycolysis to promote the M2 phenotype polarization
of tumor-associated macrophages in hepatocellular carcinoma,
thereby promoting the proliferation and migration of hepatocellular
carcinoma cells (25). Furthermore,
Rac GTPase-activating protein 1 has been regarded as a critical
driver to promote breast cancer metastasis, which is dependent on
ECT2-mediated mitochondrial quality control and aerobic glycolysis
(26). Nevertheless, to the best of
our knowledge, the association between ECT2 and prostate cancer
remains unclear. The present study demonstrated that ECT2
expression was increased in prostate cancer cells. Knockdown of
ECT2 expression exhibited significant inhibitory effects on glucose
uptake, lactate production and the expression of key glycolytic
enzymes (HK2, PKM2 and LDHA), reducing glycolytic metabolite
levels, and thus, inhibiting the proliferation, migration and
invasion of prostate cancer cells, and delaying the progression of
prostate cancer.
Transcription factors are involved in the formation
of transcription initiation complexes, and thus, serve important
roles in modulating gene expression (27). ETS1 belongs to the ETS family of
transcription factors, characterized by a DNA-binding domain
containing a GGAA/T core motif (28). Previous studies have identified that
ETS1 functions as a crucial transcription factor in various
physiological processes in living organisms, such as cell survival,
differentiation and apoptosis; therefore, it is regarded to be
involved in multiple physiological and pathological processes, such
as reproduction, diabetic nephropathy and malignant cancer types
(29–31). The regulatory role of ETS1 in
carcinoma has been extensively studied (32–34),
including in prostate cancer. The transcriptional activity of ETS1
is enhanced in advanced prostate cancer and ETS1 expression is
highest in high-grade prostate cancer. In vitro functional
experiments have demonstrated that elevated ETS1 expression
facilitated an aggressive and castrate-resistant phenotype in
prostate cancer cells, indicating the oncogenic role of ETS1
transcriptional activity in prostate cancer (34). Circular RNA_0004296 has been found
to inhibit metastasis of prostate cancer, which was largely
associated with inhibition of ETS1 mRNA expression (35). Accordingly, the present study
revealed high ETS1 expression in prostate cancer cells. A critical
binding relationship was confirmed between the transcription factor
ETS1 and the ECT2 promoter, and ETS1 could positively regulate ECT2
expression in prostate cancer cells. The subsequent rescue
experiments revealed that the inhibitory effects of knockdown of
ECT2 expression on cell proliferation, migration, invasion and
aerobic glycolysis were reversed by ETS1 overexpression, suggesting
that the effect of ECT2 expression on prostate cancer cells was
partly mediated by ETS1.
To the best of our knowledge, the present study was
the first to reveal the regulatory role of ECT2 in prostate cancer,
as well as its potential mechanism of action. The findings revealed
that knockdown of ECT2 expression may reduce aerobic glycolysis of
prostate cancer, and thus, reduce cell proliferation and invasion,
thereby inhibiting prostate cancer progression (19,36).
With regard to its mechanism of action, ECT2 was transcriptionally
activated by the transcription factor ETS1. The present study
provided a potential biomarker and therapeutic target for patients
with prostate cancer.
There were some limitations in the present study.
Although the oncogenic role of ETS1 has been confirmed previously
(34,35), re-examination of ETS1 expression in
prostate cancer cells could further validate the existing evidence,
and would be beneficial to confirm the regulation of ETS1/ECT2 in
prostate cancer. Animal experiments may be conducted in future work
to further verify the current findings, and clinical verification
should also be considered.
Acknowledgements
Not applicable.
Funding
The present study was supported by Xiamen Natural Science
Foundation (grant no. 3502Z20227422).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
BZ, KC and JC designed the study. BZ, KC, XL, ZW,
YW, LX, JX and JC performed the experiments to collect and analyze
the data. BZ and KC drafted the manuscript and JC revised the
manuscript. BZ, KC and JC confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Adamaki M and Zoumpourlis V: Prostate
cancer biomarkers: From diagnosis to prognosis and precision-guided
therapeutics. Pharmacol Ther. 228:1079322021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Uhr A, Glick L and Gomella LG: An overview
of biomarkers in the diagnosis and management of prostate cancer.
Can J Urol. 27((S3)): 24–27. 2020.PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD, Wagle NS and Jemal
A: Cancer statistics, 2023. CA Cancer J Clin. 73:17–48. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Culp MB, Soerjomataram I, Efstathiou JA,
Bray F and Jemal A: Recent global patterns in prostate cancer
incidence and mortality rates. Eur Urol. 77:38–52. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Howard N, Clementino M, Kim D, Wang L,
Verma A, Shi X, Zhang Z and DiPaola RS: New developments in
mechanisms of prostate cancer progression. Semin Cancer Biol.
57:111–116. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bhoir S and De Benedetti A: Targeting
prostate cancer, the ‘tousled way’. Int J Mol Sci. 24:111002023.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sehrawat A, Gao L, Wang Y, Bankhead A III,
McWeeney SK, King CJ, Schwartzman J, Urrutia J, Bisson WH, Coleman
DJ, et al: LSD1 activates a lethal prostate cancer gene network
independently of its demethylase function. Proc Natl Acad Sci USA.
115:E4179–E4188. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fields AP and Justilien V: The guanine
nucleotide exchange factor (GEF) Ect2 is an oncogene in human
cancer. Adv Enzyme Regul. 50:190–200. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saito S, Liu XF, Kamijo K, Raziuddin R,
Tatsumoto T, Okamoto I, Chen X, Lee CC, Lorenzi MV, Ohara N and
Miki T: Deregulation and mislocalization of the cytokinesis
regulator ECT2 activate the Rho signaling pathways leading to
malignant transformation. J Biol Chem. 279:7169–7179. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schneid S, Wolff F, Buchner K, Bertram N,
Baygun S, Barbosa P, Mangal S and Zanin E: The BRCT domains of ECT2
have distinct functions during cytokinesis. Cell Rep.
34:1088052021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cook DR, Kang M, Martin TD, Galanko JA,
Loeza GH, Trembath DG, Justilien V, Pickering KA, Vincent DF,
Jarosch A, et al: Aberrant expression and subcellular localization
of ECT2 drives colorectal cancer progression and growth. Cancer
Res. 82:90–104. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang H, Geng Y, Sun C and Yu J:
Upregulation of ECT2 predicts adverse clinical outcomes and
increases 5-fluorouracil resistance in gastric cancer patients. J
Oncol. 2021:21028902021.PubMed/NCBI
|
|
13
|
Sun BY, Wei QQ, Liu CX, Zhang L, Luo G, Li
T and Lü MH: ECT2 promotes proliferation and metastasis of
esophageal squamous cell carcinoma via the RhoA-ERK signaling
pathway. Eur Rev Med Pharmacol Sci. 24:7991–8000. 2020.PubMed/NCBI
|
|
14
|
Yi M, Zhang D, Song B, Zhao B, Niu M, Wu
Y, Dai Z and Wu K: Increased expression of ECT2 predicts the poor
prognosis of breast cancer patients. Exp Hematol Oncol. 11:1072022.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guo Z, Chen X, Du T, Zhu D, Lai Y, Dong W,
Wu W, Lin C, Liu L and Huang H: Elevated levels of epithelial cell
transforming sequence 2 predicts poor prognosis for prostate
cancer. Med Oncol. 34:132017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chandrashekar DS, Karthikeyan SK, Korla
PK, Patel H, Shovon AR, Athar M, Netto GJ, Qin ZS, Kumar S, Manne
U, et al: UALCAN: An update to the integrated cancer data analysis
platform. Neoplasia. 25:18–27. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y and Weinberg RA:
Epithelial-to-mesenchymal transition in cancer: Complexity and
opportunities. Front Med. 12:361–373. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cai K, Chen S, Zhu C, Li L, Yu C, He Z and
Sun C: FOXD1 facilitates pancreatic cancer cell proliferation,
invasion, and metastasis by regulating GLUT1-mediated aerobic
glycolysis. Cell Death Dis. 13:7652022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
DeBerardinis RJ, Lum JJ, Hatzivassiliou G
and Thompson CB: The biology of cancer: Metabolic reprogramming
fuels cell growth and proliferation. Cell Metab. 7:11–20. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chelakkot C, Chelakkot VS, Shin Y and Song
K: Modulating glycolysis to improve cancer therapy. Int J Mol Sci.
24:26062023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li L, Liang Y, Kang L, Liu Y, Gao S, Chen
S, Li Y, You W, Dong Q, Hong T, et al: Transcriptional regulation
of the warburg effect in cancer by SIX1. Cancer Cell. 33:368–385.
e72018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu Z, Wu J, Zhao Q, Fu S and Jin J:
Emerging roles of aerobic glycolysis in breast cancer. Clin Transl
Oncol. 22:631–646. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu D, Wang Y, Wu J, Zhang Z, Chen J, Xie
M, Tang R, Chen C, Chen L, Lin S, et al: ECT2 overexpression
promotes the polarization of tumor-associated macrophages in
hepatocellular carcinoma via the ECT2/PLK1/PTEN pathway. Cell Death
Dis. 12:1622021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ren K, Zhou D, Wang M, Li E, Hou C, Su Y,
Zou Q, Zhou P and Liu X: RACGAP1 modulates ECT2-Dependent
mitochondrial quality control to drive breast cancer metastasis.
Exp Cell Res. 400:1124932021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhi T, Jiang K, Xu X, Yu T, Zhou F, Wang
Y, Liu N and Zhang J: ECT2/PSMD14/PTTG1 axis promotes the
proliferation of glioma through stabilizing E2F1. Neuro Oncol.
21:462–473. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang S, Linde MH, Munde M, Carvalho VD,
Wilson WD and Poon GM: Mechanistic heterogeneity in site
recognition by the structurally homologous DNA-binding domains of
the ETS family transcription factors Ets-1 and PU.1. J Biol Chem.
289:21605–21616. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Geng XD, Wang WW, Feng Z, Liu R, Cheng XL,
Shen WJ, Dong ZY, Cai GY, Chen XM, Hong Q and Wu D: Identification
of key genes and pathways in diabetic nephropathy by bioinformatics
analysis. J Diabetes Investig. 10:972–984. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chakraborty S and Banerjee S:
Multidimensional computational study to understand non-coding RNA
interactions in breast cancer metastasis. Sci Rep. 13:157712023.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang F, Liu Y and Wang P, Wang X, Chu M
and Wang P: Mutation of the ETS1 3′UTR interacts with miR-216a-3p
to regulate granulosa cell apoptosis in sheep. Theriogenology.
210:133–142. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dittmer J: The role of the transcription
factor Ets1 in carcinoma. Semin Cancer Biol. 35:20–38. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen Y, Peng C, Chen J, Chen D, Yang B, He
B, Hu W, Zhang Y, Liu H, Dai L, et al: WTAP facilitates progression
of hepatocellular carcinoma via m6A-HuR-dependent epigenetic
silencing of ETS1. Mol Cancer. 18:1272019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Smith AM, Findlay VJ, Bandurraga SG,
Kistner-Griffin E, Spruill LS, Liu A, Golshayan AR and Turner DP:
ETS1 transcriptional activity is increased in advanced prostate
cancer and promotes the castrate-resistant phenotype.
Carcinogenesis. 33:572–580. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mao S, Zhang W, Yang F, Guo Y, Wang H, Wu
Y, Wang R, Maskey N, Zheng Z, Li C, et al: Hsa_circ_0004296
inhibits metastasis of prostate cancer by interacting with EIF4A3
to prevent nuclear export of ETS1 mRNA. J Exp Clin Cancer Res.
40:3362021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu W, Zeng F, Li S, Li G, Lai X, Wang QJ
and Deng F: Crosstalk of protein kinase C ε with Smad2/3 promotes
tumor cell proliferation in prostate cancer cells by enhancing
aerobic glycolysis. Cell Mol Life Sci. 75:4583–4598. 2018.
View Article : Google Scholar : PubMed/NCBI
|