Introduction
Gastric cancer (GC) is the fourth leading cause of
cancer death worldwide, and 1,089,103 new cases and 768,793 deaths
were reported in 2020 (1). The
incidence of GC varies by region; with the highest prevalence
detected in eastern and central Asia, and Latin America (2). Notably, this type of cancer has no
symptoms in the early stages and patients diagnosed at advanced
stages have unfavorable prognoses (3,4). GC
survival varies depending on the stage of the disease during
surgical intervention; for early advanced stages, the 5-year
survival rate is 18% (5). Due to
the poor prognosis of GC, its therapeutic resistance and the side
effects of chemotherapy, it is essential to develop new strategies
for the treatment of GC.
Metformin has attracted attention as an antitumor
agent that may be used as an alternative treatment to improve the
survival and quality of life of patients with GC (6,7).
Metformin (1,1 dimethylbiguanide) is an antihyperglycemic drug used
to treat diabetes mellitus type 2 (DM2) (8). Epidemiological, in vitro and
in vivo studies, and clinical trials have demonstrated the
ability of metformin as an antitumor agent in various types of
cancer, such as lung, endometrial, breast and prostate cancer
(9,10). The antitumor mechanism of action of
metformin is not fully understood; however, two mechanisms have
been proposed: A direct effect through activating AMPK, and an
indirect effect by reducing blood glucose and insulin levels
(9). Metformin is internalized into
cells primarily by organic cation transporter 1 (11). Once inside the cell, metformin acts
directly on the mitochondria, inhibiting the first complex of the
electron transport chain (NADH dehydrogenase). This process
decreases the amount of ATP and increases the AMP/ATP ratio. An
increased concentration of cytosolic AMP leads to AMPK activation,
and liver kinase B1 is the enzyme that activates AMPK via
phosphorylation of the Thr172 residue of the α subunit (12). Subsequently, the metabolism of the
cell changes from an anabolic state to a catabolic state to restore
cellular homeostasis; therefore, processes such as gluconeogenesis,
lipogenesis and protein synthesis are inhibited (13).
Previous studies have demonstrated that the
antitumor effect of metformin alone on GC cell lines is capable of
decreasing cell viability (14–17),
cell proliferation (14,15,17)
and cell survival (17), and
increasing apoptosis (15,16,18).
In addition, in vivo models of metformin have shown its
potential antitumor effect reducing the expression of cyclin D,
cyclin-dependent kinase (CDK)4 and CDK6, and the levels of
retinoblastoma protein (Rb) phosphorylation (14), as well as inhibiting survivin and
mTOR (18).
To the best of our knowledge, the effect of
metformin in combination with chemotherapeutic drugs has yet to be
sufficiently studied; cisplatin, adriamycin, paclitaxel,
oxaliplatin, docetaxel, 5-fluorouracil, rapamycin, carboplatin,
epirubicin or methotrexate in combination with metformin have been
analyzed for the treatment of GC in vitro (17,19–22).
However, the combination of metformin with chemotherapy regimens
has not been carried out for GC treatment, which involves the
combination of different drugs and the activation of multiple
pathways, including apoptosis, proliferation and resistance. The
present study aimed to evaluate the mechanisms underlying the
effects of metformin, alone or in combination with chemotherapy, on
the apoptosis, mitochondrial membrane potential (ΔΨm), caspase-3,
−8 and −9 activity, cell cycle progression, proliferation,
senescence and clonogenic capacity of NCI-N87 GC cells.
Materials and methods
Cell culture and reagents
The NCI-N87 GC cell line was obtained form from
American Type Culture Collection (CRL-5822™). Cells were maintained
in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% inactivated fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin-neomycin (PSN; Gibco; Thermo Fisher
Scientific, Inc.) at 37°C in a humidified atmosphere containing 95%
air and 5% CO2. The cells were passaged once they
reached 75–85% confluence. Before the initiation of all
experiments, cell viability was determined with Trypan Blue
(MilliporeSigma) and viability was >95%. This study was approved
by the National Scientific Research Committee of Mexican Social
Security Institute (approval number: R-2019-785-050; Guadalajara,
Mexico).
Cytogenetic characterization of the GC
cell line
The cytogenetic characteristics of NCI-N87 cells
were assessed by fluorescence in situ hybridization (FISH)
using commercially available direct labeled FISH probes. NCI-N87
cells were harvested using Accutase (cat. no. 423201; Biolegend,
Inc.) and were washed twice with PBS. The cells were then
resuspended in 1 ml RPMI-1640 supplemented with 10% inactivated FBS
and 1% PSN, and those harvested cells were treated with 0.075 M
potassium chloride solution at 37°C for 20 min, centrifuged at 240
× g for 10 min at 25°C, fixed in methanol:acetic acid (3:1)
solution at 2°C, resuspended in the same fixing solution, and
dropped onto cleaned microscope slides for FISH. The cells were
dehydrated and hybridized according to the FISH probe
manufacturer's protocol. The following probes were obtained from
Cytocell (Oxford Gene Technology IP Limited):
DXZ1/DYZ3 (cat no. LPE 0XY), CKS1B/CDKN2C
(cat. no. LPH 039), EGFR amplification (cat. no. LPS 003),
MYC breakapart (cat. no. LPH 010), CDKN2A-B/D9Z3
(cat. no. LPH 009), IGH/CCND1 (cat. no. LPH 072),
RB1/LAMP1 (cat. no. LPS 011), IGH/BCL2 (cat. no. LPH
071), TP53/ATM (cat. no. LPH 052), ERBB2/D17Z1 (cat.
no. LPS 001), PML/RARA (cat. no. LPH 023) and TCRAD
breakapart (cat no. LPH 047). Samples were observed under a
fluorescence microscope and microscopic observations were
interpreted following the International System for Human
Cytogenomic Nomenclature 2020 recommendations (23). Only FISH assays with abnormal
results are presented in the present study.
Drugs
Before performing the experiments in GC cells,
various solutions were prepared. Metformin (cat. no. 317240; Merck
KGaA) was dissolved in RPMI and stored at −20°C until use.
Epirubicin (cat. no. E9406; MilliporeSigma), 5-fluorouracil (cat.
no. F6627; MilliporeSigma) and cisplatin (cat. no. P4394;
MilliporeSigma) were dissolved in sterile saline and maintained at
4°C, with the exception of cisplatin, which was stored at room
temperature. Docetaxel (cat. no. 01885; MilliporeSigma) was
dissolved in DMSO and stored at −80°C.
Cell treatments
NCI-N87 cells were treated for 48 h at 37°C with
metformin (10 mM) and four chemotherapy drugs: epirubicin (0.5 µM),
cisplatin (15 µM), docetaxel (0.5 µM) or 5-fluorouracil (30 µM). In
addition, metformin was used in combination with each of the
chemotherapy drugs, as well as with the three chemotherapy
regimens: Epirubicin (0.5 µM) + cisplatin (15 µM) + 5-fluorouracil
(30 µM) (ECF), docetaxel (0.5 µM) + cisplatin (15 µM) +
5-fluorouracil (30 µM) (DCF) and cisplatin (15 µM) + 5-fluorouracil
(30 µM) (CF). The control group consisted of cells without
treatment.
Determination of apoptosis in GC
cells
NCI-N87 cells (5×105 cells/well) were
seeded in 24-well plates and cultured in 1 ml RPMI-1640
supplemented with 10% inactivated FBS and 1% PSN for apoptosis
determination. The next day, the seeded cells were treated with the
drugs for 48 h at 37°C. Subsequently, cells were harvested using
Accutase and were washed twice with PBS. The cells were then
resuspended in 400 µl Annexin V Binding Buffer, and FITC Annexin V
(cat. no. 640922; Biolegend) and SYTOX® (cat. no.
S34859; Invitrogen, Carlsbad, CA, USA) were added. The cells were
incubated for 15 min in the dark at 25°C. Finally, 10,000 events
were acquired for each sample using the Attune Acoustic Focusing
Cytometer (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
data were analyzed using Kaluza V2.1 software (Beckman Coulter,
Inc.). The results of apoptosis analysis were expressed as the mean
± SD of live, apoptotic and necrotic cells. Etoposide (100 µM)
(PiSA Farmacéutica) was used as a positive control for the cell
death assay (data not shown).
Determination of ΔΨm in GC cells
The loss of ΔΨm was determined using JC-10 reagent
(cat. no. ab112134; Abcam). NCI-N87 cells (4×104
cells/well) were seeded in 96-well plates with black wells and
clear bottoms and were cultured in 100 µl RPMI-1640 supplemented
with 10% inactivated FBS and 1% PSN. The next day, cells were
exposed to the different treatments for 48 h. For JC-10 staining,
50 µl JC-10 was diluted in 5 ml Assay Buffer A, and 50 µl of the
mix was added to each sample and incubated at 37°C for 1 h.
Subsequently, 50 µl Assay Buffer B was added before measuring
fluorescence intensity. Finally, fluorescence was measured at
excitation 488 nm and emission ratio 530/590 nm in a plate reader
(Biotek Synergy HT; Biotek; Agilent Technologies, Inc.). Data are
presented as the mean ± SD. The positive control for the ΔΨm assay
was etoposide (100 µM) (data not shown).
Determination of caspase activity in
GC cells
To evaluate activated caspase-1, −3, −4, −5, −6, −7,
−8 and −9 in apoptotic cells, a Generic Caspase Activity Assay kit
(cat. no. ab112130; Abcam) was used. NCI-N87 cells
(5×105 cells/well) were seeded in 24-well plates and
cultured in 1 ml RPMI-1640 supplemented with 10% inactivated FBS
and 1% PSN. The next day, cells were exposed to different
treatments for 48 h. Cells were harvested using Accutase and were
washed twice with PBS. Then, cells were resuspended in 0.5 ml
culture medium, and 1 µl TF2-VAD-FMK (obtained from the Generic
Caspase Activity Assay kit) was added to each sample and the cells
were incubated at 37°C for 2 h. Subsequently, the cells were washed
once with PBS. Finally, cells were resuspended in 0.5 ml Assay
Buffer. A total of 10,000 events were acquired for each sample
using the Attune Acoustic Focusing Cytometer. The data were
analyzed using Kaluza V2.1 software. Data are presented as the mean
± SD of cell percentage. The positive control for the general
caspase activity assay was etoposide (100 µM) (data not shown).
Determination of caspase-3, −8 and −9
activity in GC cells
To evaluate the activity of caspases, the NCI-N87
cells (5×105 cells/well) were seeded in 24-well plates
and cultured in 1 ml RPMI-1640 supplemented with 10% inactivated
FBS and 1% PSN. The next day, cells were exposed to different
treatments for 48 h. Three different kits were used to determine
the activity of each caspase (caspase-3, caspase-8 and caspase-9,
cat. no. ab65613, ab65614 and ab65615, respectively; Abcam). Cells
were harvested using Accutase and were washed twice with PBS. Then,
cells were resuspended in 300 µl culture medium, and 1 µl of the
corresponding substrate was added (FITC-DEVD-FMK/caspase-3,
FITC-IETD-FMK/caspase-8 and FITC-LEHD-FMK/caspase-9) for 1 h at
37°C. Subsequently, the cells were washed once with 500 µl Wash
Buffer, the supernatant was removed and the cells were resuspended
in 300 µl Wash Buffer. At least 10,000 events were acquired using
the Attune Acoustic Focusing Cytometer and the data were analyzed
using Kaluza V2.1 software. Data are presented as the mean ± SD of
cell percentage. The positive control for the caspase activity
assay was etoposide (100 µM) (data not shown).
Cell cycle assessment in GC cells
Cell cycle progression was determined using the BD
Cycletest™ Plus DNA kit (cat. no. 340242; BD Biosciences). To
synchronize the cells, they were depleted of serum in a
step-by-step manner: Cells were cultured with RPMI-1640
supplemented with 5% FBS for 12 h; after which, cells were cultured
with RPMI-1640 supplemented with 1% FBS for 12 h; finally, cells
were cultured with serum-free RPMI-1640 for 18 h. After
synchronization, NCI-N87 cells (5×105 cells/well) were
seeded in 24-well plates and cultured in 1 ml RPMI-1640
supplemented with 10% inactivated FBS and 1% PSN. The next day,
seeded cells were treated with different drugs and combinations for
48 h. Subsequently, the cells were harvested using Accutase and
were washed twice with PBS, before the DNA staining procedure was
performed. First, trypsin buffer was added to each sample and
incubated at room temperature for 10 min; after which, a trypsin
inhibitor and RNase buffer were added and incubated for 10 min at
room temperature. Finally, propidium iodide solution was added and
incubated on ice for 10 min in the dark. At least 30,000 events
were acquired for each sample using the Attune Acoustic Focusing
Cytometer. Data were analyzed using ModFit LT 5.0 software (Verity
Software House, Inc.). Data are presented as the mean ± SD of the
percentage of cells in the G1, S and G2
phases. The DNA QC particles kit (cat. no. 349523; BD Biosciences)
was used to check the calibration and linearity of the equipment
(data not shown).
Proliferation assay in GC cells
Proliferation was determined using the BrdU Cell
Proliferation ELISA Kit (cat. no. ab126556; Abcam). The NCI-N87
cells (4×104 cells/well) were seeded in 96-well plates
and cultured in 200 µl RPMI-1640 supplemented with 10% inactivated
FBS and 1% PSN. The next day, cells were exposed to the different
treatments for 96 h, and BrdU was incubated for 24 h at 37°C. The
cell culture medium was aspirated, 200 µl/well fixing solution was
added to denature the DNA and the cells were incubated for 30 min
at room temperature. Subsequently, the first wash was performed
using Wash Buffer; after which, 100 µl/well Anti-BrdU Monoclonal
Detector Antibody was added and the cells were incubated for 1 h at
room temperature. A second wash was then performed and 100 µl/well
Peroxidase Goat Anti-Mouse IgG Conjugate was used to incubate the
cells for 30 min at room temperature. Subsequently, a third wash
was performed, 100 µl/well TMB Peroxidase Substrate was added and
the cells were incubated for 30 min at room temperature in the
dark. Finally, 100 µl/well Stop Solution was added and the optical
density was determined at 450 nm using a plate reader (Biotek
Synergy HT). Data are presented as the mean ± SD of the percentage
of proliferation. The positive control for the cell proliferation
assay was etoposide (100 µM) (data not shown).
Senescence assessment in GC cells
Senescence was evaluated using the Senescence
Detection Kit (cat. no. ab65351; Abcam). The NCI-N87 cells
(1×106 cells/well) were seeded in 12-well plates and
cultured in 2 ml RPMI-1640 supplemented with 10% inactivated FBS
and 1% PSN. The next day, seeded cells were treated with the
different drugs for 48 h. The culture medium was then removed and
the cells were washed once with 1 ml PBS. Subsequently, the cells
were fixed with 0.5 ml Fixative Solution for 15 min at room
temperature, washed twice with PBS, and incubated with 0.5 ml
Staining Solution Mix (Staining Solution, Staining Supplement and
β-galactosidase) inside a sealable bag overnight at 37°C. Cells
were observed under a light microscope to determine senescent
cells. Data were analyzed using ImageJ software (Version 1.8.0_172;
National Institutes of Health). Data are presented as the mean ± SD
of β-galactosidase-stained surface. The positive control for the
senescence assay was doxorubicin (1 µM) (PiSA Farmacéutica) (data
not shown).
Clonogenic assay in GC cells
Cell survival was determined using a clonogenic
assay. The NCI-N87 cells (15×105 cells/well) were seeded
in 6-well plates and cultured in 3 ml RMPI-1640 supplemented with
10% inactivated FBS and 1% PSN. The following day, cells were
exposed to the different treatments for 24 h. Subsequently, the
cells were harvested using Accutase and were washed twice with PBS.
Then, 4,000 cells/well were seeded in 6-well plates in 3 ml culture
medium. To determine the ability to form colonies, the cells were
incubated for 15 days at 37°C (during this period, the culture
medium was changed every 3 days). To stain colonies, the cells were
first fixed with 1 ml/well formaldehyde (3.7% diluted in PBS) for
15 min at 25°C, washed twice with 2 ml PBS and dried overnight.
Colonies were stained with 1 ml/well sulforhodamine (0.4% diluted
in 1% acetic acid) for 30 min at 25°C and were finally washed three
times with acetic acid (1% diluted in H2O). Colonies
(>60 cells) were viewed under a light microscope at ×40
magnification and images were captured using Zen 2012 blue edition
v1.1.2.0 software (Zeiss GmbH). The colony count was performed with
ImageJ software. The positive control for the clonogenic assay was
etoposide (100 µM) (data not shown).
Statistical analysis
All data are presented as the mean ± SD of three
independent experiments performed in triplicate. To assess
normality, the Shapiro-Wilk test was performed. Two-way ANOVA was
used for statistical analysis, followed by Tukey post hoc test to
compare all cell treatments. P<0.05 was considered to indicate a
statistically significant difference. Data were analyzed using
GraphPad Prism v8.0.2 software (Dotmatics).
Results
Treatment with metformin alone, and in
combination with chemotherapy, increases apoptosis and promotes
loss of ΔΨm in NCI-N87 GC cells
The results of the present study showed that
metformin alone induced apoptosis compared with in the untreated
cells control (P<0.05; Figs. 1A
and S1B). Furthermore, epirubicin,
cisplatin, and 5-fluorouracil increased the percentage of apoptotic
cells (P<0.05), and when metformin was combined with each
chemotherapy drug, it was observed that the tendency was for it to
improve the effectiveness of chemotherapy drug-induced apoptosis.
However, only the combination of metformin + 5-fluorouracil was
significant compared with 5-fluorouracil alone (P<0.05).
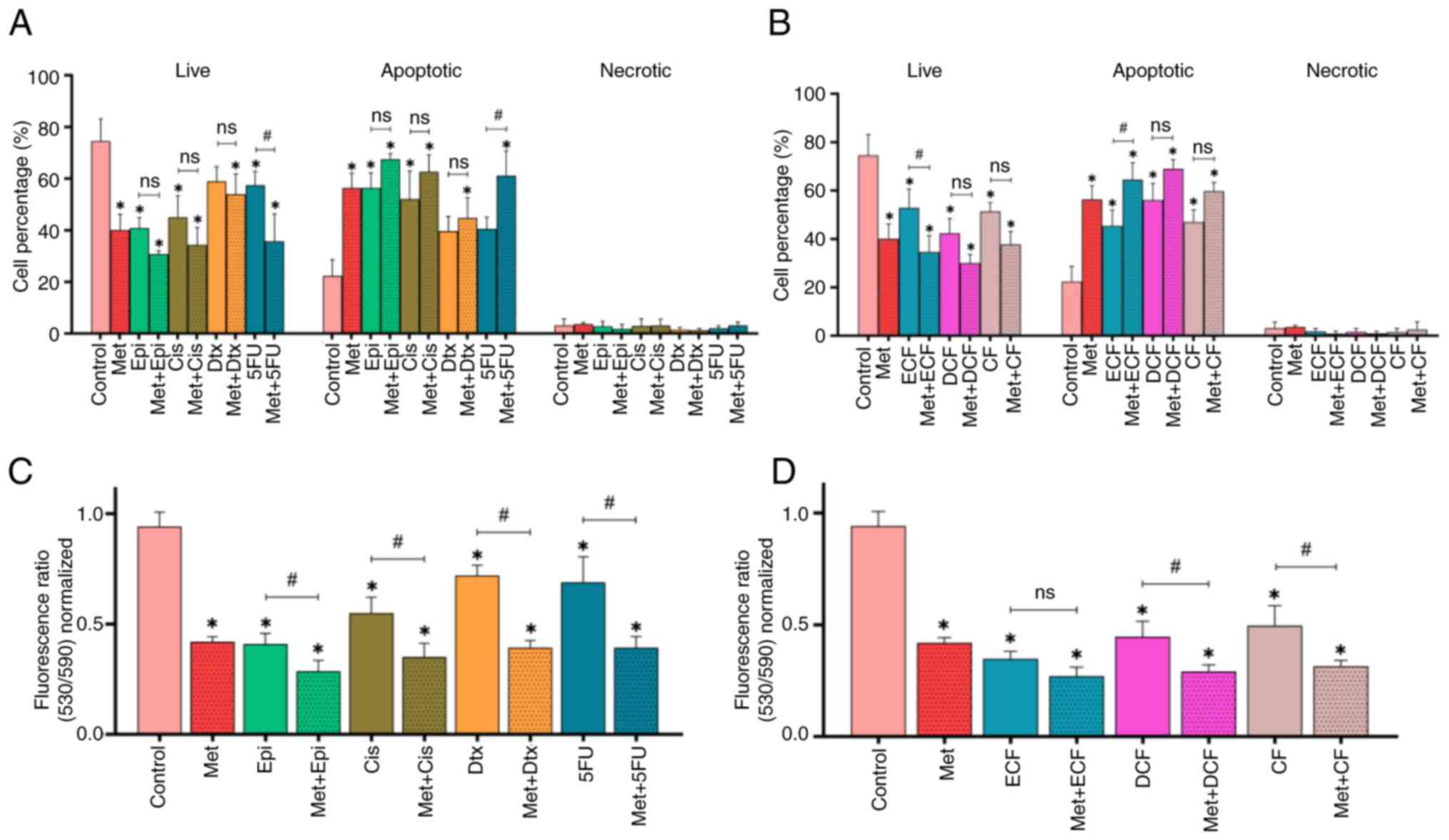 | Figure 1.Effect of Met in combination with
chemotherapy on apoptosis and loss of mitochondrial membrane
potential. Percentage of live, apoptotic and necrotic cells treated
with Met in combination with (A) chemotherapy drugs and (B)
chemotherapy regimens. Loss of mitochondrial membrane potential of
NCI-N87 gastric cancer cells treated with Met in combination with
(C) chemotherapy drugs and (D) chemotherapy regimens. Data are
presented as the mean ± SD from three independent experiments
performed in triplicate. *P<0.05 vs. control;
#P<0.05. Met, metformin; Epi, epirubicin; Cis,
cisplatin; Dtx, docetaxel; 5FU, 5-fluorouracil; ECF, epirubicin +
cisplatin + 5-fluorouracil; DCF, docetaxel + cisplatin +
5-fluorouracil; CF, cisplatin + 5-fluorouracil; ns, not
significant. |
The chemotherapy regimens ECF, DCF and CF induced
the apoptosis of NCI-N87 cells (P<0.05), and the combination of
metformin with the ECF regimen significantly enhanced the apoptosis
of GC cells compared with the regimen alone (P<0.05) (Figs. 1B and S1C).
Notably, metformin alone significantly induced a
loss of ΔΨm in GC cells (P<0.05), as did the four
chemotherapeutic drugs (P<0.05), when compared with the control
group (Fig. 1C). When metformin was
combined with each of the chemotherapeutic drugs, a greater effect
on the loss of ΔΨm in GC cells was observed (P<0.05) compared
with chemotherapy drugs alone. In addition, the ECF, DCF and CF
chemotherapy regimens decreased the ΔΨm of NCI-N87 cells
(P<0.05), and when metformin was combined with DCF and CF
regimens, that effect was amplified in comparison with the
chemotherapy regimens alone (P<0.05) (Fig. 1D).
Apoptosis is induced through caspase
activity
Metformin increased caspase activity in comparison
with untreated cells (P<0.05; Figs.
S2 and 2A). In addition, the
four chemotherapy drugs alone significantly increased caspase
activity compared with that in the control group (P<0.05).
Additionally, the combinations of metformin + epirubicin and
metformin + 5-fluorouracil induced significantly increased caspase
activity compared with that in the cells treated with chemotherapy
drugs alone (P<0.05). By contrast, the combination of metformin
+ cisplatin and metformin + docetaxel had no significant
effect.
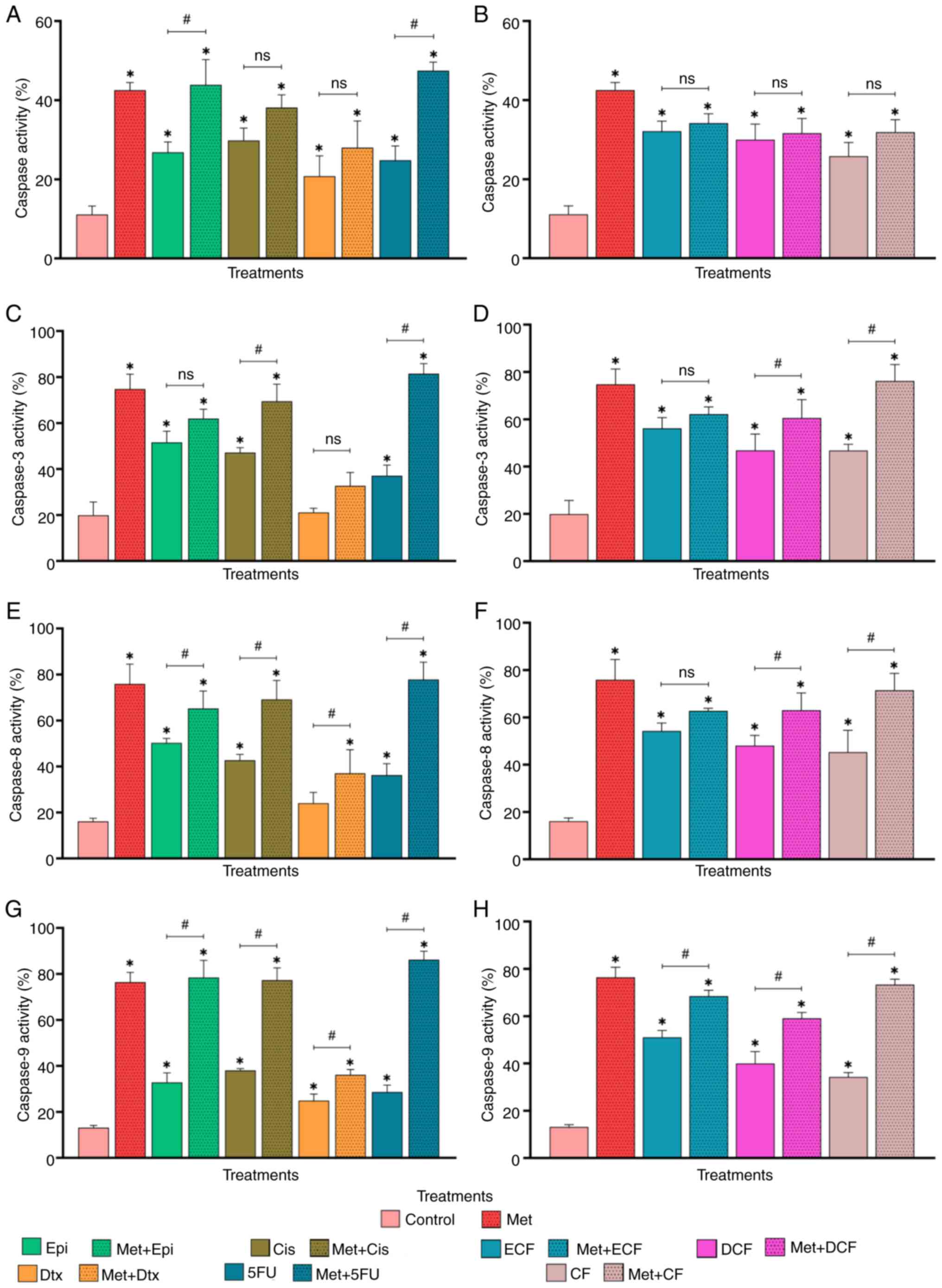 | Figure 2.Effect of Met in combination with
chemotherapy drugs on caspase activity. Percentage of caspase-1,
−3, −4, −5, −6, −7, −8, and −9 activity (TF2-VAD-FMK) in cells
treated with Met in combination with (A) chemotherapy drugs and (B)
chemotherapy regimens. Percentage of caspase-3 activity
(FITC-DEVD-FMK) in cells treated with Met in combination with (C)
chemotherapy drugs and (D) chemotherapy regimens. Percentage of
caspase-8 activity (FITC-IETD-FMK) in cells treated with Met in
combination with (E) chemotherapy drugs and (F) chemotherapy
regimens. Percentage of caspase-9 activity (FITC-LEHD-FMK) in cells
treated with Met in combination with (G) chemotherapy drugs and (H)
chemotherapy regimens. Data are presented as the mean ± SD from
three independent experiments performed in triplicate. *P<0.05
vs. control; #P<0.05. Met, metformin; Epi,
epirubicin; Cis, cisplatin; Dtx, docetaxel; 5FU, 5-fluorouracil;
ECF, epirubicin + cisplatin + 5-fluorouracil; DCF, docetaxel +
cisplatin + 5-fluorouracil; CF, cisplatin + 5-fluorouracil; ns, not
significant. |
The chemotherapy regimens ECF, DCF and CF also
increased caspase activity compared with that in the control group
(P<0.05); however, when metformin was combined with each
chemotherapy regimen, there was no significant difference compared
with the regimen alone (Fig.
2B).
After confirming that all treatments induced caspase
activity, the present study further investigated the participation
of the executioner caspase-3 and the initiator caspases-8 and −9.
Metformin alone increased the activities of caspases-3, −8 and −9
in GC cells compared with those in the control group (P<0.05;
Fig. 2C-H). In addition,
epirubicin, cisplatin and 5-fluorouracil increased the activities
of the three caspases (P<0.05), whereas docetaxel only
significantly increased caspase-9 activity (P<0.05). Metformin
in combination with cisplatin and 5-fluorouracil increased
caspase-3 activity (P<0.05) compared with each drug alone.
Whereas metformin in combination with all chemotherapy drugs
increased caspase-8 and −9 activities compared with the
chemotherapy drugs alone (P<0.05). These findings indicated that
metformin may enhance the effectiveness of chemotherapeutic drugs
by increasing caspase activity.
The chemotherapy regimens ECF, DCF and CF increased
the activities of caspases-3, −8 and −9 compared with that in the
control group (P<0.05; Fig. 2D, F
and H). In addition, metformin in combination with DCF and CF
increased caspase-3, −8, and −9 activity compared with the regimens
alone (P<0.05). By contrast, metformin combined with ECF only
significant increased caspase-9 activity compared to the ECF
regimen alone (P<0.05).
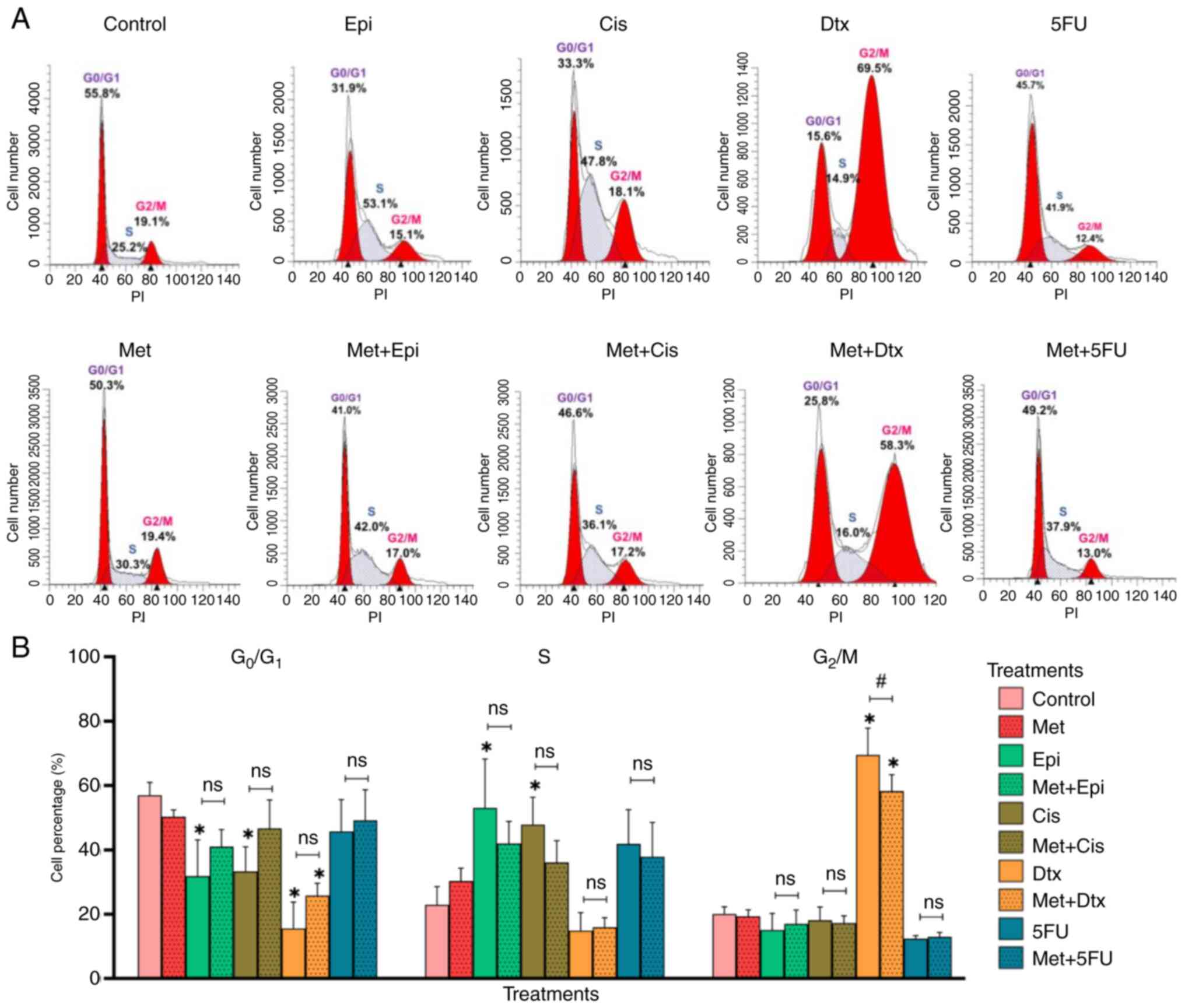
Cell cycle progression in NCI-N87 GC
cells treated with metformin alone and in combination with
chemotherapy drugs
Most NCI-N87 cells treated with metformin were
accumulated in the G0/G1 phase of the cell
cycle (50.31%) (Fig. 3A and B).
Epirubicin and cisplatin are agents non-specific to the cell cycle
phase, which can affect cells in all cell cycle phases (24). It was observed that most GC cells
accumulated in the S phase of the cell cycle when they were treated
with epirubicin (53.08%) or cisplatin (47.82%). On the other hand,
docetaxel and 5-fluorouracil are cell cycle-specific agents, which
act in one particular phase of the cell cycle (G2/M and
S, respectively) (24). Most of the
GC cells treated with docetaxel were in the G2/M phase
(69.53%), and cells treated with 5-fluorouracil accumulated in both
G0/G1 (45.73%) and S phases (41.87%).
GC cells exposed to chemotherapy regimens ECF and CF
accumulated in the G0/G1 phase (56.9 and
51.9%, respectively), whereas cells treated with DCF accumulated in
both the G0/G1 (40.2%) and G2/M
phases (31.7%) (Fig. S3).
Metformin alone decreases the
proliferation of NCI-N87 GC cells
Some genes involved in different pathways that
promote cancer progression were assessed in the present study. The
results revealed that NCI-N87 cells showed deletion of BCL2
and TP53 genes (Fig. 4A and
B), indicating that other proteins may participate in the
apoptosis process, caspase pathways and proliferation. Notably,
uncontrolled proliferation is a hallmark of cancer cells. NCI-N87
GC cells showed amplification of ERBB2 and duplication of
MYC (Fig. 4C and D), which
may favor the proliferation of these cells. Therefore, it was
considered essential to evaluate the effect of metformin on cell
proliferation. NCI-N87 GC cells were exposed to the treatments for
96 h. The results revealed that metformin alone significantly
reduced the proliferation of GC cells compared with that in the
control group (P<0.05; Fig. 4E).
Similarly, epirubicin, cisplatin, docetaxel and 5-fluorouracil
exhibited an antiproliferative effect on GC cells (P<0.05).
Notably, the combination of metformin with each chemotherapy drug
did not significantly affect cell proliferation compared with the
chemotherapy drugs alone. Similar effects were observed in response
to chemotherapy regimens. Treatments with ECF, DCF and CF
significantly reduced cell proliferation compared with that in the
control group (P<0.05), whereas no differences were observed
when metformin was combined with each chemotherapy regimen
(Fig. 4F).
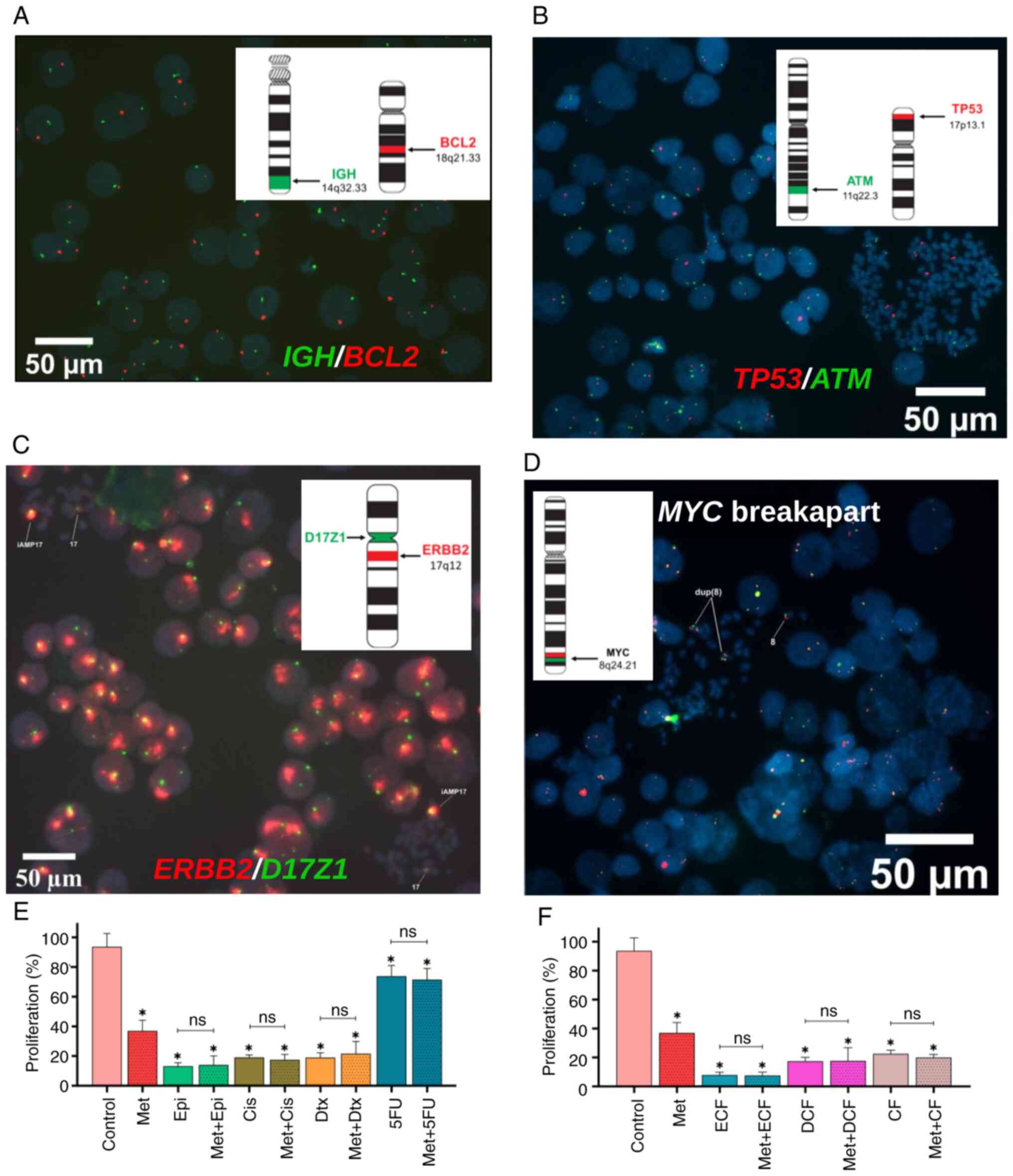 | Figure 4.Effect of metformin in combination
with chemotherapy on proliferation. (A) FISH with IGH/BCL2
probe showed a deletion of the BCL2 gene, (B) FISH with TP53/ATM
disclosed a deletion of the TP53 gene, (C) FISH with
ERBB2/D17Z1 probe showed an amplification of the
ERBB2 gene in all cells and (D) FISH with MYC breakapart probe
revealed a gain of this gene. Proliferation of cells treated
with metformin in combination with (E) each chemotherapeutic drug
and (F) chemotherapy regimen. Data are presented as the mean ± SD
from three independent experiments performed in triplicate.
*P<0.05 vs. control. FISH, Fluorescence in situ
hybridization; Met, metformin; Epi, epirubicin; Cis, cisplatin;
Dtx, docetaxel; 5FU, 5-fluorouracil; ECF, epirubicin + cisplatin +
5-fluorouracil; DCF, docetaxel + cisplatin + 5-fluorouracil; CF,
cisplatin + 5-fluorouracil; ns, not significant. |
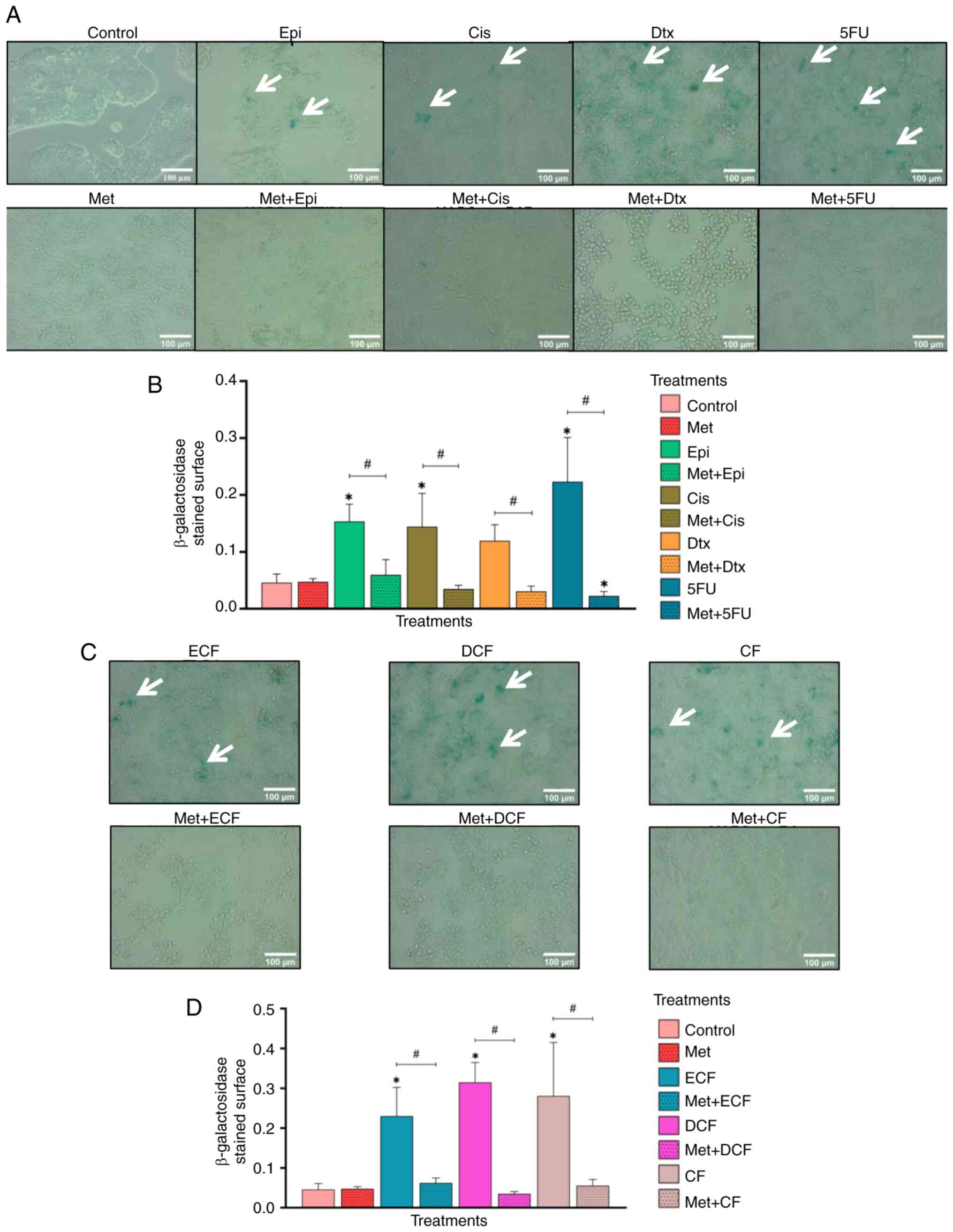
Metformin alone does not induce
senescence and counteracts the effect of chemotherapy-induced
senescence in NCI-N87 GC cells
The present study demonstrated that metformin did
not induce senescence; however, epirubicin, cisplatin and
5-fluorouracil alone significantly induced senescence compared with
that in the control group (P<0.05; Fig. 5A and B), as did the chemotherapy
regimens ECF, DCF and CF (P<0.05; Fig. 5C and D). Conversely, combining
metformin with each chemotherapeutic drug or regimen significantly
decreased cellular senescence (P<0.05).
Metformin alone and in combination
with chemotherapy decreases the clonogenic capacity of NCI-N87 GC
cells
The present study demonstrated that metformin alone
resulted in a significantly reduced number of GC cell colonies
(P<0.05; Fig. 6A and B). In
addition, the four chemotherapy drugs had the same effect as
metformin (P<0.05). Notably, no significant differences were
detected when metformin was combined with any of the chemotherapy
drugs, in comparison to the chemotherapy drugs alone. No colonies
were observed after GC cells were treated with each chemotherapy
regimen alone or when combined with metformin (data not shown).
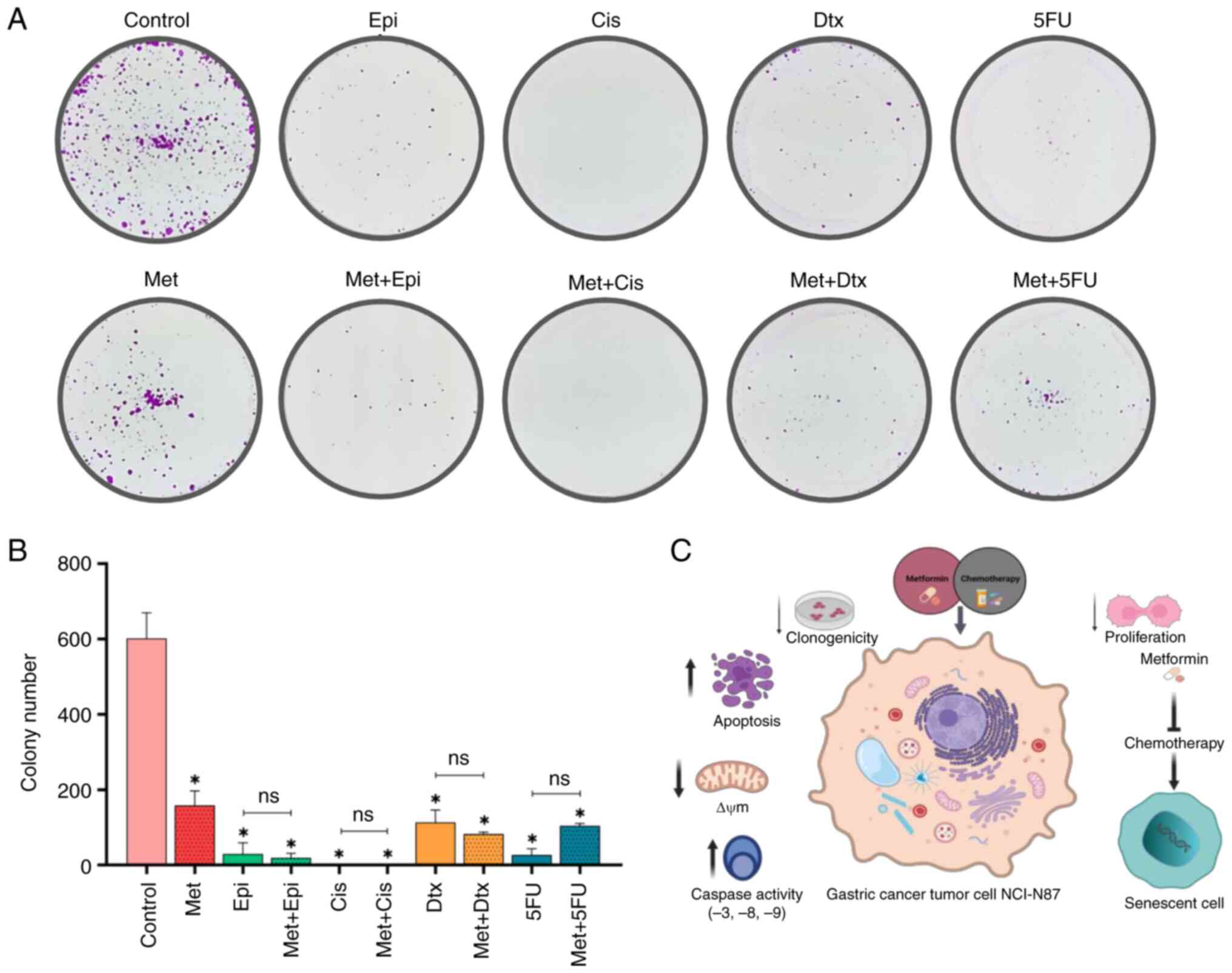 | Figure 6.Effect of Met in combination with
chemotherapy on clonogenic capacity. (A) Surviving cells after 15
days. (B) Number of colonies of surviving cells. (C) Integrative
figure on the effect of metformin in combination with chemotherapy
on NCI-N87 gastric cancer cells. Data are presented as the mean ±
SD from three independent experiments performed in triplicate.
*P<0.05 vs. control;. Met, metformin; Epi, epirubicin; Cis,
cisplatin; Dtx, docetaxel; 5FU, 5-fluorouracil; ECF, epirubicin +
cisplatin + 5-fluorouracil; DCF, docetaxel + cisplatin +
5-fluorouracil; CF, cisplatin + 5-fluorouracil; ns, not
significant; ΔΨm, mitochondrial membrane potential. |
Discussion
Even though the incidence and mortality of GC have
decreased in recent years (1), the
prognosis of patients remains unfavorable due to chemoresistance.
Developing new strategies to improve GC treatment is crucial, and
metformin has attracted attention in the last few years as an
antitumor agent (7). To the best of
our knowledge, the present study was the first to demonstrate that
metformin, in combination with chemotherapy regimens, induced cell
death through increasing apoptosis accompanied by caspase activity
and loss of ΔΨm. Furthermore, metformin significantly counteracted
senescence induced by chemotherapy (Fig. 6C).
Apoptosis is considered the main mechanism of
chemotherapy-induced cell death (25). This type of cell death is
characterized by cell shrinkage, chromatin condensation, membrane
budding, phosphatidylserine externalization and caspase activity
(26). The present study revealed
that NCI-N87 cells exhibited TP53 deletion. Deletions and
variants in this gene have been reported in ~50% of different types
of tumors, including breast, lung and ovarian cancer (27). In addition, deletion of the
BCL2 gene was detected in NCI-N87 cells, which is
interesting because this protein is anti-apoptotic and is generally
overexpressed in tumor cells as a mechanism of cell death
resistance (28). However, it is
important to note that the present results indicated that
ERBB2 and MYC were amplified in NCI-N87 cells,
suggesting that these cells have sustained proliferation, as has
been observed in patients with GC (29).
The present results showed that metformin alone had
a significant apoptotic effect on NCI-N87 GC cells. The same effect
has been observed in previous studies; for example, treatment with
metformin alone has been shown to significantly increase the
apoptosis of GC cell lines (SGC7901 and BGC-823), and this effect
was revealed to be correlated with inhibition of the HIF1α/PKM2
signaling pathway (15). The
primary mechanism of action of metformin is through AMPK
activation, resulting in mTOR inhibition. This effect has been
demonstrated in three GC cell lines (MKN28, SGC-7901 and BGC-823)
and confirmed in a xenograft mouse model. In addition, metformin
has been reported to decrease survivin expression (an
anti-apoptotic protein) and increase apoptotic cells (18).
Additionally, AMPK activation and mTOR inhibition
have been confirmed in AGS GC cells (16). Although the present study did not
evaluate AMPK and mTOR, the present data are consistent with
previous studies that support the idea that metformin induces
apoptosis in GC models (15,16,18).
Notably, one of the limitations of the present study is that the
effects of metformin on non-tumor cells were not detected. However,
it has been demonstrated in previous studies that metformin does
not induce apoptosis in a human gastric epithelial cell line
(GES-1) (18). Another study also
demonstrated that metformin does not affect the viability of
various non-tumor cells (normal human colon CCD 841 CoN cells,
embryonic lung HEL 299 cells and 293 cells) (16).
The present study observed that metformin increased
the apoptotic effect of 5-fluorouracil on NCI-N87 cells. A similar
result was reported when metformin was combined with cisplatin,
adriamycin or paclitaxel in AGS CG cells (19), and metformin combined with
oxaliplatin in GC cells (SGC7901 and SNU-16) (20). Thus, this evidence suggests that
metformin potentiates the apoptotic effect of some chemotherapy
drugs in GC. Only one previous study combined metformin with more
than one chemotherapy drug; metformin was shown to enhance tumor
reduction when combined with cisplatin and rapamycin in mice, and
this effect was revealed to be dependent on AMPK activation and
mTOR inhibition (17). Notably, to
the best of our knowledge, the present study is the first to assess
the combination of metformin with chemotherapy regimens in an in
vitro GC study model. The present results indicated that
metformin can enhance the apoptotic effect of chemotherapy regimens
ECF, DCF and CF, which are currently used for the treatment of
patients with GC. These results may encourage future in vivo
studies and clinical trials to determine the antitumor effect of
metformin in the treatment of GC.
ΔΨm is a reflection of mitochondrial functional
status (30), and a higher ΔΨm has
been shown to be correlated with increased proliferation rate and
tumorigenicity in 47DT human breast cancer cells (31). It has also been reported that cells
with high ΔΨm are resistant to apoptotic inducers (32). In the present study, it was
demonstrated that metformin alone induced the loss of ΔΨm in
NCI-N87 GC cells. This effect was consistent with a previous report
where metformin decreased ΔΨm in AGS cells, and increased reactive
oxygen species (ROS) levels and cytochrome c (16). These data indicated that loss of ΔΨm
is a key process in metformin-induced apoptosis. The four
chemotherapy drugs that were used in the present study induced loss
of ΔΨm in NCI-N87 cells. Moreover, metformin in combination with
chemotherapy drugs and DCF and CF regimens decreased ΔΨm.
Therefore, metformin in combination with chemotherapy could
potentiate this effect, confirming the results observed in
apoptosis.
Caspase activation indicates the beginning of
apoptosis (26). It was
hypothesized that metformin-induced apoptosis depends on caspase
activity, as treatments with this drug resulted in a significant
increase in caspase activity. Metformin alone significantly
increased caspase activity, mainly that of caspases-3, −8 and −9.
Similarly, in previous reports, metformin has been shown to
increase caspase-3/7 activity in MKN-28, SGC-7901, BGC-823 and AGS
GC cell lines (16,18). It has been proposed that
metformin-induced apoptosis is via the intrinsic pathway, since
increased caspase-9 activity has been shown to be correlated with
loss of ΔΨm, ROS levels and increased cytochrome c in AGS GC
cells (16). However, the present
findings suggested that metformin-induced apoptosis occurs by both
extrinsic and intrinsic pathways since it activated both initiator
caspases (caspase-8 and −9 for extrinsic and intrinsic pathways,
respectively), which indicated that metformin-induced apoptosis
could also be activated by the extrinsic pathway. This reinforces
the apoptotic effect that metformin has on tumor cells. The fact
that metformin can activate both apoptosis pathways is of
importance since this mechanism would ensure that the cell will
enter the apoptosis process through either of the two pathways,
inhibiting resistance to cell death.
As aforementioned, only a few studies have
investigated the effect of metformin in combination with
chemotherapy. The present study observed a strong tendency of
metformin to increase the caspase activity in combination with
chemotherapy drugs and chemotherapy regimens. A recent study
reported that metformin combined with oxaliplatin increased
caspase-3 activity in SGC7901 and SNU-16 GC cell lines (20). Taken together, these findings
indicated that in the process of apoptosis, metformin may help to
enhance the apoptotic effect of both the individual drugs and the
three chemotherapy regimens by increasing the loss of ΔΨm, and the
activation of initiator and effector caspases. However, it is
essential to continue conducting studies to evaluate the
participation of proapoptotic and anti-apoptotic proteins.
Tumor cells are characterized by uncontrolled
proliferation due to the evasion of growth suppressors (33). The present study evaluated the
progression of the cell cycle and proliferation. When cells were
exposed to metformin, they accumulated in the
G0/G1 phase, and proliferation was
significantly decreased. These findings were similar to those
reported in the MKN74 GC cell line, where most cells also
accumulated in the G0/G1 phase. In addition,
decreased expression of cyclin D1 and CDK4/6 was observed, as was
decreased Rb phosphorylation; these are critical markers in the
transition from G0/G1 to S phase (14). Other studies have also observed that
GC cells accumulate in G0/G1 when exposed to
metformin (15,17). Therefore, it may be inferred that
metformin could induce cell cycle arrest in the
G0/G1 phase in GC cells. To the best of our
knowledge, the effect of metformin on cell cycle progression and
proliferation in combination with chemotherapy in GC has not yet
been evaluated. The present results showed that metformin does not
modify the cell cycle phase in which the individual chemotherapy
drugs act, nor were there any changes in the chemotherapy regimens;
and no differences were detected regarding cell proliferation.
Cellular senescence is a state that can be triggered
by stress or developmental signals, and is characterized by growth
arrest, active metabolism, resistance to cell death and secretion
of extracellular factors (34). The
senescence-associated secretory phenotype (SASP) acts dually in
cancer progression. On the one hand, it has an anti-neoplastic
effect by recruiting immune system cells to premalignant lesions
and promoting the repair of damaged tissues. By contrast, it has a
pro-neoplastic effect that promotes proliferation, angiogenesis and
inflammation due to the secretion of proinflammatory factors, such
as IL-6, IL-8, MMPs and VEGF (35).
Chemotherapy has been reported to induce the SASP, which has been
shown to be correlated with chemoresistance; factors secreted by
senescent cells can influence neighboring cells and promote tumor
progression (36).
The present study investigated the effect of
metformin and chemotherapy on senescence using the biomarker
β-galactosidase. To the best of our knowledge, no studies have
evaluated senescence in GC in vitro or in vivo, in
response to a combination of metformin with conventional
chemotherapy. No senescent cells were observed in response to
treatment with metformin; however, as expected, chemotherapy
induced senescence in NCI-N87 cells. A previous study suggested
that the SASP could participate in chemoresistance, reducing
therapy efficacy (37). Metformin
could be proposed as an agent to suppress SASP as it is able to
block the master transcription factor NF-κB, which is required for
the expression of numerous proinflammatory genes expressed in
senescent cells (38). In addition,
metformin decreased the mRNA expression of proinflammatory
cytokines, such as CXCL5, IL-1B, IL-6 and IL-8 in fibroblasts and
macrophages (38). Taken together,
it may be suggested that metformin, besides not inducing
senescence, can counteract the SASP induced by chemotherapy and
consequently reduce one of the main obstacles in cancer treatment,
chemoresistance.
Not all cells enter a cell death process after
exposure to chemotherapeutic treatments; some become resistant or
senescent. As aforementioned, surviving cells may contribute to
tumor progression (39). Therefore,
the present study evaluated the effect of the cells that survived
after treatment. Although metformin is not an antitumor drug, it
significantly decreased the clonogenic capacity of NCI-N87 GC
cells. Similarly, this effect has been reported in N87 and MKN45 GC
cells when exposed only to metformin (17). This indicates that metformin alone
may decrease the clonogenic capacity of GC cells. The present study
showed that metformin, when combined with 5-fluorouracil tended to
increase the number of colonies, although this finding was not
significant. A previous study reported that combining metformin
with docetaxel or 5-fluorouracil can decrease the clonogenic
capacity of AGS GC cells (21).
These discrepancies could be due to the cell line evaluated.
Although both cell lines are GC, each has its own genetic
characteristics and, therefore, differences in gene expression. In
addition, it is important to mention that in GC treatments,
5-fluorouracil is not administered as a monotherapy; it is combined
with other antineoplastic drugs (40). Hence, it would be unlikely for the
combination of metformin with 5-fluorouracil to be administered to
patients as treatment for GC. Both the chemotherapy regimens and
the combinations with metformin did not allow the formation of
colonies of NCI-N87 GC cells. Therefore, these findings suggested
that metformin does not interfere with the decrease in clonogenic
capacity caused by chemotherapy.
The use of metformin in GC has not been sufficiently
studied because clinical investigations are mainly observational
studies in patients with DM2. There are studies that have evaluated
the effect of metformin and the risk of developing GC, but the
results are controversial. Different studies have detected no
impact on the risk of developing GC of patients with DM2 when
comparing those taking metformin with another type of treatment,
such as sulfonylurea and insulin (41,42).
However, other studies have reported that metformin reduces the
risk of GC in patients who are prescribed metformin (43–45).
These discrepancies may be related to biases and study design. In
addition, previous studies have investigated the effect of
metformin on survival; metformin has been reported to promote
survival and decrease recurrence in patients with DM2 and GC after
gastrectomy (46,47). It has also been reported that
metformin improves overall survival in patients with DM2 and GC
(48). However, another study
reported that metformin had no impact on the survival of patients
with GC and DM2 (49). Shuai et
al (50) performed a systematic
review and a meta-analysis to evaluate the effect of metformin on
GC in patients with DM2 and revealed that the reduction in the
incidence of GC was 21% (HR 0.790; 95% CI 0.624–1.001) (50). In order to provide more information
about the effect of metformin on this type of cancer, prospective
studies of patients with DM2 and clinical trials in patients with
GC without DM2 are required.
The evidence from previous observational studies in
patients indicates that metformin may reduce the risk of developing
cancer (51), promote survival and
could act as an adjuvant agent (52). In addition, in vitro and
in vivo studies have reported that metformin alone, and in
combination with chemotherapy may inhibit cellular growth and
proliferation, suppress epithelial-mesenchymal transition, target
stem cells, increase apoptosis and reduce tumor size (7,53).
Important considerations must be made in preclinical
and clinical studies. The dose of metformin (10 mM) used in the
present study was the median inhibitory concentration
(IC50), which is one of the parameters established in
vitro studies (54,55). The IC50 is a measure
commonly used in in vitro studies to evaluate the potency of
a compound in inhibiting a certain biological response. It is also
important to mention that the main objective of the present study
was to investigate the molecular effects of the apoptosis process,
and the desired effect was observed at this concentration.
Currently, the doses of metformin used in a number of oncological
trials have been shown to be effective for glucose control
(56–58). Establishing the appropriate dose of
metformin for use in cancer is necessary to safeguard patient
safety and well-being.
In conclusion, the results of the present study
indicated that metformin could be used as an adjuvant agent, since
it could enhance the efficacy of chemotherapy regimens, increase
apoptosis of tumor cells and counteract senescence induced by
chemotherapy treatment, which may prevent or combat the
chemoresistance that is associated with an unfavorable prognosis in
this type of cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
KCVI, JRCL, BGOT and LAPM performed the in
vitro experiments. RMGA and JRGG performed the FISH
experiments. KCVI, JYSL and PCOL analyzed data and wrote the
manuscript. JYSL, TDPR and PCOL designed the study. JYSL and PCOL
performed the final review and editing. PCOL supervised the study.
PCOL, JYSL and KCVI confirm the authenticity of all raw data. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
This study was approved by the National Scientific
Research Committee of Mexican Social Security Institute (approval
number: R-2019-785-050; Guadalajara, Mexico).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GC
|
gastric cancer
|
|
FISH
|
fluorescence in situ hybridization
|
|
ΔΨm
|
mitochondrial membrane potential
|
|
DM2
|
diabetes mellitus type 2
|
References
|
1
|
Ferlay J, Colombet M, Soerjomataram I,
Parkin DM, Piñeros M, Znaor A and Bray F: Cancer statistics for the
year 2020: An overview. Int J Cancer. Apr 5–2021.(Epub ahead of
print). View Article : Google Scholar
|
|
2
|
Balakrishnan M, George R, Sharma A and
Graham DY: Changing trends in stomach cancer throughout the world.
Curr Gastroenterol Rep. 19:362017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Smyth EC, Verheij M, Allum W, Cunningham
D, Cervantes A and Arnold D; ESMO Guidelines Committee, : Gastric
cancer: ESMO clinical practice guidelines for diagnosis, treatment
and follow-up. Ann Oncol. 27 (Suppl 5):v38–v49. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chiurillo MA: Role of the Wnt/β-catenin
pathway in gastric cancer: An in-depth literature review. World J
Exp Med. 5:84–102. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rawla P and Barsouk A: Epidemiology of
gastric cancer: Global trends, risk factors and prevention. Prz
Gastroenterol. 14:26–38. 2019.PubMed/NCBI
|
|
6
|
Rena G, Hardie DG and Pearson ER: The
mechanisms of action of metformin. Diabetologia. 60:1577–1585.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Courtois S, Lehours P and Bessède E: The
therapeutic potential of metformin in gastric cancer. Gastric
Cancer. 22:653–662. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scarpello JHB and Howlett HCS: Metformin
therapy and clinical uses. Diab Vasc Dis Res. 5:157–167. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Daugan M, Dufaÿ Wojcicki A, d'Hayer B and
Boudy V: Metformin: An anti-diabetic drug to fight cancer.
Pharmacol Res. 113:675–685. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hua Y, Zheng Y, Yao Y, Jia R, Ge S and
Zhuang A: Metformin and cancer hallmarks: Shedding new lights on
therapeutic repurposing. J Transl Med. 21:4032023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Graham GG, Punt J, Arora M, Day RO, Doogue
MP, Duong JK, Furlong TJ, Greenfield JR, Greenup LC, Kirkpatrick
CM, et al: Clinical pharmacokinetics of metformin. Clin
Pharmacokinet. 50:81–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Morales DR and Morris AD: Metformin in
cancer treatment and prevention. Annu Rev Med. 66:17–29. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Viollet B, Guigas B, Sanz Garcia N,
Leclerc J, Foretz M and Andreelli F: Cellular and molecular
mechanisms of metformin: An overview. Clin Sci (Lond). 122:253–270.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kato K, Gong J, Iwama H, Kitanaka A, Tani
J, Miyoshi H, Nomura K, Mimura S, Kobayashi M, Aritomo Y, et al:
The antidiabetic drug metformin inhibits gastric cancer cell
proliferation in vitro and in vivo. Mol Cancer Ther. 11:549–560.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen G, Feng W, Zhang S, Bian K, Yang Y,
Fang C, Chen M, Yang J and Zou X: Metformin inhibits gastric cancer
via the inhibition of HIF1α/PKM2 signaling. Am J Cancer Res.
5:1423–1434. 2015.PubMed/NCBI
|
|
16
|
Lu CC, Chiang JH, Tsai FJ, Hsu YM, Juan
YN, Yang JS and Chiu HY: Metformin triggers the intrinsic apoptotic
response in human AGS gastric adenocarcinoma cells by activating
AMPK and suppressing mTOR/AKT signaling. Int J Oncol. 54:1271–1281.
2019.PubMed/NCBI
|
|
17
|
Yu G, Fang W, Xia T, Chen Y, Gao Y, Jiao
X, Huang S, Wang J, Li Z and Xie K: Metformin potentiates rapamycin
and cisplatin in gastric cancer in mice. Oncotarget. 6:12748–12762.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Han G, Gong H, Wang Y, Guo S and Liu K:
AMPK/mTOR-mediated inhibition of survivin partly contributes to
metformin-induced apoptosis in human gastric cancer cell. Cancer
Biol Ther. 16:77–87. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu X: Effect of metformin combined with
chemotherapeutic agents on gastric cancer cell line AGS. Pak J
Pharm Sci. 30:1833–1836. 2017.PubMed/NCBI
|
|
20
|
Zhu M, Wang J and Zhou R: Combination of
metformin and oxaliplatin inhibits gastric cancer cell
proliferation and induces apoptosis. Acta Biochim Pol. 69:321–326.
2022.PubMed/NCBI
|
|
21
|
Fatehi-Agdam M, Vatankhah MA, Panahizadeh
R, Jeddi F and Najafzadeh N: Efficacy of metformin and
chemotherapeutic agents on the inhibition of colony formation and
Shh/Gli1 pathway: Metformin/docetaxel versus
metformin/5-fluorouracil. Drug Res (Stuttg). 71:17–25. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zarei E, Sefidi-Heris Y and Saadat I:
Synergistic effects of metformin and curcumin on cytotoxicity of
chemotherapy drugs using a gastric cancer cell line model. EXCLI J.
20:1488–1498. 2021.PubMed/NCBI
|
|
23
|
Nomenclature ISCoHC: ISCN 2020: An
International System for Human Cytogenomic Nomenclature. S. Karger
AG; London: 2020
|
|
24
|
Sun Y, Liu Y, Ma X and Hu H: The influence
of cell cycle regulation on chemotherapy. Int J Mol Sci.
22:69232021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ricci MS and Zong WX: Chemotherapeutic
approaches for targeting cell death pathways. Oncologist.
11:342–357. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duffy MJ, Synnott NC, McGowan PM, Crown J,
O'Connor D and Gallagher WM: p53 as a target for the treatment of
cancer. Cancer Treat Rev. 40:1153–1160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Strasser A and Vaux DL: Cell death in the
origin and treatment of cancer. Mol Cell. 78:1045–1054. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nevisi F, Yaghmaie M, Pashaiefar H,
Alimoghaddam K, Iravani M, Javadi G and Ghavamzadeh A: Correlation
of HER2, MDM2, c-MYC, c-MET, and TP53 copy number alterations in
circulating tumor cells with tissue in gastric cancer patients: A
pilot study. Iran Biomed J. 24:47–53. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang BB, Wang DG, Guo FF and Xuan C:
Mitochondrial membrane potential and reactive oxygen species in
cancer stem cells. Fam Cancer. 14:19–23. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu M, Shi Y, Wei X, Yang Y, Zhou Y, Hao X,
Zhang N and Niu R: Depletion of mitochondrial DNA by ethidium
bromide treatment inhibits the proliferation and tumorigenesis of
T47D human breast cancer cells. Toxicol Lett. 170:83–93. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pietilä M, Lehtonen S, Närhi M, Hassinen
IE, Leskelä HV, Aranko K, Nordström K, Vepsäläinen A and Lehenkari
P: Mitochondrial function determines the viability and osteogenic
potency of human mesenchymal stem cells. Tissue Eng Part C Methods.
16:435–445. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lopes-Paciencia S, Saint-Germain E, Rowell
MC, Ruiz AF, Kalegari P and Ferbeyre G: The senescence-associated
secretory phenotype and its regulation. Cytokine. 117:15–22. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Campisi J: Aging, cellular senescence, and
cancer. Annu Rev Physiol. 75:685–705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gilbert LA and Hemann MT: DNA
damage-mediated induction of a chemoresistant niche. Cell.
143:355–366. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chambers CR, Ritchie S, Pereira BA and
Timpson P: Overcoming the senescence-associated secretory phenotype
(SASP): A complex mechanism of resistance in the treatment of
cancer. Mol Oncol. 15:3242–3255. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Moiseeva O, Deschênes-Simard X, St-Germain
E, Igelmann S, Huot G, Cadar AE, Bourdeau V, Pollak MN and Ferbeyre
G: Metformin inhibits the senescence-associated secretory phenotype
by interfering with IKK/NF-κB activation. Aging Cell. 12:489–498.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee M and Lee JS: Exploiting tumor cell
senescence in anticancer therapy. BMB Rep. 47:51–59. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ajani JA, D'Amico TA, Bentrem DJ, Chao J,
Cooke D, Corvera C, Das P, Enzinger PC, Enzler T, Fanta P, et al:
Gastric cancer, version 2.2022, NCCN clinical practice guidelines
in oncology. J Natl Compr Canc Netw. 20:167–192. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
de Jong RG, Burden AM, de Kort S, van
Herk-Sukel MP, Vissers PA, Janssen PK, Haak HR, Masclee AA, de
Vries F and Janssen-Heijnen ML: No decreased risk of
gastrointestinal cancers in users of metformin in the netherlands;
A time-varying analysis of metformin exposure. Cancer Prev Res
(Phila). 10:290–297. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zheng J, Xie SH, Santoni G and Lagergren
J: Metformin use and risk of gastric adenocarcinoma in a Swedish
population-based cohort study. Br J Cancer. 121:877–882. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kim YI, Kim SY, Cho SJ, Park JH, Choi IJ,
Lee YJ, Lee EK, Kook MC, Kim CG, Ryu KW and Kim YW: Long-term
metformin use reduces gastric cancer risk in type 2 diabetics
without insulin treatment: A nationwide cohort study. Aliment
Pharmacol Ther. 39:854–863. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tseng CH: Metformin reduces gastric cancer
risk in patients with type 2 diabetes mellitus. Aging (Albany NY).
8:1636–1649. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dulskas A, Patasius A, Kaceniene A,
Linkeviciute-Ulinskiene D, Zabuliene L and Smailyte G: A cohort
study of antihyperglycemic medication exposure and gastric cancer
risk. J Clin Med. 9:4352020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee CK, Jung M, Jung I, Heo SJ, Jeong YH,
An JY, Kim HI, Cheong JH, Hyung WJ, Noh SH, et al: Cumulative
metformin use and its impact on survival in gastric cancer patients
after gastrectomy. Ann Surg. 263:96–102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Seo HS, Jung YJ, Kim JH, Lee HH and Park
CH: The effect of metformin on prognosis in patients with locally
advanced gastric cancer associated with type 2 diabetes mellitus.
Am J Clin Oncol. 42:909–917. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lacroix O, Couttenier A, Vaes E, Cardwell
CR, De Schutter H and Robert A: Impact of metformin on gastric
adenocarcinoma survival: A Belgian population based study. Cancer
Epidemiol. 53:149–155. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Baglia ML, Cui Y, Zheng T, Yang G, Li H,
You M, Xu L, Murff H, Gao YT, Zheng W, et al: Diabetes medication
use in association with survival among patients of breast,
colorectal, lung, or gastric cancer. Cancer Res Treat. 51:538–546.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shuai Y, Li C and Zhou X: The effect of
metformin on gastric cancer in patients with type 2 diabetes: A
systematic review and meta-analysis. Clin Transl Oncol.
22:1580–1590. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Evans JMM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang J, Wen L, Zhou Q, He K and Teng L:
Preventative and therapeutic effects of metformin in gastric
cancer: A new contribution of an old friend. Cancer Manag Res.
12:8545–8554. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cunha Júnior AD, Bragagnoli AC, Costa FO
and Carvalheira JBC: Repurposing metformin for the treatment of
gastrointestinal cancer. World J Gastroenterol. 27:1883–1904. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sebaugh JL: Guidelines for accurate
EC50/IC50 estimation. Pharm Stat. 10:128–134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Brooks EA, Galarza S, Gencoglu MF,
Cornelison RC, Munson JM and Peyton SR: Applicability of drug
response metrics for cancer studies using biomaterials. Philos
Trans R Soc Lond B Biol Sci. 374:201802262019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Drzewoski J and Hanefeld M: The current
and potential therapeutic use of metformin-the good old drug.
Pharmaceuticals (Basel). 14:1222021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chae YK, Arya A, Malecek MK, Shin DS,
Carneiro B, Chandra S, Kaplan J, Kalyan A, Altman JK, Platanias L
and Giles F: Repurposing metformin for cancer treatment: Current
clinical studies. Oncotarget. 7:40767–40780. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Saraei P, Asadi I, Kakar MA and Moradi-Kor
N: The beneficial effects of metformin on cancer prevention and
therapy: A comprehensive review of recent advances. Cancer Manag
Res. 11:3295–3313. 2019. View Article : Google Scholar : PubMed/NCBI
|