Introduction
Renal cell carcinoma (RCC), a malignant tumor of the
urinary system, is one of the 15 most common cancers worldwide
(1). Nearly 400,000 people are
affected by RCC, resulting in >100,000 deaths each year globally
(2). At present, RCC is the second
most common genitourinary tumor in China, second only to bladder
cancer. The incidence of RCC has been on the rise annually and is
notably higher in males than that in females (3,4). Early
diagnosis of RCC is closely associated with improved survival rates
(5). Therefore, it is the focus of
RCC research to explore the mechanisms and landmark targets of
RCC.
P53, a tumor suppressor protein, can regulate the
cell cycle, DNA replication and cell division (6). Normally, when cells undergo
uncontrolled division and proliferation, the P53 protein is
activated, which in turn induces P21 expression. This process
results in cell cycle arrest and inhibition of cell proliferation;
however, when cell damage cannot be repaired, P53 triggers
apoptosis-related genes (such as Bcl-2-associated X-protein) and
programmed cell death (6–8). In addition, when P53 proteins are
mutated or aggregated, their functions are lost, leading to
abnormal cell proliferation and eventually inducing the occurrence
of cancer (9). As a key tumor
suppressor, the P53 protein is also regulated by several genes.
Specifically, a large number of genes modulate the occurrence and
development of RCC by regulating the P53 protein. For instance,
tripartite motif 47 can promote the malignant progression of RCC by
mediating the ubiquitination and degradation of P53 protein
(10), and RNA-binding motif 4
inhibits the proliferation of RCC cells by enhancing the stability
of P53 mRNA (11).
Triggering receptor expressed on myeloid cells-2
(TREM2), a transmembrane receptor protein of the immunoglobulin
superfamily, is primarily expressed in microglia and myeloid cells
(12). TREM2 has several biological
functions, including cell proliferation, cell survival,
inflammatory regulation and phagocytosis (13). Disease research initially focused on
the association between TREM2 and Alzheimer's disease. In the
central nervous system, TREM2 promotes microglial transition to
disease-associated microglia by affecting lipid metabolism
(cholesterol, myelin and phospholipid metabolism), which may
contribute to the occurrence of Alzheimer's disease. Therefore,
TREM2 is considered a promising therapeutic target for the
treatment of the disease (14). In
addition, TREM2 has been reported to serve an important role in
cancer. For example, Katzenelenbogen et al (15) reported the novel Arg1+
Trem2+ regulatory myeloid cells, which can mediate
immunosuppression. The study reported that the elimination of these
cells activated the natural killer cells and inhibited tumor
growth. Furthermore, TREM2+ monocyte-derived macrophages
have been reported to inhibit the accumulation of natural killer
cells in tumor tissues and reduce oncolytic activity, thereby
promoting the malignant development of lung cancer (16). Moreover, although a study has
suggested that TREM2 can promote the occurrence and development of
RCC through the phosphatase and tensin homolog-phosphatidylinositol
3-kinase/protein kinase B signaling (17), whether there are additional
mechanisms remains to be further explored.
The present study aimed to investigate the role of
TREM2 in the progression of RCC and to explore its underlying
mechanisms, particularly its interaction with the P53 signaling
pathway, using methods such as reverse transcription-quantitative
PCR (RT-qPCR), cell proliferation, migration and invasion assays,
and western blotting, with the goal of providing novel therapeutic
targets for RCC.
Materials and methods
Tissue samples
RCC tumor and adjacent normal tissues were collected
from patients with RCC (n=10) admitted to Wujin Hospital Affiliated
with Jiangsu University (Changzhou, China) between January 2022 and
June 2022. The tissue samples were stored at −80°C prior to use.
Patients were divided into the high TREM2 expression group or the
low TREM2 expression group based on the median expression of TREM2.
The clinicopathological data of the patients in each group are
presented in Table I. The TNM
staging system used in this study was the American Joint Committee
on Cancer 8th edition system (18).
The present study was approved by the Ethics Committee of Wujin
Hospital Affiliated with Jiangsu University (approval no.
2022-SR-092), and written informed consent was obtained from all
patients according to the requirements of The Declaration of
Helsinki (19).
 | Table I.Clinicopathological data of the
patients. |
Table I.
Clinicopathological data of the
patients.
| Variable | Total (n=10) | High expression of
TREM2 (n=5) | Low expression of
TREM2 (n=5) |
t/χ2 | P-value |
|---|
| Age, years | 59.00±10.20 | 59.00±14.75 | 59.00±4.06 | 0.000 | >0.999 |
| Sex |
|
|
| 0.400 | >0.999 |
|
Male | 5 (50) | 2 (40) | 3 (60) |
|
|
|
Female | 5 (50) | 3 (60) | 2 (40) |
|
|
| BMI,
kg/m2 | 22.78±1.67 | 22.60±1.51 | 22.90±2.00 | −0.286 | 0.782 |
| Tumor size, cm | 4.29±2.38 | 5.48±3.00 | 3.10±0.42 | 1.759 | 0.117 |
| Tumor side |
|
|
| 0.400 | >0.999 |
|
Left | 5 (50) | 2 (40) | 3 (60) |
|
|
|
Right | 5 (50) | 3 (60) | 2 (40) |
|
|
| TNM stage |
|
|
| 1.111 | >0.999 |
| T1 | 9 (90) | 4 (80) | 5 (100) |
|
|
| T2 | 0 (0) | 0 (0) | 0 (0) |
|
|
| T3 | 0 (0) | 0 (0) | 0 (0) |
|
|
| T4 | 1 (10) | 1 (20) | 0 (0) |
|
|
| Lymph node
metastasis |
|
|
| 1.111 | >0.999 |
|
Negative | 9 (90) | 4 (80) | 5 (100) |
|
|
|
Positive | 1 (10) | 1 (20) | 0 (0) |
|
|
| Pathology type |
|
|
| 3.143 | >0.999 |
| Clear
cell | 7 (70) | 4 (80) | 3 (60) |
|
|
|
Papillary | 2 (20) | 0 (0) | 2 (40) |
|
|
|
Chromophobe | 0 (0) | 0 (0) | 0 (0) |
|
|
|
Others | 1 (10) | 1 (20) | 0 (0) |
|
|
Patients diagnosed with RCC, aged between 18 and 75
years, who provided written informed consent, and had sufficient
tissue available for analysis were included in the study. The
exclusion criteria included: Patients with other types of cancer,
those who received chemotherapy or radiotherapy prior to sample
collection, and patients with severe systemic diseases or
infections.
Cell culture
Human RCC ACHN (cat. no. SCSP-5063), human renal
tubular epithelial HK-2 (cat. no. SCSP-511) and 293T (cat. no.
SCSP-502) cell lines were purchased from the National Collection of
Authenticated Cell Cultures (Cell Bank of Type Culture Collection
of The Chinese Academy of Sciences). The 293T, ACHN and HK-2 cells
were cultured in Roswell Park Memorial Institute 1640 Medium (cat.
no. 22400071; Gibco; Thermo Fisher Scientific, Inc.) containing 10%
fetal bovine serum (cat. no. 10099158; Gibco; Thermo Fisher
Scientific, Inc.) in a thermostatic incubator (Thermo Fisher
Scientific, Inc.) at 37°C with 5% CO2 for 48 h.
Lentivirus packaging and
infection
To assess the role of RCC, lentiviral vectors were
used to modulate the expression of TREM2 in RCC cells. The
constructs for these experiments were generated by cloning the
short hairpin (sh)RNA-targeting TREM2 (sh-TREM2, sense:
5′-GATCCGAGCCTCTTGGAAGGAGAAATCTCGAGATTTCTCCTTCCAAGAGGCTCTTTTTG-3′
and anti-sense:
5′-AATTCAAAAAGAGCCTCTTGGAAGGAGAAATCTCGAGATTTCTCCTTCCAAGAGGCTCG-3′)
into a pSicoR vector, resulting in a lenti-shTREM2 construct for
knockdown. A lentivirus vector containing scrambled shRNA (sh-NC,
sense:
5′-GATCCCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTTG-3′
and anti-sense:
5′-AATTCAAAAACAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGG'3′)
was constructed as a negative control. For overexpression, the
full-length TREM2 coding sequence was incorporated into a
pCDH-CMV-MCS-EF1-copGFP vector, resulting in a lenti-TREM2
construct. A control vector lacking any insert (empty vector) was
used as a negative control. For transfection, a total of 5 µg of
each plasmid (Sangon Biotech Co., Ltd.) was used, with a 3:2:1
ratio of the lentivirus, packaging (psPAX2) and envelope (pMD2.G)
plasmids. A third-generation lentiviral packaging system was used.
The plasmids were transfected into 293T cells using
Lipofectamine™ 2000 (cat. no. 11668019;
Invitrogen™; Thermo Fisher Scientific, Inc.) at 37°C for
48 h. After transfection, the medium was replaced with fresh growth
medium to facilitate viral production. Viral particles were
harvested by collecting the medium every 8 h, subsequently
filtering it through a 0.45 µm filter (cat. no. HVLP02500;
MilliporeSigma; Merck KGaA) to remove cellular debris. The purified
viral particles were then aliquoted and stored at −80°C.
The MOI used to infect ACHN cells was 10, with a
transduction duration of 24 h. After the 24-h transduction period,
the cells were washed with PBS and incubated with fresh medium for
an additional 48 h. Following this incubation, ACHN cells were
selected using puromycin at a concentration of 2 µg/ml for 7 days
to create a stable cell line. For maintenance, the concentration of
puromycin was reduced to 1 µg/ml. After the stable cell lines
continued to be cultured for 3–7 days, total RNA was extracted, and
the knockdown or overexpression of TREM2 was detected by
RT-qPCR.
RT-qPCR
Total RNA was extracted from RCC tissues, ACHN cells
and HK-2 cells using a TRIzol™ kit (cat. no. 15596026CN;
Invitrogen; Thermo Fisher Scientific, Inc.). The extracted RNA was
reverse transcribed into complementary DNA using the
SuperScript™ III Reverse Transcriptase kit (cat. no.
18080085; Invitrogen; Thermo Fisher Scientific, Inc.) with the
following temperature protocol: 25°C for 10 min, 50°C for 50 min
and 85°C for 5 min. Quantitative PCR was performed using the
Hieff® qPCR SYBR® Green Master Mix (No Rox;
cat. no. 11201ES08; Shanghai Yeasen Biotechnology Co., Ltd.) under
the following thermocycling conditions: Initial denaturation at
95°C for 5 min, followed by 40 cycles of 95°C for 10 sec and 60°C
for 30 sec. The sequences of the primers used are listed in
Table II. GAPDH served as a
standardized control. The expression of TREM2 was quantified using
the 2−ΔΔCq method (20).
| Table II.Primer sequences. |
Table II.
Primer sequences.
| RNA | Direction | Sequence
(5′-3′) |
|---|
| TREM2 | Forward |
5′-AGGGCCCATGCCAGCGTGTGGT-3′ |
|
| Reverse |
5′-CCAGAGATCTCCAGCATC-3′ |
| GAPDH | Forward |
5′-GTCTCCTCTGACTTCAACAGCG-3′ |
|
| Reverse |
5′-ACCACCCTGTTGCTGTAGCCAA-3′ |
Western blotting
ACHN cells were lysed with radioimmunoprecipitation
assay buffer (cat. no. 89900; Thermo Fisher Scientific, Inc.) at
4°C for 30 min. Subsequently, the lysed cells were centrifuged at
12,000 × g at 4°C for 15 min using a high-speed microcentrifuge,
and the supernatant was collected. Bicinchoninic acid was used to
determine the protein concentration in the supernatant. Upon
denaturation at 100°C for 5 min, protein samples (20 µg/lane) were
subjected to 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis. The protein samples were then transferred to
polyvinylidene fluoride membranes (cat. no. IPFL00005;
MilliporeSigma; Merck KGaA) using the wet transfer method. The
membranes were blocked with 5% skim milk at room temperature for 2
h and then primary antibodies were added for incubation overnight
at 4°C. The antibodies used included β-actin (1:5,000; cat. no.
13E5; CST Biological Reagents Co., Ltd.), TREM2 (1:1,000; cat. no.
91068; CST Biological Reagents Co., Ltd.), P53 (1:2,000; cat. no.
2527; CST Biological Reagents Co., Ltd.), p-P53 (1:1,000; cat. no.
82530; CST), P21 (1:2,000; cat. no. 2947; CST Biological Reagents
Co., Ltd.) and p-P21 (1:500; cat. no. ab47300; Abcam).
Subsequently, the membranes were washed three times with
Tris-buffered saline with 0.1% Tween 20. The membranes were then
incubated with HRP-conjugated goat anti-rabbit IgG secondary
antibodies (1:5,000; cat. no. 7074; CST Biological Reagents Co.,
Ltd.) at room temperature for 2 h, followed by rinsing again with
Tris-buffered saline with Tween 20 in triplicate. Subsequently, the
Pierce™ ECL Western Blotting Substrate (cat. no. 32209;
Thermo Fisher Scientific, Inc.) was evenly dripped onto the
membrane. The proteins were developed using the ChemiDoc Imaging
System (Bio-Rad Laboratories, Inc.) and the gray values were
calculated using ImageJ software (V1.8.0; National Institutes of
Health).
Cell Counting Kit-8 (CCK-8) assay
The proliferation ability of RCC cells was assessed
by CCK-8 assay (cat. no. HY-K0301; MedChemExpress). Briefly, ACHN
cells infected with the corresponding lentivirus were seeded into
96-well plates at a density of 5×103 cells/well, with
six replicates per group. After 48 h, the cells were supplemented
with 10 µl of the CCK-8 reagent and incubated for another 2 h. The
absorbance of each well at 450 nm was detected using a microplate
reader.
5-ethynyl-2′-deoxyuridine (EdU)
incorporation assay
To further assess cell proliferation, an EdU
incorporation assay was performed. EdU, a nucleoside analog of
thymidine (21), was used to track
active DNA synthesis. Cells were incubated with 10 µM EdU for 2 h
at 37°C and then fixed with 4% paraformaldehyde at room temperature
for 15 min. After fixation, cells underwent a click reaction with
Alexa Fluor™ 488 Azide (cat. no. A10266; Invitrogen;
Thermo Fisher Scenic, Inc.) to stain the incorporated EdU according
to the manufacturer's instructions at room temperature for 30 min.
Subsequently, cells were counterstained with DAPI at room
temperature for 10 min to label all DNA, providing a control for
cell nuclei identification. Fluorescence microscopy was used to
capture images of the labeled cells. The proportion of
EdU+ cells was quantified by comparing them to the total
number of DAPI-stained nuclei.
Wound healing assay
ACHN cells cultured to 100% confluence in a 6-well
plate were subjected to starvation treatment with a serum-free
medium. After 24 h, a tip was used to scratch the cells. The
scratched cells were then washed with phosphate-buffered saline and
cultured in a serum-free medium for 24 h at 37°C. Subsequently,
wound healing of the cells was observed using an inverted
microscope and photographed at 0 and 24 h. Two black lines were
drawn on the images to facilitate calculations. The black line at 0
h represented the initial scratch or wound created in the cell
monolayer, serving as the baseline for the measurements. The black
line at 24 h illustrated the boundary of cell migration into the
scratch area after 24 h of incubation. This line was determined
based on the leading edge of the migrating cells, which is a
standard practice in scratch assays to assess cell migration and
wound closure rate (22). The
criteria for determining this line involved identifying the
foremost edge of the cells that moved closest to the original wound
edge without crossing it.
Transwell assay
The invasion assay was performed with a Transwell
chamber coated with Matrigel (BD Biosciences) at 37°C for 1 h. The
upper chamber was supplemented with ACHN cells (5×104)
resuspended in serum-free medium, whilst the lower chamber was
covered with Dulbecco's Modified Eagle Medium containing 10% fetal
bovine serum (cat. no. 10099158; Gibco; Thermo Fisher Scientific,
Inc.). The chambers were cultured in an incubator at 37°C and 5%
CO2 for 24 h, during which time a certain number of
cells attached to the upper chamber passed through the membrane to
the lower chamber. Subsequently, the chambers were rinsed with
phosphate-buffered saline, fixed with a 4% formaldehyde solution at
room temperature for 15 min, and stained with crystal violet
(0.01%) at room temperature for 15 min. The cells that passed
through the membrane into the lower chamber were observed using a
light microscope (BX53; Olympus Corporation) and images were
captured.
Statistical analysis
These aforementioned experiments were independently
repeated three times. All data were analyzed using SPSS 23.0 (IBM
Corp.) and GraphPad Prism V.7.01 (Dotmatics). Continuous data from
biological replicates are expressed as mean ± standard deviation.
The paired or unpaired Student's t-test was used for comparisons
between two groups, and one-way analysis of variance followed
Tukey's post hoc test was used to compare differences among
multiple groups. Categorical data are expressed as n (%) and
Fisher's exact test was used for analysis. P<0.05 was considered
to indicate a statistically significant difference.
Results
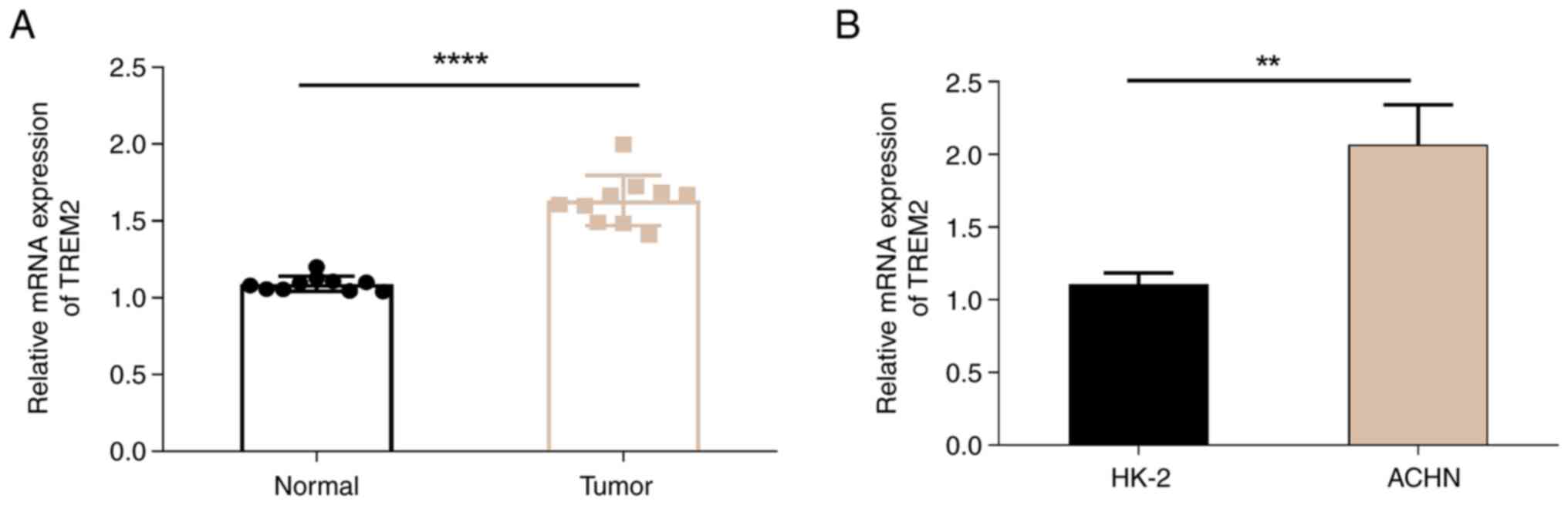
TREM2 is highly expressed in patients
with RCC
To assess the effect of TREM2 expression on RCC, 10
pairs of RCC and adjacent normal tissues were obtained. RNA was
extracted from the tissue samples, and the expression level of
TREM2 was determined by reverse transcription-quantitative PCR. The
results demonstrated that the mRNA expression level of TREM2 was
significantly increased in tumor tissue compared with that in
normal tissues (Fig. 1A). To
further confirm the higher expression level of TREM2 in cancer
cells, the expression level of TREM2 in the RCC ACHN cell line and
the normal renal cortex/proximal tubular HK-2 cell line were
evaluated. The results revealed that the expression level of TREM2
was significantly higher in ACHN cells than that in HK-2 cells
(Fig. 1B). These findings indicate
that TREM2 is a potential target of RCC, and the mechanism of TREM2
in RCC deserves further exploration.
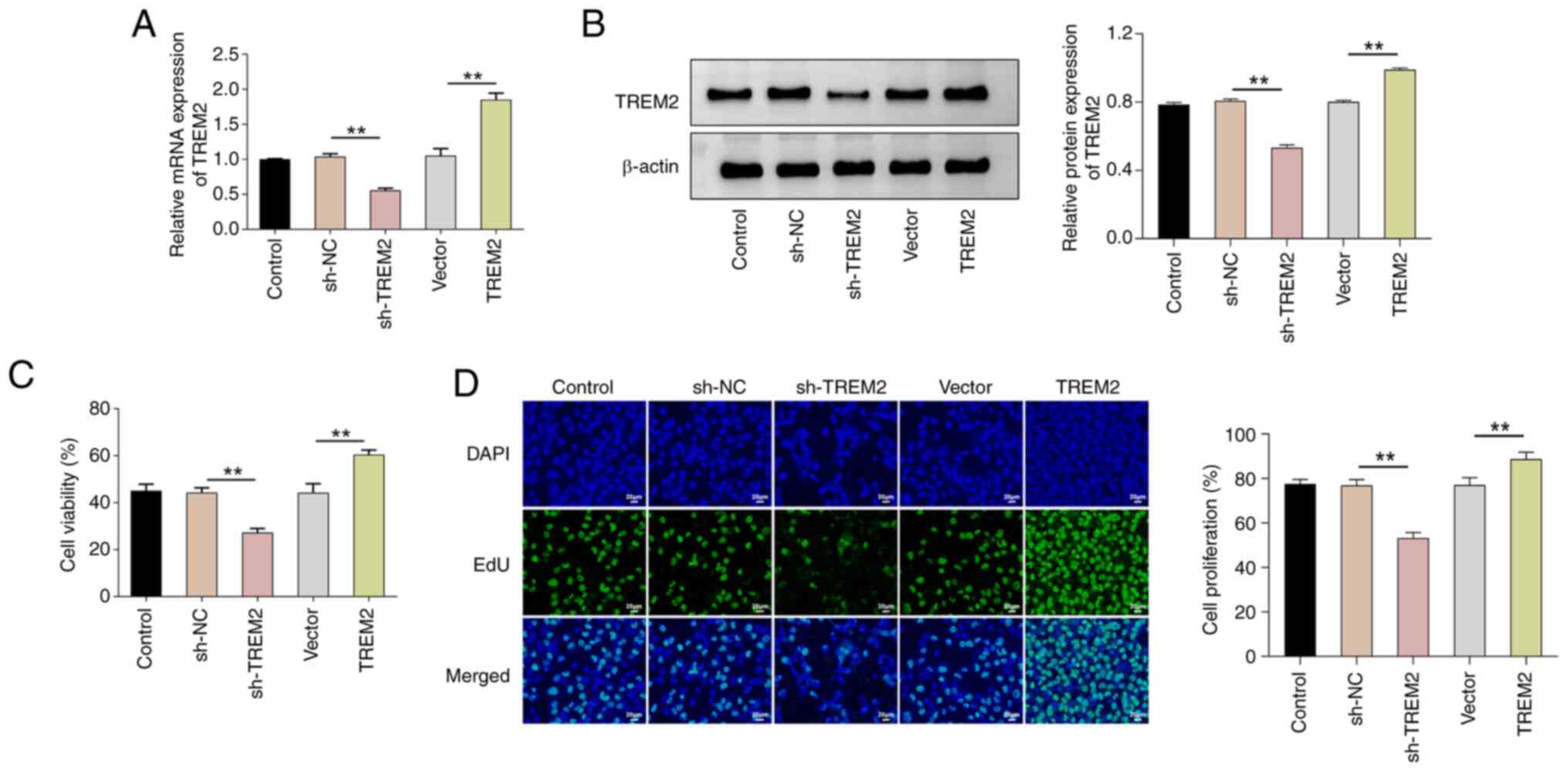
TREM2 promotes the proliferation of
RCC cells
To assess the function of TREM2 in regulating the
phenotype of ACHN cells, lentiviral transfection was used to
knockdown or overexpress TREM2 in ACHN cells. After 48 h of
infection, the cells were collected, and the expression of TREM2
was assessed using reverse transcription-quantitative PCR and
western blotting (Fig. 2A and B).
The results showed that TREM2 mRNA and protein levels were
significantly lower in the sh-TREM2 group compared with the sh-NC
group and significantly higher in the TREM2 overexpression group
compared with the vector group. Furthermore, the results of the
CCK-8 and EdU staining assays demonstrated that the knockdown of
TREM2 significantly decreased cell viability and proliferation in
comparison with the sh-NC group; whilst overexpression of TREM2
significantly increased cell viability and proliferation in
comparison with the vector group (Fig.
2C and D). Therefore, the findings indicate that TREM2 could
promote the proliferation of RCC cells.
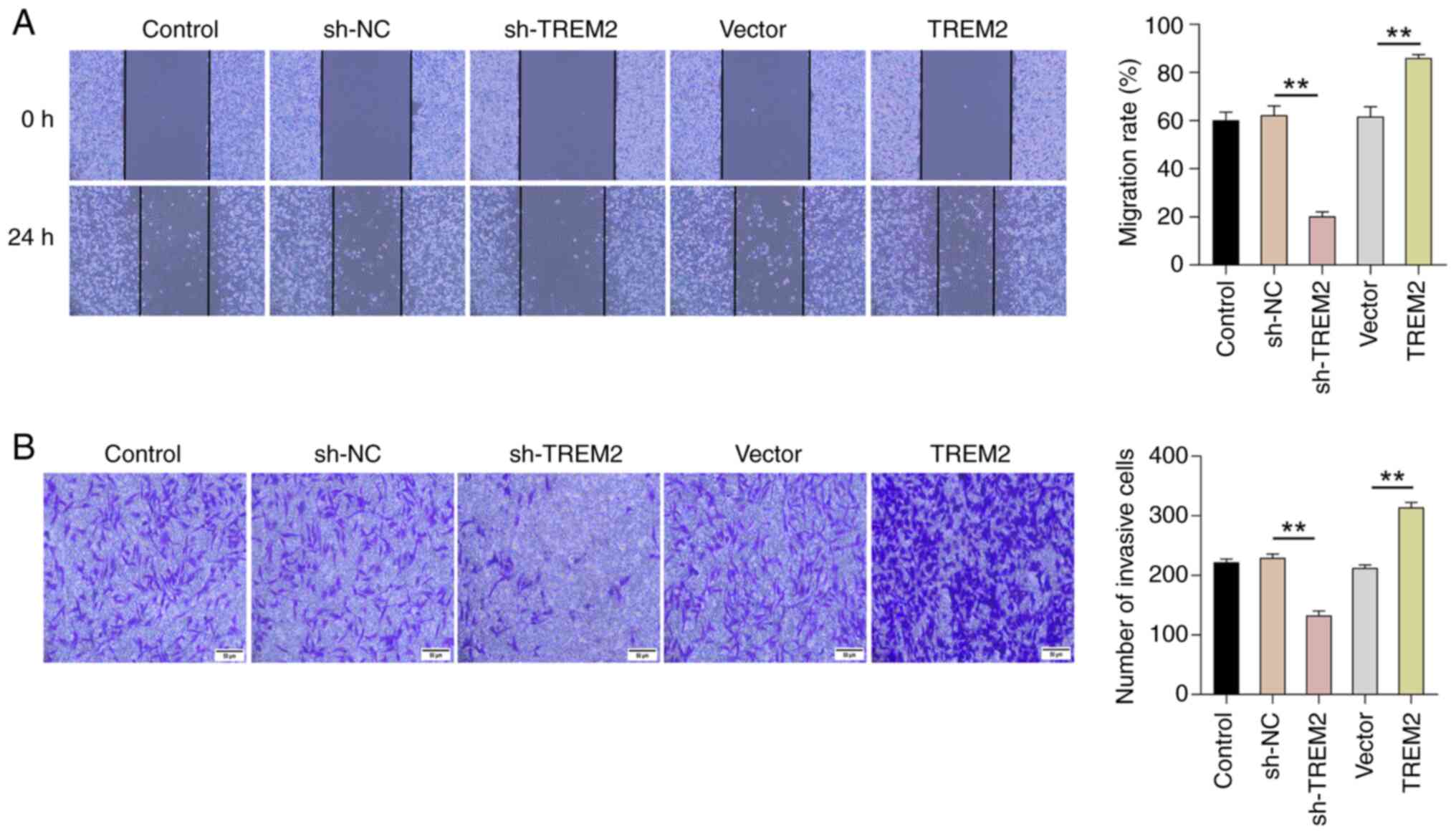
TREM2 promotes the migration and
invasion of RCC cells
Migration and invasion are an important indices to
assess the malignant phenotype of cancer cells (23). To evaluate the effect of TREM2 on
the migration of RCC cells, a wound healing assay was performed to
observe the wound healing of cells at 0 and 24 h. Briefly, in
comparison with the sh-NC group, the knockdown of TREM2
significantly inhibited the wound healing rate of ACHN in RCC
cells. Conversely, overexpression of TREM2 promoted the wound
healing rate of ACHN in RCC cells compared with the vector group
(Fig. 3A). These results suggest
that TREM2 could enhance the migration of RCC cells. A Transwell
assay further revealed that knockdown of TREM2 significantly
inhibited the cell invasion compared with that in the sh-NC group,
while overexpression of TREM2 resulted in a significantly higher
cell invasion compared with the vector group (Fig. 3B). Overall, the findings indicate
that TREM2 promotes the migration and invasion of RCC cells.
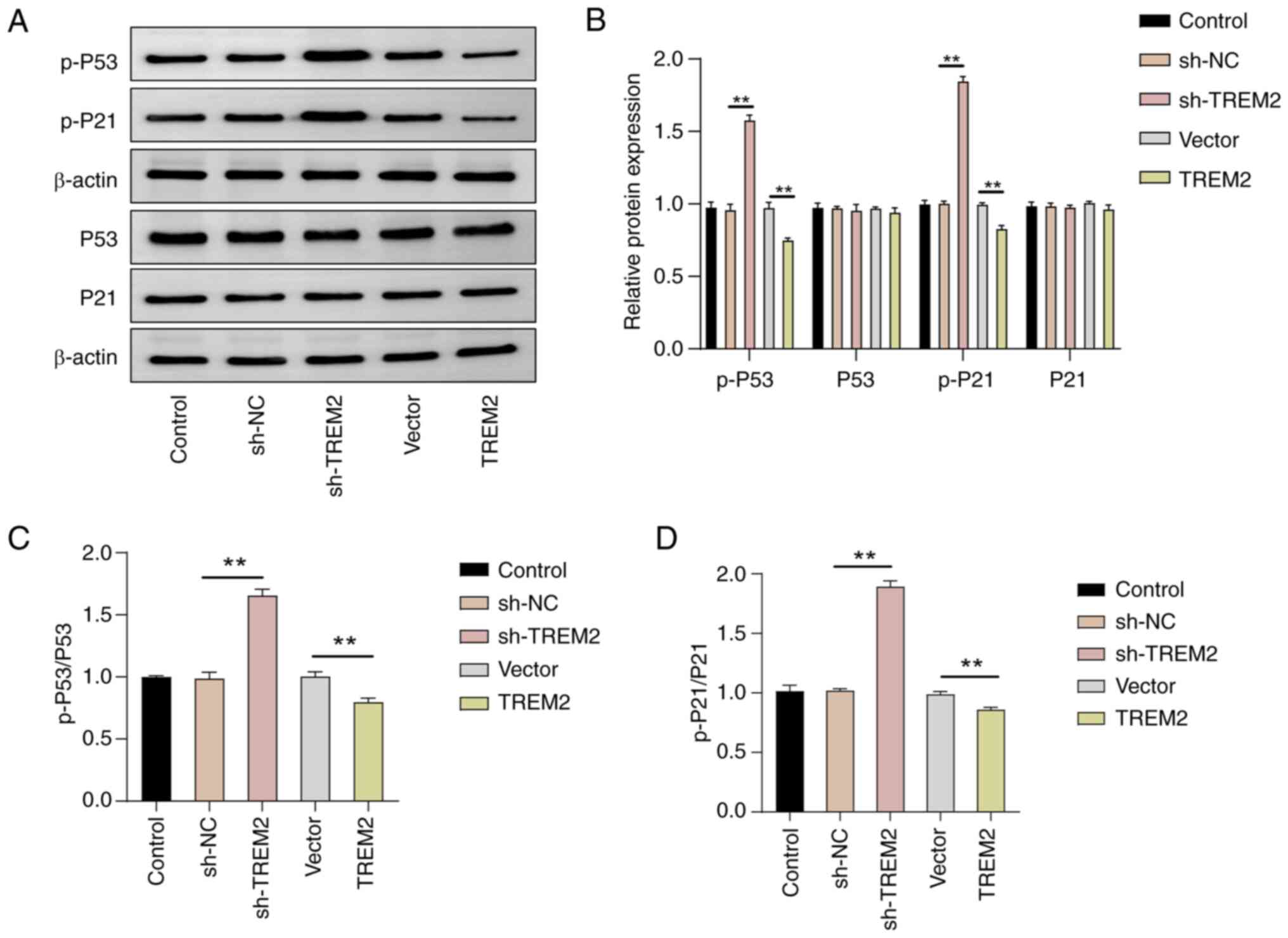
TREM2 promotes the proliferation of
RCC cells by inhibiting the P53 signaling pathway
The P53 signaling pathway serves an important role
in regulating the cell cycle and the proliferation, invasion and
migration of cells (9). The effect
of TREM2 on the activation of the P53 signaling pathway was
evaluated to assess whether TREM2 regulates tumor occurrence and
development through the P53 signaling pathway. Western blotting
revealed that knockdown or overexpression of TREM2 had no
significant effect on the total protein levels of P53 and P21;
however, in comparison with the sh-NC group, knockdown of TREM2
significantly increased the phosphorylation levels of P53 and P21,
whilst overexpression of TREM2 significantly decreased the
phosphorylation levels of P53 and P21 (P<0.01, Fig. 4). In summary, the results indicated
that TREM2 may promote the proliferation, invasion and migration of
RCC cells by inhibiting the activation of the P53 signaling
pathway.
Discussion
In the present study, TREM2 was significantly
upregulated in RCC tissues, which is consistent with the findings
of a previous study (17). Such a
result strengthens the hypothesis that TREM2 serves a crucial role
in the pathophysiology of RCC. Furthermore, the results of the
present study align with emerging research indicating that TREM2 is
associated with several cancers, including lung cancer, colorectal
cancer, thyroid cancer and RCC (16,17,24,25),
highlighting its potential as a common mediator in oncogenic
processes. Notably, the findings of the present study extend the
current understanding by demonstrating that TREM2 was not only
elevated in RCC but also served a significant role in regulating
key molecular pathways. A novel layer of complexity to its role in
cancer was added in the observed TREM2 upregulation in RCC tissues,
particularly in how it influenced malignant cell proliferation, a
pivotal characteristic of cancer cells (26). The present study made a unique
contribution to the oncological field by providing evidence of the
regulatory effects of TREM2 on the phosphorylation levels of P53
and P21. P53 and P21 are critical regulators of cell cycle and
apoptosis (27), suggesting a
mechanism by which TREM2 could promote oncogenesis and tumor
progression. These insights into the molecular interactions of
TREM2 not only offer a deeper understanding of its function in RCC
but also underscore its potential as a therapeutic target based on
its significant impact on tumor biology.
In the present study, the results of the CCK-8 assay
revealed that TREM2 could promote the proliferation of ACHN cells,
which aligns with the findings of Zhang et al (17). The invasion and migration ability of
cancer cells is an important index to evaluate the malignancy of
tumors (26,28) and it is of importance to explore the
molecular regulatory mechanism of cancer cell invasion and
migration for identifying therapeutic targets for cancer. In the
present study, the wound healing and Transwell assays were used to
determine the regulatory effect of TREM2 on the migration and
invasion of RCC cells. The results revealed that knockdown of TREM2
expression significantly inhibited the migration and invasion of
ACHN cells, whereas overexpression of TREM2 promoted migration and
invasion. These results indicate that TREM2 may promote the
development of RCC by facilitating the migration and invasion of
RCC cells. Moreover, as reported in several previous studies, TREM2
can regulate the invasion and migration of many cancer cells, but
its function varies in different tumors (24,29,30).
For example, TREM2 promotes the invasion and migration of gastric
cancer cells (24), but inhibits
the viability of colorectal and liver cancer cells (24,29,30).
Therefore, the role of TREM2 in different cancers must be carefully
evaluated when selecting it as a target for cancer treatment.
In the present study, the relationship between TREM2
and RCC progression was evaluated, focusing on the P53/P21
signaling axis. This signaling axis serves a crucial role in cell
cycle regulation, DNA replication and cell division (31). The cell cycle of cancer cells is
frequently abnormal, accelerating cell division and leading to
uncontrollable cell proliferation and tumor formation (32). P53, a key regulator of the cell
cycle, is often altered in several forms of cancer, marking a
significant step in carcinogenesis (6,33,34);
however, the present study revealed that the total expression
levels of the P53 protein were not affected by TREM2, suggesting
that the regulatory role of TREM2 in cancer proliferation is not
directly involved in altering P53 expression. Protein
phosphorylation, a vital post-translational modification of the P53
protein, serves a crucial role in its nuclear localization and
activation of downstream signaling pathways (35). In the present study, it was observed
that TREM2 knockdown could increase the phosphorylation levels of
P53, enhancing its tumor-suppressive functions; however, TREM2
overexpression decreased P53 phosphorylation, potentially
facilitating oncogenic activities. Additionally, P21, as a
downstream effector of the P53 pathway, serves a key role in
halting the cell cycle by inhibiting cyclin D1 and cyclin-dependent
kinase (36). The results of the
present study indicate that TREM2 could influence the progression
of RCCs by modulating the phosphorylation states of P53 and P21,
thereby affecting their functional states without altering their
protein expression levels. This modulation suggests that TREM2
could exert its effects on tumor biology through a specific,
post-translational mechanism. According to Lei et al
(37), such mechanisms of
TREM2-mediated post-translational mechanism serve a significant
role in regulating cancer progression, supporting the pivotal roles
of TREM2 in modulating key signaling pathways in cancer. For
example, TREM2-mediated phosphorylation affected Akt in prostate
adenocarcinoma and RCC, and NF-κB in papillary thyroid carcinoma
and gastric cancer (38). To the
best of our knowledge, the present study is the first to
demonstrate that TREM2 regulates RCC progression by specifically
targeting the P53/P21 signaling axis, particularly through the
modulation of protein phosphorylation rather than through changes
in protein quantity. Further in-depth investigation is needed to
elucidate exactly how TREM2 interacts with the molecular mechanisms
regulating the phosphorylation of P53 and P21, thereby providing a
deeper understanding of its role in cancer pathology and its
potential as a therapeutic target.
Although the present study elucidated the role of
TREM2 in promoting the proliferation, and migration of RCC cells
through controlled in vitro experiments, it is crucial to
recognize the limitations posed by the lack of in vivo
studies and comprehensive clinical data. The complexity of the
tumor microenvironment in living organisms, including multiple cell
types and dynamic signaling interactions, serves a crucial role in
influencing cancer cell behaviors. These intricacies are often not
fully replicated in in vitro models. Additionally, the
present study did not include longitudinal clinical data or
follow-up data of patients with RCC, which are essential for
assessing outcomes, such as survival rates and therapy responses.
Furthermore, TREM2 serves a crucial role in modulating immune
responses within the tumor microenvironment, influencing not only
inflammation and tissue remodeling but also the broader immune
landscape in cancers like RCC. Specifically, TREM2 has been
associated with immunosuppressive functions, including the
regulation of myeloid cell activity and the suppression of
CD8+ T-cell infiltration (16,38,39).
Future studies should use flow cytometry, immune assays, RNA
sequencing, animal models and clinical trials to thoroughly explore
the immunosuppressive impact of TREM2 on RCC, highlighting its
potential as a therapeutic target. Finally, the interactions
between TREM2 and other genes and signaling pathways involved in
RCC remain underexplored. Understanding these interactions could
reveal complex regulatory networks that contribute to the
pathogenesis of RCC and may identify novel therapeutic targets. To
thoroughly explore these interactions, a comprehensive experimental
approach is necessary, which should include the following: RNA
sequencing to analyze gene expression patterns; protein-protein
interaction assays to detect direct interactions; genetic
manipulation to observe effects of TREM2 modulation;
phosphorylation studies to assess signaling changes; reporter
assays for evaluating transcription factor activity; pathway
inhibition tests to determine pathway dependencies; and animal
model analyses to study the in vivo relevance. These methods
will collectively enhance the understanding of the role of TREM2 in
RCC and its potential as a therapeutic target.
In conclusion, the present study demonstrated that
TREM2 may promote the malignant biological behavior of RCC cells
through the P53 signaling pathway. Furthermore, the results
revealed the relationship between TREM2 and RCC, providing new
insights into the regulation of RCC by TREM2 and further
demonstrating the potential of TREM2 as a therapeutic target for
RCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by Jiangsu University Clinical
Medical Science and Technology Development Fund (grant no.
JLY2021021).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LZ, QX and BW designed the study. ZL collated the
data, performed data analyses. QX and BW drafted the manuscript. QX
and BW confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Wujin Hospital Affiliated with Jiangsu University
(Changzhou, China; approval no. 2022-SR-092), and written informed
consent was obtained from all patients according to the
requirements of the approved guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Miller KD, Nogueira L, Devasia T, Mariotto
AB, Yabroff KR, Jemal A, Kramer J and Siegel RL: Cancer treatment
and survivorship statistics, 2022. CA Cancer J Clin. 72:409–436.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Linehan WM and Ricketts CJ: The cancer
genome atlas of renal cell carcinoma: Findings and clinical
implications. Nat Rev Urol. 16:539–552. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Campi R, Rebez G, Klatte T, Roussel E,
Ouizad I, Ingels A, Pavan N, Kara O, Erdem S, Bertolo R, et al:
Effect of smoking, hypertension and lifestyle factors on kidney
cancer-perspectives for prevention and screening programmes. Nat
Rev Urol. 20:669–681. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ge W, Song S, Qi X, Chen F, Che X, Sun Y,
Wang J, Li X, Liu N, Wang Q and Wu G: Review and prospect of immune
checkpoint blockade therapy represented by PD-1/PD-L1 in the
treatment of clear cell renal cell carcinoma. Oncol Res.
31:255–270. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harrison H, Thompson RE, Lin Z, Rossi SH,
Stewart GD, Griffin SJ and Usher-Smith JA: Risk prediction models
for kidney cancer: A systematic review. Eur Urol Focus.
7:1380–1390. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Joerger AC and Fersht AR: Structural
biology of the tumor suppressor p53. Annu Rev Biochem. 77:557–582.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu D and Prives C: Relevance of the
p53-MDM2 axis to aging. Cell Death Differ. 25:169–179. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zavileyskiy L and Bunik V: Regulation of
p53 function by formation of non-nuclear heterologous protein
complexes. Biomolecules. 12:3272022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang H, Guo M, Wei H and Chen Y: Targeting
p53 pathways: Mechanisms, structures, and advances in therapy.
Signal Transduct Target Ther. 8:922023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen JX, Xu D, Cao JW, Zuo L, Han ZT, Tian
YJ, Chu CM, Zhou W, Pan XW and Cui XG: TRIM47 promotes malignant
progression of renal cell carcinoma by degrading P53 through
ubiquitination. Cancer Cell Int. 21:1292021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jian W, Xue W, Wang T, Yu Y, Cai L, Meng
Y, Xia Z and Zhang C: RBM4 inhibits the growth of clear cell renal
cell carcinoma by enhancing the stability of p53 mRNA. Mol
Carcinog. 62:464–478. 2023. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fassler M, Benaim C and George J: TREM2
agonism with a monoclonal antibody attenuates tau pathology and
neurodegeneration. Cells. 12:15492023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Molgora M, Liu YA, Colonna M and Cella M:
TREM2: A new player in the tumor microenvironment. Semin Immunol.
67:1017392023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li RY, Qin Q, Yang HC, Wang YY, Mi YX, Yin
YS, Wang M, Yu CJ and Tang Y: TREM2 in the pathogenesis of AD: A
lipid metabolism regulator and potential metabolic therapeutic
target. Mol Neurodegener. 17:402022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Katzenelenbogen Y, Sheban F, Yalin A, Yofe
I, Svetlichnyy D, Jaitin DA, Bornstein C, Moshe A, Keren-Shaul H,
Cohen M, et al: Coupled scRNA-Seq and intracellular protein
activity reveal an immunosuppressive role of TREM2 in cancer. Cell.
182:872–885.e19. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park MD, Reyes-Torres I, LeBerichel J,
Hamon P, LaMarche NM, Hegde S, Belabed M, Troncoso L, Grout JA,
Magen A, et al: TREM2 macrophages drive NK cell paucity and
dysfunction in lung cancer. Nat Immunol. 24:792–801. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang H, Sheng L, Tao J, Chen R, Li Y, Sun
Z and Qian W: Depletion of the triggering receptor expressed on
myeloid cells 2 inhibits progression of renal cell carcinoma via
regulating related protein expression and PTEN-PI3K/Akt pathway.
Int J Oncol. 49:2498–2506. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Amin MB, Edge SB, Greene FL, Byrd DR,
Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR,
Sullivan DC, et al: AJCC cancer staging manual. 8th edition. New
York: Springer; 2017
|
|
19
|
World Medical Association, . World medical
association declaration of Helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Salic A and Mitchison TJ: A chemical
method for fast and sensitive detection of DNA synthesis in vivo.
Proc Natl Acad Sci USA. 105:2415–2420. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: A convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leber MF and Efferth T: Molecular
principles of cancer invasion and metastasis (review). Int J Oncol.
34:881–895. 2009.PubMed/NCBI
|
|
24
|
Li C, Hou X, Yuan S, Zhang Y, Yuan W, Liu
X, Li J, Wang Y, Guan Q and Zhou Y: High expression of TREM2
promotes EMT via the PI3K/AKT pathway in gastric cancer:
Bioinformatics analysis and experimental verification. J Cancer.
12:3277–3290. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang J and Li Z: TREM2 is a prognostic
biomarker and correlated with an immunosuppressive microenvironment
in thyroid cancer. Dis Markers. 2022:18073862022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jung Y, Kraikivski P, Shafiekhani S,
Terhune SS and Dash RK: Crosstalk between Plk1 and p53, cell cycle,
and G2/M DNA damage checkpoint regulation in cancer: Computational
modeling and analysis. NPJ Syst Biol Appl. 7:462021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Saha T, Solomon J, Samson AO and Gil-Henn
H: Invasion and metastasis as a central hallmark of breast cancer.
J Clin Med. 10:34982021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim SM, Kim EM, Ji KY, Lee HY, Yee SM, Woo
SM, Yi JW, Yun CH, Choi H and Kang HS: TREM2 Acts as a tumor
suppressor in colorectal carcinoma through Wnt1/β-catenin and Erk
signaling. Cancers (Basel). 11:13152019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tang W, Lv B, Yang B, Chen Y, Yuan F, Ma
L, Chen S, Zhang S and Xia J: TREM2 acts as a tumor suppressor in
hepatocellular carcinoma by targeting the PI3K/Akt/β-catenin
pathway. Oncogenesis. 8:92019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Engeland K: Cell cycle regulation:
p53-p21-RB signaling. Cell Death Differ. 29:946–960. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu J, Peng Y and Wei W: Cell cycle on the
crossroad of tumorigenesis and cancer therapy. Trends Cell Biol.
32:30–44. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sabapathy K and Lane DP: Corrigendum to
‘understanding p53 functions through p53 antibodies’. J Mol Cell
Biol. 11:11052019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hernández Borrero LJ and El-Deiry WS:
Tumor suppressor p53: Biology, signaling pathways, and therapeutic
targeting. Biochim Biophys Acta Rev Cancer. 1876:1885562021.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen L, Liu S and Tao Y: Regulating tumor
suppressor genes: Post-translational modifications. Signal
Transduct Target Ther. 5:902020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shamloo B and Usluer S: p21 in cancer
research. Cancers (Basel). 11:11782019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lei X, Gou YN, Hao JY and Huang XJ:
Mechanisms of TREM2 mediated immunosuppression and regulation of
cancer progression. Front Oncol. 14:13757292024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tan J, Fan W, Liu T, Zhu B, Liu Y, Wang S,
Wu J, Liu J, Zou F, Wei J, et al: TREM2+ macrophages
suppress CD8+ T-cell infiltration after transarterial
chemoembolisation in hepatocellular carcinoma. J Hepatol.
79:126–140. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun R, Han R, McCornack C, Khan S, Tabor
GT, Chen Y, Hou J, Jiang H, Schoch KM, Mao DD, et al: TREM2
inhibition triggers antitumor cell activity of myeloid cells in
glioblastoma. Sci Adv. 9:eade35592023. View Article : Google Scholar : PubMed/NCBI
|