Introduction
Succinate dehydrogenase (SDH) is an enzyme complex
located on the inner mitochondrial membrane that is composed of
four subunits (SDHA, SDHB, SDHC and SDHD). This complex serves a
dual role in converting succinate to fumarate during the Krebs
cycle and participates in the electron transport chain (1). SDH deficiency leads to the
accumulation of succinate, which inhibits proline hydroxylase
activity and induces the accumulation of the hypoxia-inducible
factor (HIF)-1α, thus activating vascular endothelial growth factor
(VEGF) and insulin-like growth factor-1, and ultimately causing
tumorigenesis (2). Germline
mutations in any of the four subunits of SDH result in deficiency
of the SDH complex associated with a group of hereditary tumors,
including paraganglioma, phaeochromocytoma, gastrointestinal
stromal tumor, pituitary adenoma and renal cell carcinoma (RCC)
(3).
SDH-deficient RCC is a specific type of RCC that was
first proposed in 2004 by Vanharanta et al (4), recognized by the International Society
for Urological Pathology (ISUP) Vancouver in 2013 (5), and formally included as a subtype of
RCC by the World Health Organization (WHO) in 2016 (6). SDH-deficient RCC is a rare malignancy
with a high genetic correlation that accounts for 0.05–0.2% of all
RCC cases. This subtype is usually caused by germline mutations
with the addition of a somatic second hit, which leads to
dysfunction of the SDH complex (7,8). The
SDHB gene harbors most mutations, followed by SDHC and SDHD
(9), while SDHA-deficient RCC is
even rarer; to the best of our knowledge, only four patients have
been reported in the literature to date (10–13).
The present study reports the clinical,
morphological and molecular features of a new patient diagnosed
with SDHA-deficient RCC harboring a novel SDHA mutation, and
reviews the data of the four previously reported patients.
Next-generation sequencing (NGS) and Sanger sequencing identified a
novel heterozygous frameshift variant (NM_004168.4: c.992_999dup)
in the SDHA gene, which has not been previously reported to be
associated with RCC in the literature. This new case with a
heterozygous SDHA frameshift variant expands the phenotypic
spectrum of the SDHA gene, and provides further clinical,
morphological and molecular data of SDHA-deficient RCC.
Case report
A 22-year-old female patient presented with a left
kidney tumor and was admitted to Hubei Cancer Hospital (Tongji
Medical College, Huazhong University of Science and Technology;
Wuhan, China) in July 2021. An abdominal computerized tomography
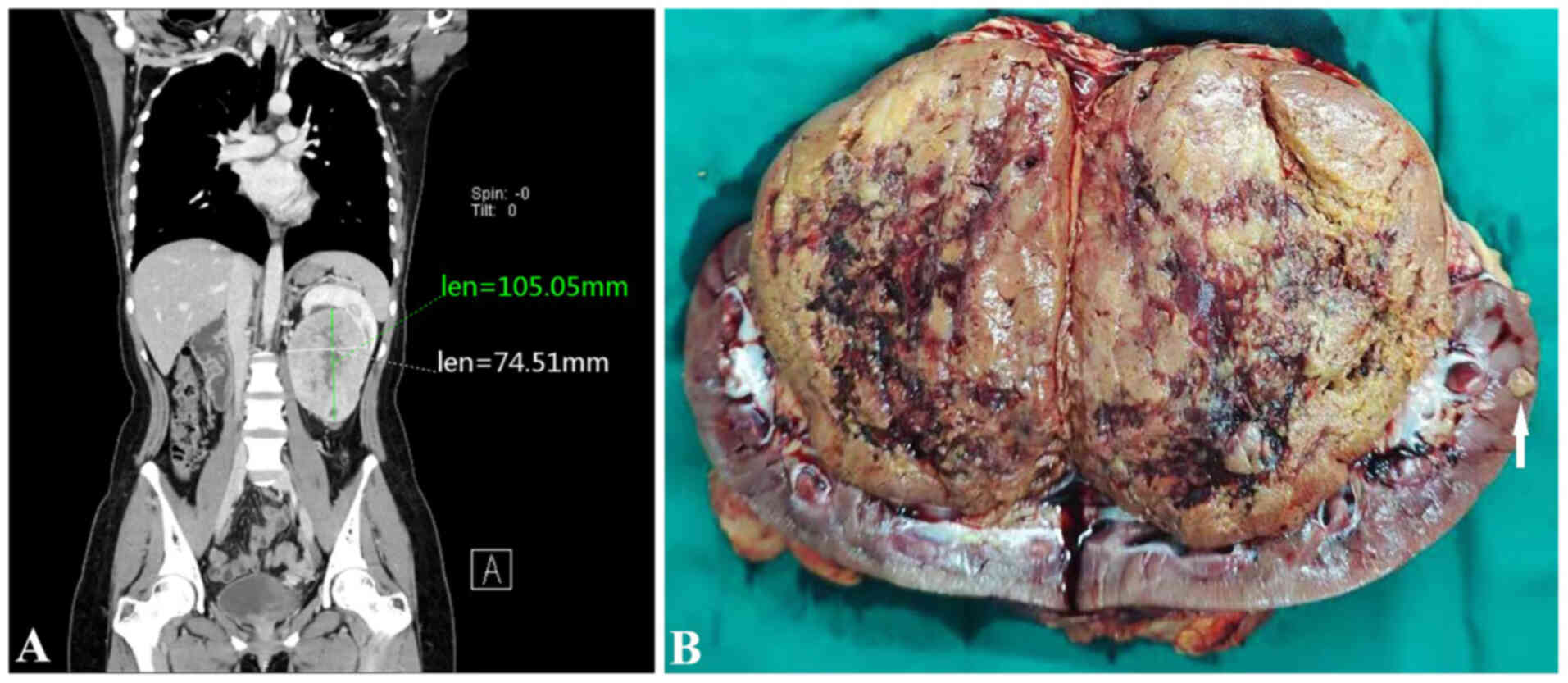
(CT) scan showed multiple nodular masses in the left kidney, with
the larger one being ~10.5×7.5 cm in size (Fig. 1A). The left kidney was pushed
upward, with no clear boundary between the mass and the renal
pelvis. The patient had no family history of RCC or other tumors.
After evaluation, radical nephrectomy was performed.
Gross examination revealed a 10×8×7 cm solid tan
non-encapsulated tumor in the hilum of the kidney. The cut surface
of the tumor was gray-brown with hemorrhagic and cystic foci.
Several small nodules 1–2 cm in diameter were observed in the renal
parenchyma adjacent to the largest mass (Fig. 1B). Examination of the
histopathological staining with hematoxylin and eosin (H&E) as
described in the supplementary information indicated that the tumor
showed multiple nodules with a focally pushing border (Fig. 2A) and protruded into the renal
pelvis. Single native renal tubules were entrapped at the periphery
of the tumor. The tumor was mainly composed of dense tubular and
vesicular structures (Fig. 2B),
some of which were solid (Fig. 2C).
In some areas, the tubules gradually expanded and tumor cells
protruded into the lumen, forming characteristic annular tubular
structures around eosinophilic hyaline bodies. Calcification of the
eosinophilic hyaline bodies could also be detected (Fig. 2D-F). The tumor cells contained
low-grade (ISUP grade 2) nuclei, which were round or oval, and
relatively uniform in size, with abundant eosinophilic, somewhat
flocculent cytoplasm, as well as small nucleoli. The tumor stroma
was rich in thin-walled vascular networks, and portions of the
stroma showed signs of loose edema and bleeding. Single tumor cells
with intracellular mucus and small to medium-sized round, or
dilated and twisted glandular ducts, were present in the
surrounding area. The glandular lumen was filled with gray-blue
mucus (Fig. 2G-I). The morphology
of the small nodules around the largest mass in the renal
parenchyma was consistent with that of the largest mass itself.
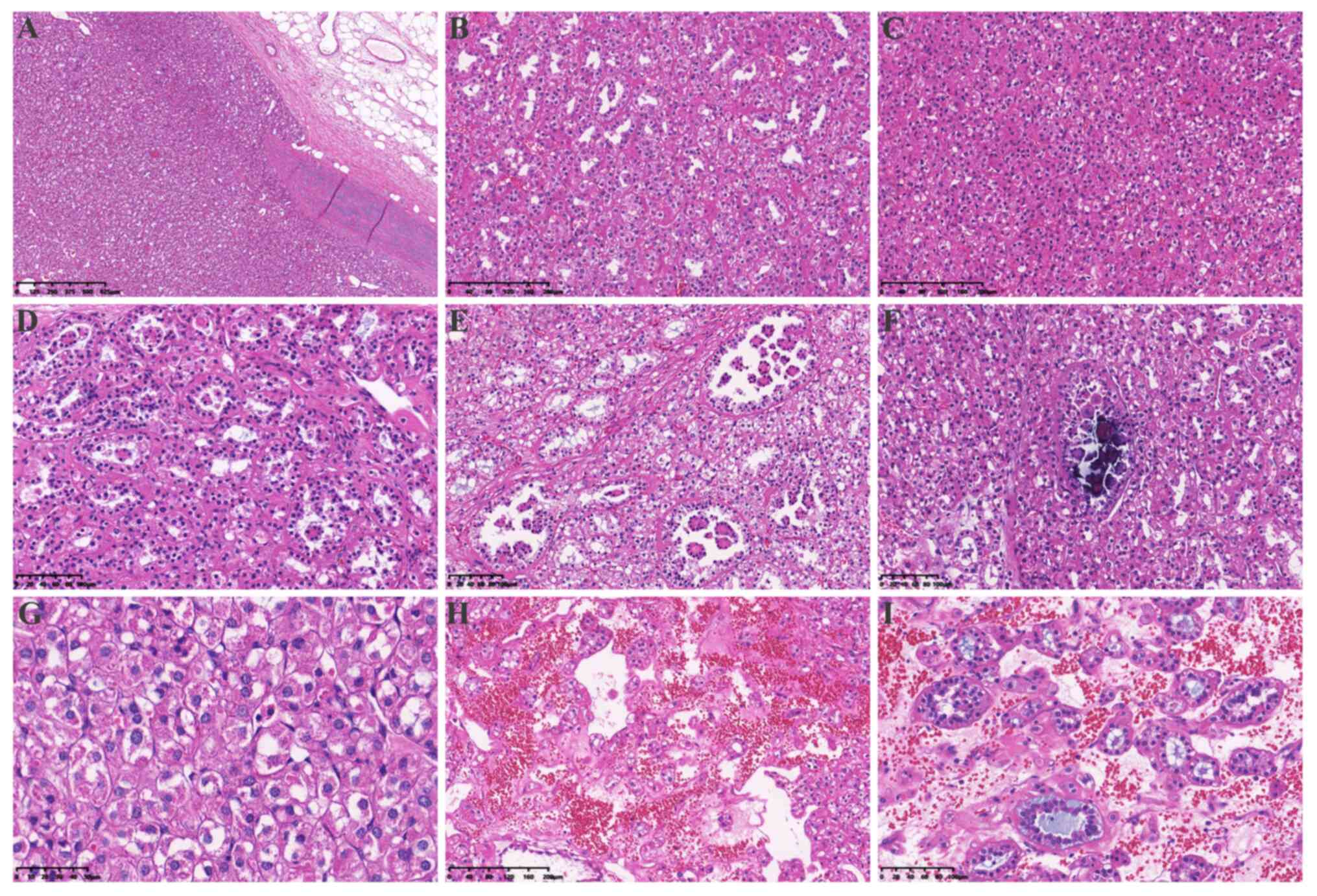 | Figure 2.Hematoxylin and eosin staining of the
tumor tissue. (A) A pushing border surrounded the tumor
(magnification, 4×). (B) The tumor was mainly composed of dense
tubular and vesicular structures (magnification, 10×). (C) The
tumor was arranged in solid vesicles (magnification, 10×). (D) Some
tubules were dilated (magnification, 20×), with (E) tumor cells
protruding into the lumen and forming characteristic annular
tubular structures around eosinophilic hyaline bodies
(magnification, 20×) or (F) calcifying in the lumen (magnification,
20×). (G) The morphology of the tumor cells was characterized by
eosinophilic and flocculent cytoplasm (magnification, 40×). (H) The
tumor stroma was rich in a thin-walled vascular network and some
stroma showed signs of loose edema and bleeding (magnification,
10×). (I) Tumor cells in and around the stroma were diverse,
including single cells rich in intracellular mucus, small to
medium-sized round or dilated and twisted glandular ducts, and
glandular lumen filled with gray-blue mucus magnification,
(20×). |
Immunohistochemistry (IHC) was performed on 4-µm
thick 4% neutral formaldehyde solution fixed and paraffin-embedded
(FFPE) tumor tissue blocks using the automated immunostained
Autostainer Link48 (Dako; Agilent Technologies, Inc.) according to
the manufacturer's protocol. The IHC protocol was described in the
supplementary information and the information of primary antibodies
was stated in detail in Table SI.
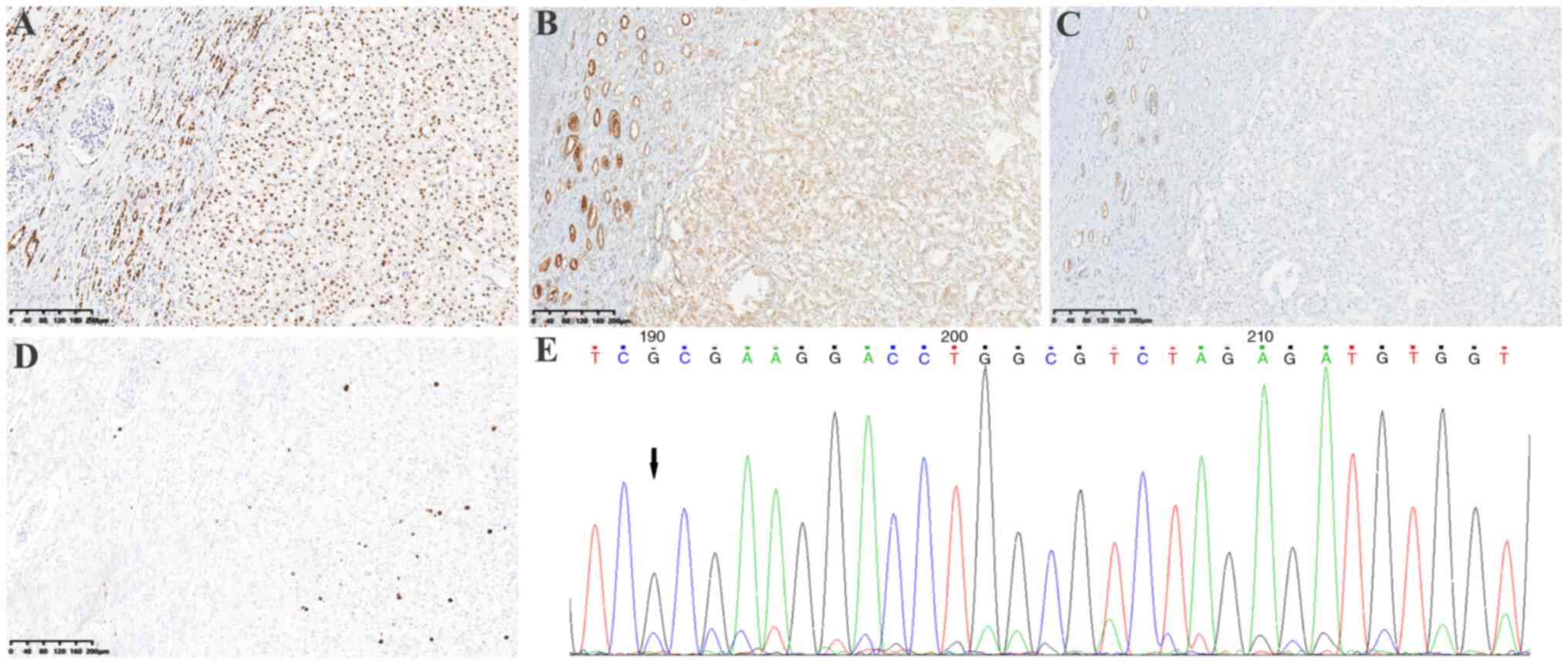
Results showed that PAX8 was positive (Fig. 3A), SDHA was weakly positive
(Fig. 3B) and SDHB was negative
(Fig. 3C). In addition, CAIX, CK7,
CD10, CD117, P504S, Vimentin, E-Cadherin, HMB45, Cathepsin K,
Melan-A, S-100, TFE3 and TFEB were negative; fumarate hydratase
(FH) was positive (data not shown); and the Ki67 index was ~5%
(Fig. 3D). Fluorescence in
situ hybridization (FISH) was performed on 4 µm-thick
formalin-fixed paraffin-embedded tissues to detect TFE3 (Xp11.2)
and TFEB (6p21) gene rearrangement using a dual-color TFE3 (Xp11.2)
and TFEB (6p21) break-apart rearrangement probe (IBP Group)
according to the manufacturer's protocols. Results showed that TFE3
(Xp11.2) and TFEB (6p21) rearrangement were both negative (data not
shown).
NGS analysis was performed as described in the
previous study (14) using the 4%
neutral formaldehyde formalin-fixed and paraffin-embedded tumor
tissue to detect 425 cancer-relevant genes (Geneseeq Technology
Inc.), including SDHA, SDHB, SDHC, SDHD, FH, TSC1/2, ARID1A, POLE,
CHEK2 and GATA2, and a novel SDHA (RefSeq accession number: SDHA
NM_004168.4) frameshift variant: c.992_999dup (p.A334Pfs*17) was
identified. This variant of SDHA has been submitted to the ClinVar
database (http://www.ncbi.nlm.nih.gov/clinvar), under the
accession number SCV004035231. The NGS analysis also examined copy
number alterations of the genes, and no copy number alterations
were detected. Bioinformatics analysis using the bcl2fastq (v2.19)
software (Illumina, Inc.) revealed it was most likely a somatic
event in the SDHA gene. Sanger sequencing also confirmed the novel
SDHA frameshift variant (NM_004168.4): c.992_999dup (CCCCTGTC)
(Fig. 3E).
The patient did not receive adjuvant chemotherapy
and was followed-up by CT examinations every 6 months in the first
2 years and then once a year after resection. There was no apparent
recurrence or metastasis in July 2024 (Fig. S1).
Discussion
SDH-deficient RCC is a specific type of RCC
generally caused by a pathogenic germline variant with the addition
of a somatic second hit in every one of the four SDH subunits,
which leads to dysfunction of the SDH complex. A germline mutation
of the SDHB gene is usually detected, and somatic mutation of SDHB
is rarely reported in SDH-deficient RCC (3). In addition, a few cases of SDHC or
SDHD gene mutations have been reported (9), whereas SDHA-deficient RCC is rare.
SDH-deficient RCC often occurs in young adults (age range, 14–76
years; median age, 35 years), with a slight male predominance
(7–8). The majority of SDH-deficient RCC cases
are well-circumscribed or lobulated with a pushing border, and are
sometimes characterized by the presence of a pseudocapsule
(15). The tumors consist of sheets
or compact nests of bland cells with eosinophilic cytoplasm, which
may have a pale, bubbly appearance, and lack prominent cell borders
(16). The most distinctive feature
is the presence of cytoplasmic inclusions containing eosinophilic
or pale flocculent material, despite being absent in some cases
(15,16). Most cases of SDH-deficient RCC are
consistent with a low-grade morphology, but an increasing number of
high-grade tumors have been reported (15,16).
Cases associated with SDHA mutations more commonly show a higher
nuclear grade, and demonstrate papillary, solid, cribriform or
desmoplastic architecture (10–12).
The clinicopathological features of the four cases
of SDHA-deficient RCC previously reported in the literature
(10–13) are summarized in Table I. All four patients were male, aged
23–62 years, with a median age of 49.5 years. The tumors were
usually large, ranging between 8 and 11 cm in diameter. In three
out of the four patients, the tumor was arranged in papillary,
tubular, vesicular or solid structures with eosinophilic cytoplasm.
The tumor cells contained high-grade nuclei (ISUP grade 3/4), with
large pleomorphic nuclei, coarse chromatin and prominent nucleoli,
even with sarcomatoid dedifferentiation. The fourth case exhibited
a low-grade morphology, similar to chromophobe RCC. The patient
described in the present study was a young woman, with low-grade
nuclear morphology, which were different from the four previously
reported patients with SDHA-deficient RCC. The distinctive
morphological features of SDH-deficient RCC, such as the tumor
cells with abundant eosinophilic, somewhat flocculent cytoplasm,
were seen in the present patient. The tumor was mainly arranged in
tubular, vesicular and solid structures, without an obvious
papillary structure. Elongated papillae tumor cells protruded into
the dilated tubular lumen and formed a characteristic annular
tubule structure surrounding the eosinophilic hyaline bodies, with
focal calcification. The morphology of the small nodules in the
renal parenchyma outside the tumor body was consistent with that of
the main tumor. A wide panel of IHC studies was performed in order
to rule out other renal neoplasms, and as loss of SDHB expression
is a prerequisite for the diagnosis of SDH-deficient RCC.
Furthermore, the tumors usually show positive staining for PAX8,
and negative staining for CD117 and CK7, and FH expression is
consistently preserved. To the best of our knowledge, the present
study is the first to detect annular tubule structures surrounding
the eosinophilic hyaline bodies in SDH-deficient RCC.
 | Table I.Clinical and molecular features of
SDHA-deficient RCC cases. |
Table I.
Clinical and molecular features of
SDHA-deficient RCC cases.
|
|
|
|
| IHC |
|
|
|
|
|
|---|
| First author,
year | Sex/Age, years | Tumor location | Tumor size, cm |
|
|
|
|
|
|
|---|
| SDHA | SDHB | Molecular
genetics | Germline/somatic | Treatment | Outcome | (Refs.) |
|---|
| McEvoy, 2018 | Male/45 | NA | 11 | - | - | SDHA biallelic
mutations | Germline and
somatic | Laparoscopic
nephrectomy + PD-1 inhibitor and TKIs | Recurrence at 10
months | (10) |
| Yakirevich, 2015 | Male/54 | Right | 10 | - | - | SDHA homozygous
deletion | Somatic | Radical
nephrectomy | NA | (11) |
| Ozluk, 2015 | Male/62 | Right | 8 | - | - | Single-nucleotide
splice site deletion in SDHA gene | Somatic | Radical nephrectomy
and lymphadenectomy | Local recurrence at
10 months | (12) |
| Jiang, 2015 | Male/23 | Left | 4 | +, weak | - | SDHA heterozygous
mutation | Germline | Partial nephrectomy +
sunitinib | No recurrence within
2 years | (13) |
| Present study | Female/22 | Left | 11 | +, weak | - | SDHA frameshift
mutation | Somatic | Radical
nephrectomy | No recurrence 36
months after resection | - |
A previous study has shown that pathogenic mutations
of SDH complex subunits lead to loss of expression of the SDHB
protein (9). SDHA-deficient RCC is
caused by a mutation in the SDHA gene resulting in the dysfunction
of the SDH complex and loss of expression of both SDHA and SDHB
proteins. The four cases of SDHA-deficient RCCs reported in the
literature contained SDHA gene pathogenic mutations, including one
case with a germline truncating variant in conjunction with a
somatic missense variant; another case with biallelic homozygous
deletion; and one case with a single nucleotide splice site
deletion. These three cases expressed neither SDHB nor SDHA
protein. The last low-grade chromophobe cell carcinoid RCC
consisted of a germline heterozygous mutation in SDHA, with IHC
showing a weak positive expression of the SDHA protein and the loss
of SDHB. Similarly, in the present case report, the tumor presented
with a frameshift variant in the SDHA gene that showed a weak
positive expression of SDHA and led to the loss of SDHB expression.
These two cases were both low-grade, with a heterozygous mutation
in the SDHA gene, which might explain the decreased (but still
present) expression of the SDHA protein and the defective SDH
complex with negative SDHB expression. This finding enriches the
known molecular mechanisms associated with SDHA-deficient RCC.
Among the four cases of SDHA-deficient RCCs reported in the
literature, three were high-grade (two of which resurged 10 months
after surgery) and one low-grade. The present case was low-grade,
with no recurrence and/or metastasis 36 months after surgery.
Notably, not all mutations in the SDHA gene lead to
SDH complex deficiency. In a recent cohort study (17), NGS and Sanger sequencing were used
to detect SDHA gene status in 107 cases of clear cell RCC (ccRCC),
17 cases of papillary RCC type 2 (pRCC2), 3 cases of chromophobe
cell carcinoma and 2 cases of collecting duct carcinoma. Single
nucleotide variants (SNVs) of the SDHA gene causing amino acid
sequence variants (missense mutations) were detected in six pRCC2
and five ccRCC cases, but no mutations of the SDHB, SDHC or SDHD
genes were detected. In addition, IHC indicated a decrease in SDHA
and SDHB protein expression in the six pRCC2 and five ccRCC cases,
but none showed a complete loss of SDHA or SDHB. In this previous
study, the authors suggested that SDHA SNVs could lead to decreased
expression of the SDHA protein, but not cause SDH complex
dysfunction as manifested by the complete loss of SDHB protein
expression. Consequently, the 11 RCC cases were not classified as
SDHA-deficient RCCs. In addition, the diagnosis of pRCC2 is no
longer recommended according to the 5th edition of the WHO
Classification of Tumors of Urinary System and Male Genital Organs
published in 2022 (16).
Accordingly, the 17 cases of type 2 pRCC2s in this previous study
require further molecular characterization for unbiased
classification.
The present case should be distinguished from other
similar tumors. Specifically, TFE3-rearranged/TFEB-altered RCC
represents a group of RCC with a variety of morphologies that
characteristically include single native renal tubules at their
periphery (16). The most
distinctive pattern of TFEB-rearranged RCC is a biphasic structure
composed of nests of larger epithelioid cells and smaller cells
clustered around the hyaline basement membrane in the center
(18). TFE3-rearranged/TFEB-altered
RCC cases consistently express melanocytic markers, such as HMB45,
Melan A and Cathepsin K (19). In
addition, strong nuclear labeling for TFE3 and TFEB, TFE3/TFEB
arrangement identified by break-apart FISH, or TFE3/TFEB gene
fusion identified by RNA sequencing, can be used to easily
distinguish them from other neoplasms (20). Although in the present case, the
structure of the tumor mimics the biphasic appearance, with some
cells present at the periphery of the nests and other cells
clustering around the hyaline basement membrane, TFE3, TFEB, HMB45,
Melan A and Cathepsin K were negative, and TFE3/TFEB was not
identified by break-apart FISH in the present study. Accordingly, a
diagnosis of TFE3-rearranged/TFEB-altered RCC could not be made for
the present case.
FH-deficient RCC typically demonstrates multiple
admixed morphological patterns and characteristic prominent
eosinophilic nucleoli. A previous study has reported a few cases of
low-grade FH-deficient RCC with oncocytic morphology, which
resemble SDH-deficient RCC, but retain SDHB expression (21). Negative immunohistochemical staining
for FH in tumor cells is highly specific, and positive staining for
2SC is highly sensitive for FH-deficient RCC. In the case presented
in the current study, FH was positive and SDHB was negative,
supporting a diagnosis of SDH-deficient RCC. Oncocytoma commonly
grows in solid nests with a central stellate scar, and is composed
of round to polygonal cells with densely granular eosinophilic
cytoplasm and round uniform nuclei with a central small nucleolus.
In addition, these tumors are typically positive for CD117, whereas
CD117 was negative in the present case. RCC with TSC/mTOR gene
mutations belongs to a group of tumors with eosinophilic cytoplasm
and often contains vacuolar structures and prominent nucleoli;
these tumors also typically contain TSC2/mTOR gene mutations
(22). The aforementioned tumors
overlap morphologically with the present case, but the specific
immunophenotype and molecular genetics can be well
differentiated.
The majority of SDH-deficient RCC cases demonstrate
low-grade morphology and have a favorable prognosis with a low
metastatic rate; however, for a few cases with high-grade features
(i.e. coagulative necrosis and sarcomatoid transformation), the
rate of metastasis is as high as 70% (7), and adjuvant treatment is necessary for
advanced patients. As previously mentioned (2), tumorigenesis caused by SDH deficiency
is achieved via a pseudohypoxic pathway involving HIFs and VEGF;
therefore, targeted therapy could be the first-line therapy for
advanced RCC. Tyrosine kinase inhibitors (TKIs) are involved in the
inhibition of VEGF-induced angiogenesis, and remain the mainstay
first-line treatment for advanced RCC (23,24).
Immunotherapy, such as immune checkpoint inhibitors against
programmed cell death protein 1 or its ligand (25), has become the standard first-line
treatment for a number of patients with RCC. For a substantial
proportion of patients who are not suitable for immunotherapy, TKI
treatment remains an appropriate first-line therapy, and is widely
used as second-line and subsequent-line therapy. In a recent large,
multicenter, phase 2/3 trial (STAR), which aimed to assess the
potential benefits of a treatment break strategy compared with a
conventional treatment continuation strategy in patients with RCC
receiving TKI therapy, the results demonstrated that a drug-free
interval strategy was non-inferior to a conventional continuation
strategy for first-line treatment with TKIs, and treatment breaks
may be a feasible and cost-effective option with lifestyle benefits
for patients during TKI therapy (26). Furthermore, in a recently published
phase 3, multicenter, open-label, active-controlled study,
belzutifan, a HIF-2α inhibitor, exhibited a significant benefit
over everolimus, an inhibitor of mammalian target of rapamycin,
with respect to progression-free survival and objective response in
participants with advanced RCC who had previously received
antiangiogenic and immune checkpoint therapies (27).
In conclusion, SDH-deficient RCC is a specific type
of RCC with genetic associations. SDHA-deficient RCC is rare, and
the present case enriches the histological morphology, and
immunohistochemical and molecular characteristics of SDHA-deficient
RCC. When the protein expression of SDHA and/or SDHB is abnormal,
further molecular analysis is required to confirm a pathogenic
mutation of the SDHA gene and to avoid misdiagnosis. Future studies
regarding the mechanism of this type of cancer are needed to
strengthen the overall understanding of SDHA-deficient RCC.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was partially supported by the Special Fund for
Clinical Research of the Wu Jieping Medical Foundation (grant no.
320.6750.2021-21-15).
Availability of data and materials
The data generated in the present study may be found
in the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar) under accession
number SCV004035231 at the following URL: http://www.ncbi.nlm.nih.gov/clinvar/variation/2580151/?oq=SCV004035231&m=NM_004168.4(SDHA):c.992_999dup%20(p.Ala334fs).
All other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
JQY and LFF conceived and designed the study. MH
acquired and analyzed the data. XTW, XXX and QR performed the
research and experiments, and interpreted the data. MH and JQY
performed the writing, review and revision of the paper. MH and JQY
confirm the authenticity of all the raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent was obtained from the
patient.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Aldera AP and Govender D: Gene of the
month: SDH. J Clin Pathol. 71:95–97. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bardella C, Pollard PJ and Tomlinson I:
SDH mutations in cancer. Biochim Biophys Acta. 1807:1432–1443.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gill AJ: Succinate dehydrogenase
(SDH)-deficient neoplasia. Histopathology. 72:106–116. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vanharanta S, Buchta M, McWhinney SR,
Virta SK, Peçzkowska M, Morrison CD, Lehtonen R, Januszewicz A,
Järvinen H, Juhola M, et al: Early-onset renal cell carcinoma as a
novel extraparaganglial component of SDHB-associated heritable
paraganglioma. Am J Hum Genet. 74:153–159. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Srigley JR, Delahunt B, Eble JN, Egevad L,
Epstein JI, Grignon D, Hes O, Moch H, Montironi R, Tickoo SK, et
al: The international society of urological pathology (ISUP)
vancouver classification of renal neoplasia. Am J Surg Pathol.
37:1469–1489. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moch H, Cubilla AL, Humphrey PA, Reuter VE
and Ulbright TM: The 2016 WHO classification of tumours of the
urinary system and male genital organs-part A: Renal, penile, and
testicular tumours. Eur Urol. 70:93–105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gill AJ, Hes O, Papathomas T, Šedivcová M,
Tan PH, Agaimy A, Andresen PA, Kedziora A, Clarkson A, Toon CW, et
al: Succinate dehydrogenase (SDH)-deficient renal carcinoma: A
morphologically distinct entity: a clinicopathologic series of 36
tumors from 27 patients. Am J Surg Pathol. 38:1588–1602. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Williamson SR, Eble JN, Amin MB, Gupta NS,
Smith SC, Sholl LM, Montironi R, Hirsch MS and Hornick JL:
Succinate dehydrogenase-deficient renal cell carcinoma: Detailed
characterization of 11 tumors defining a unique subtype of renal
cell carcinoma. Mod Pathol. 28:80–94. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tsai TH and Lee WY: Succinate
dehydrogenase-deficient renal cell carcinoma. Arch Pathol Lab Med.
143:643–647. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McEvoy CR, Koe L, Choong DY, Leong HS, Xu
H, Karikios D, Plew JD, Prall OW, Fellowes AP and Fox SB:
SDH-deficient renal cell carcinoma associated with biallelic
mutation in succinate dehydrogenase A: Comprehensive genetic
profiling and its relation to therapy response. NPJ Precis Oncol.
2:92018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yakirevich E, Ali SM, Mega A, McMahon C,
Brodsky AS, Ross JS, Allen J, Elvin JA, Safran H and Resnick MB: A
novel SDHA-deficient renal cell carcinoma revealed by comprehensive
genomic profiling. Am J Surg Pathol. 39:858–863. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ozluk Y, Taheri D, Matoso A, Sanli O,
Berker NK, Yakirevich E, Balasubramanian S, Ross JS, Ali SM and
Netto GJ: Renal carcinoma associated with a novel succinate
dehydrogenase A mutation: A case report and review of literature of
a rare subtype of renal carcinoma. Hum Pathol. 46:1951–1955. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang Q, Zhang Y, Zhou YH, Hou YY, Wang
JY, Li JL, Li M, Tong HX and Lu WQ: A novel germline mutation in
SDHA identified in a rare case of gastrointestinal stromal tumor
complicated with renal cell carcinoma. Int J Clin Exp Pathol.
8:12188–12197. 2015.PubMed/NCBI
|
|
14
|
Wang Y, He P, Zhou X, Wang C, Fu J, Zhang
D, Liao D, Zhou Z, Wu C and Gong W: Gene mutation profiling and
clinical significances in patients with renal cell carcinoma.
Clinics (Sao Paulo). 78:1002592023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fuchs TL, Maclean F, Turchini J, Vargas
AC, Bhattarai S, Agaimy A, Hartmann A, Kao CS, Ellis C, Bonert M,
et al: Expanding the clinicopathological spectrum of succinate
dehydrogenase-deficient renal cell carcinoma with a focus on
variant morphologies: A study of 62 new tumors in 59 patients. Mod
Pathol. 35:836–849. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
WHO Classification of Tumours Editorial
Board, . Urinary and male genital tumours: WHO classification of
tumours. 5th edition. Vol 8. International Agency for Research on
Cancer; Lyon, France: 2022
|
|
17
|
Kamai T, Higashi S, Murakami S, Arai K,
Namatame T, Kijima T, Abe H, Jamiyan T, Ishida K, Shirataki H and
Yoshida KI: Single nucleotide variants of succinate dehydrogenase A
gene in renal cell carcinoma. Cancer Sci. 112:3375–3387. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Argani P, Hawkins A, Griffin CA, Goldstein
JD, Haas M, Beckwith JB, Mankinen CB and Perlman EJ: A distinctive
pediatric renal neoplasm characterized by epithelioid morphology,
basement membrane production, focal HMB45 immunoreactivity, and
t(6;11)(p21.1;q12) chromosome translocation. Am J Pathol.
158:2089–2096. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Akgul M, Williamson SR, Ertoy D, Argani P,
Gupta S, Caliò A, Reuter V, Tickoo S, Al-Ahmadie HA, Netto GJ, et
al: Diagnostic approach in TFE3-rearranged renal cell carcinoma: A
multi-institutional international survey. J Clin Pathol.
74:291–299. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang XM, Shao L, Xiao H, Myers JL,
Pantanowitz L, Skala SL, Udager AM, Vaishampayan U, Mannan R,
Dhanasekaran SM, et al: Lessons from 801 clinical TFE3/TFEB
fluorescence in situ hybridization assays performed on renal cell
carcinoma suspicious for MiTF family aberrations. Am J Clin Pathol.
160:549–554. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Smith SC, Sirohi D, Ohe C, McHugh JB,
Hornick JL, Kalariya J, Karia S, Snape K, Hodgson SV, Cani AK, et
al: A distinctive, low-grade oncocytic fumarate hydratase-deficient
renal cell carcinoma, morphologically reminiscent of succinate
dehydrogenase-deficient renal cell carcinoma. Histopathology.
71:42–52. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tjota M, Chen H, Parilla M, Wanjari P,
Segal J and Antic T: Eosinophilic renal cell tumors with a TSC and
MTOR gene mutations are morphologically and immunohistochemically
heterogenous: Clinicopathologic and Molecular Study. Am J Surg
Pathol. 44:943–954. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lalani AA, Heng DYC, Basappa NS, Wood L,
Iqbal N, McLeod D, Soulières D and Kollmannsberger C: Evolving
landscape of first-line combination therapy in advanced renal
cancer: A systematic review. Ther Adv Med Oncol.
14:175883592211086852022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moran M, Nickens D, Adcock K, Bennetts M,
Desscan A, Charnley N and Fife K: Sunitinib for metastatic renal
cell carcinoma: A systematic review and meta-analysis of real-world
and clinical trials data. Target Oncol. 14:405–416. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aggen DH, Drake CG and Rini BI: Targeting
PD-1 or PD-L1 in metastatic kidney cancer: Combination therapy in
the first-line setting. Clin Cancer Res. 26:2087–2095. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brown JE, Royle KL, Gregory W, Ralph C,
Maraveyas A, Din O, Eisen T, Nathan P, Powles T, Griffiths R, et
al: Temporary treatment cessation versus continuation of first-line
tyrosine kinase inhibitor in patients with advanced clear cell
renal cell carcinoma (STAR): An open-label, non-inferiority,
randomised, controlled, phase 2/3 trial. Lancet Oncol. 24:213–227.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Choueiri TK, Powles T, Peltola K, de
Velasco G, Burotto M, Suarez C, Ghatalia P, Iacovelli R, Lam ET,
Verzoni E, et al: Belzutifan versus everolimus for advanced
renal-cell carcinoma. N Engl J Med. 391:710–721. 2024. View Article : Google Scholar : PubMed/NCBI
|