Introduction
Pancreatic cancer is a particularly lethal disease
with an annual incidence rate almost identical to the mortality
rate. At the time of diagnosis, more than 80% of patients usually
display either locally advanced or metastatic disease. The median
survival of patients with locally advanced or metastatic disease is
9 or 4 months, respectively, and the 5-year survival rate is 1–4%
(1). In the past 10 years,
gemcitabine has replaced 5-fluorouracil as a standard therapy for
advanced disease; however, the median survival has only modestly
improved from 4.4 to 5.6 months (2). Since then, phase III trials of novel
cytotoxic or biologic agents combined with gemcitabine have failed
to show any survival improvement compared with gemcitabine alone
(3). A recent phase III trial (PA.3
study) compared gemcitabine plus erlotinib, an epidermal growth
factor receptor (EGFR) inhibitor, with gemcitabine alone. The
findings showed a significant improvement, with a median survival
of 6.2 vs. 5.9 months, as well as a 1-year survival of 23 vs. 17%,
respectively (4). As a result,
erlotinib in combination with gemcitabine was approved by the U.S.
Food and Drug Administration in 2005 for the treatment of locally
advanced or metastatic chemonaive pancreatic cancer.
Human EGFR is a member of the ERBB family of
receptor tyrosine kinases, consisting of four closely related
members: EGFR (ERBB1), HER2 (ERBB2), HER3 (ERBB3) and HER4 (ERBB4).
On binding to EGFR, ligands, such as epidermal growth factor (EGF)
or transforming growth factor α, cause receptor dimerization with
one of the ERBB family molecules. Dimerization of receptors
activates the tyrosine kinase located at the intracellular domain
of the receptor molecules, leading to receptor autophosphorylation
and the initiation of signal-transduction cascades involving
RAS/RAF/MAPK and PI3K/AKT, culminating in cell proliferation and
survival (5). The EGFR signal
network, one of the important processes involved in tumor
progression including cell proliferation, inhibition of apoptosis,
metastasis and angiogenesis, is often dysregulated in cancer cells
(6).
Overexpression of EGFR occurs in more than 50% of
pancreatic cancers and correlates with poor prognosis and disease
progression (7). The frequency of
the EGFR mutation is only 1.5% in pancreatic cancers (8), but 59% in lung cancers (9). On the other hand, pancreatic cancer
shows the highest frequency of K-ras oncogene (KRAS) mutations
among human cancers (10). KRAS
mutations, which constitutively activate RAF/MAPK signaling, are
detected in up to 90% of pancreatic cancers (11). In the PA.3 study, no significant
correlations were found between outcome and KRAS mutations, which
were detected in 79% of the erlotinib arm, although a favorable
trend for erlotinib in patients that carry the wild-type KRAS has
been observed (12).
Erlotinib is an orally active, reversible,
competitive inhibitor of the EGFR tyrosine kinase ATP-binding site
and blocks the downstream signal transduction of the EGFR
associated with cancer progression (13). Erlotinib reportedly enhances the
antitumor activity of gemcitabine in pancreatic cancer cell lines
with or without a KRAS mutation (14). However, there is no in vivo
evidence regarding the improvement of antitumor activity of a
combination therapy of erlotinib and gemcitabine in KRAS-mutated
pancreatic cancer cell lines (14,15).
Therefore, the present study aimed to determine whether a
combination treatment of erlotinib and gemcitabine produces an
enhanced antitumor activity in a xenograft model using a
KRAS-mutated pancreatic cancer cell line.
Materials and methods
Chemicals
Erlotinib was provided by F. Hoffman-La Roche Ltd.
(Basel, Switzerland) and dissolved in DMSO for the in vitro
study, and in 6% Captisol® (Cydex Inc., Lenexa, KS, USA)
solution for the in vivo study. Captisol and gemcitabine
(Eli Lilly, Tokyo, Japan) were dissolved in distilled water and
saline, respectively.
Tumors
The KRAS-mutated human pancreatic cancer cell lines,
HPAC and Capan-1, were purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA) and maintained in
ATCC-recommended medium at 37°C in 5% CO2. The HPAC cell
lines were maintained in DMEM/F12 (Invitrogen, Carlsbad, CA, USA)
supplemented with 5% heat-inactivated fetal bovine serum (FBS;
Japan Bioserum, Hiroshima, Japan), 0.005 mg/ml transfferin
(Invitrogen), 40 ng/ml hydrocortisone (Sigma-Aldrich, St. Louis,
MO, USA), 10 ng/ml EGF (Invitrogen) and 0.002 mg/ml insulin
(Sigma-Aldrich). The Capan-1 cells were maintained in IMDM
(Sigma-Aldrich) supplemented with 20% FBS.
In vitro cell proliferation-inhibition
assays
To evaluate cell proliferation inhibition, the
tetrazolium dye
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]
(MTT) (Dojindo, Tokyo, Japan) assay was performed. Cells were
precultured overnight at 37°C in 96-well clear plates, and the
drugs were added alone or in combination. After treatment for 4
(HPAC cells) or 6 days (Capan-1 cells) at 37°C, 10 ml of MTT was
added to each well and incubated for 2–5 h at 37°C. The optical
density of each well was measured at 450 and 600 nm with a
Benchmark Plus microplate reader (Bio-Rad, Hercules, CA, USA). Each
experiment was performed in duplicate or triplicate for each drug
concentration and was independently performed two or three times.
The percentage of cell proliferation was calculated as: [(mean
absorbance of drug-treated wells - mean absorbance of cell-free
wells)/(mean absorbance of vehicle wells − mean absorbance of
cell-free wells)] × 100. The effects of the erlotinib and
gemcitabine combination were evaluated using a combination index
(CI) interpreted as: <1.0, synergistic; 1.0, additive and
>1.0, antagonistic (16). The CI
for each fraction-affected value representing the percentage of
proliferation inhibited by a drug was calculated using the
Chou-Talalay method [the isobologram equation was used mutually
non-exclusive (α=1)] (16,17). The fraction-affected value (Fa)/CI
plots for the cell lines were constructed in Excel 2003.
Immunoblotting
Cultured cells were washed twice with ice-cold PBS,
scraped and pelleted by centrifugation at 650 × g for 2 min at 4°C.
The pellets were lysed in lysis buffer (Invitrogen) supplemented
with 1 mM PMSF, 1 mM NaF and 1 mg/ml aprotinin (all from
Sigma-Aldrich) for 5 min on ice, followed by sonication.
Supernatants were collected by centrifugation at 16,000 × g for 10
min at 4°C. Protein concentrations were determined using DC protein
assay reagent (Bio-rad). Samples of the proteins (50 mg) were
diluted with Laemmli sample buffer (Sigma-Aldrich) and applied to
7.5% XV Pantera gel (DRC, Tokyo, Japan). Separated proteins were
electrophoretically transferred to 0.22-mm Immobilon membranes
(Millipore, Tokyo, Japan) and blocked for 1 h at room temperature
in Superblock blocking buffer (Thermo Scientific, Yokohama, Japan).
Membranes were incubated overnight at 4°C with antibodies
recognizing phospho-EGFR (Y1068, and Y845), EGFR and GAPDH (Santa
Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were then
washed with TTBS (Thermo Scientific) and probed with horseradish
peroxidase-conjugated anti-rabbit or anti-mouse antibody (Santa
Cruz Biotechnology) for 2 h at room temperature. After three
additional washes with TTBS, the membranes were incubated in
Enhanced Chemiluminescence Plus reagent (Amersham Biosciences,
Piscataway, NJ, USA) and detected by an ImageQuant Imager (GE
Healthcare Bio-Sciences, Tokyo, Japan).
Human cancer xenograft models
Male 5-week-old BALB-nu/nu
(CAnN.Cg-Foxn1<nu>/CrlCrlj nu/nu) mice were purchased from
Charles River Japan, Inc. (Yokohama, Japan). The animals were
housed in a pathogen-free environment under controlled conditions
(temperature 20–26°C, humidity 40–70%, light-dark cycle 12–12 h).
Chlorinated water and irradiated food were provided ad
libitum. The animals were allowed to acclimatize and recover
from shipping-related stress for one week prior to the study. The
health of the mice was monitored daily.
A cell suspension of HPAC cells (106
viable cells/mouse) was subcutaneously inoculated into the right
flank of each mouse. Fifteen days after the tumor cell inoculation,
the mice were randomly divided into four groups of eight mice and
administered either oral erlotinib (50 mg/kg/day) on days 1–21 or
intravenous gemcitabine (20 mg/kg) on days 1, 8 and 15. In the
combination therapy, erlotinib and gemcitabine were administered at
the same dose and schedule as each drug alone. Captisol (6%) or
saline was administered as the control. The drugs were administered
following the same schedule as the clinical setting (4). Tumor diameter was measured twice a
week using calipers, and tumor volume was calculated as:
ab2/2 mm3, where a is the length and b is the
width of the tumor. Day 1 denotes the first day of treatment and
day 22, the day on which the drug effects were estimated.
The protocol was reviewed by the Institutional
Animal Care and Use Committee of Chugai Pharmaceutical Co., Ltd.
Animal experiments were performed in accordance with the
‘Guidelines for the Accommodation and Care of Laboratory Animals’
of Chugai Pharmaceutical Co., Ltd.
Statistical analysis
The Wilcoxon test was used to detect the statistical
differences in tumor volume. Probability values <0.05 were
considered to be significant. Statistical analysis was performed
using an SAS preclinical package (version 8.2; SAS Institute Inc.,
Cary, NC, USA).
Results
In vitro anti-proliferative activity of
erlotinib in combination with gemcitabine in the HPAC and Capan-1
pancreatic cancer cell lines
We first determined the proliferation inhibitory
effect of erlotinib and gemcitabine alone on pancreatic cancer cell
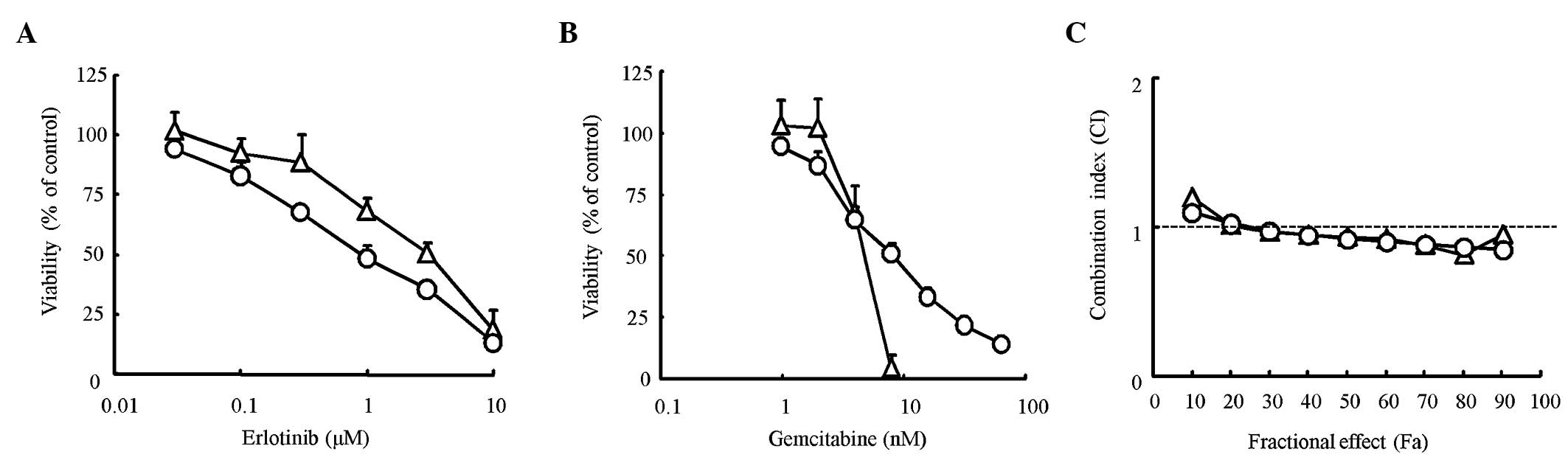
lines using the MTT assay. The IC50 value for erlotinib
was 1.1 μM for HPAC cells and 3.0 μM for Capan-1 cells. The
IC50 value for gemcitabine was 7.8 nM for HPAC cells and
4.4 nM for Capan-1 cells (Fig. 1A and
B). The difference between the IC50 values found for
the two cell lines was only slight for both erlotinib and
gemcitabine. To evaluate the combination effect of erlotinib and
gemcitabine, a combination index (CI) was determined using the
Chou-Talalay method. The CI value was nearly equal to 1 for every
dose in the two cell lines, suggesting that the combination effects
of erlotinib and gemcitabine were ‘additive’ in the two cell lines
(Fig. 1C). Thus, the effects of
erlotinib in combination with gemcitabine were considered additive
in KRAS-mutated pancreatic cancer cells.
In vitro effects of erlotinib in
combination with gemcitabine on EGFR signaling in the HPAC
pancreatic cancer cell line
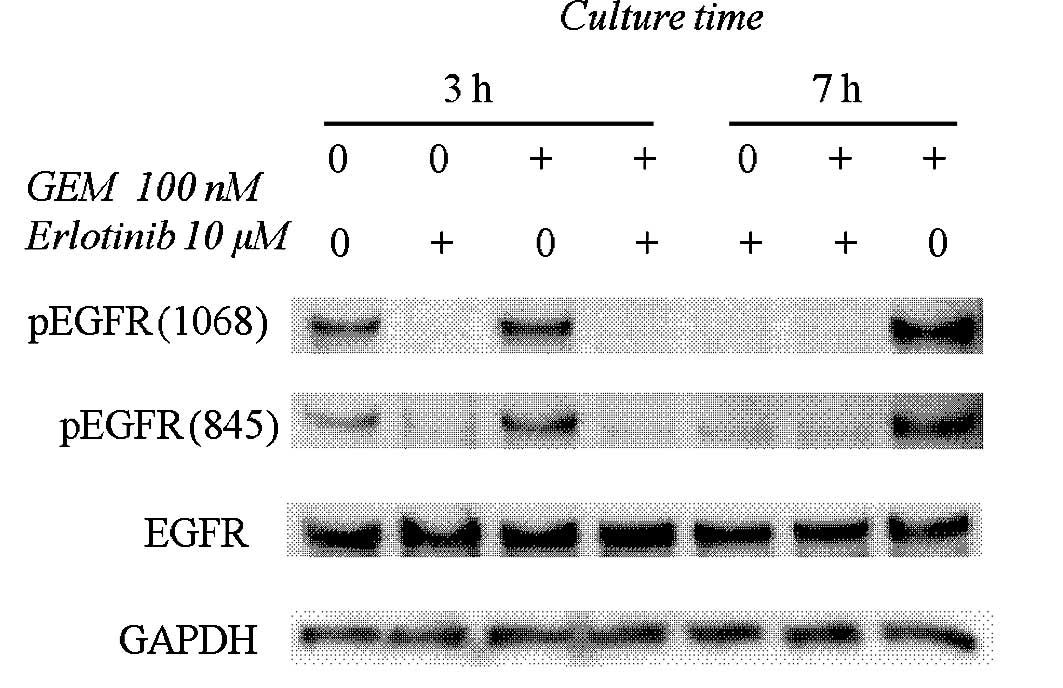
Fig. 2 shows that 10
μM erlotinib inhibited EGFR phosphorylation at the Y845
(Src-dependent phosphorylation) and Y1068 (auto-phosphorylation)
sites. Gemcitabine (100 nM) augmented the phosphorylation levels at
Y845 and Y1068 of EGFR, and these phosphorylations were completely
blocked by erlotinib (Fig. 2).
Antitumor effects of erlotinib in
combination with gemcitabine in the HPAC xenograft model
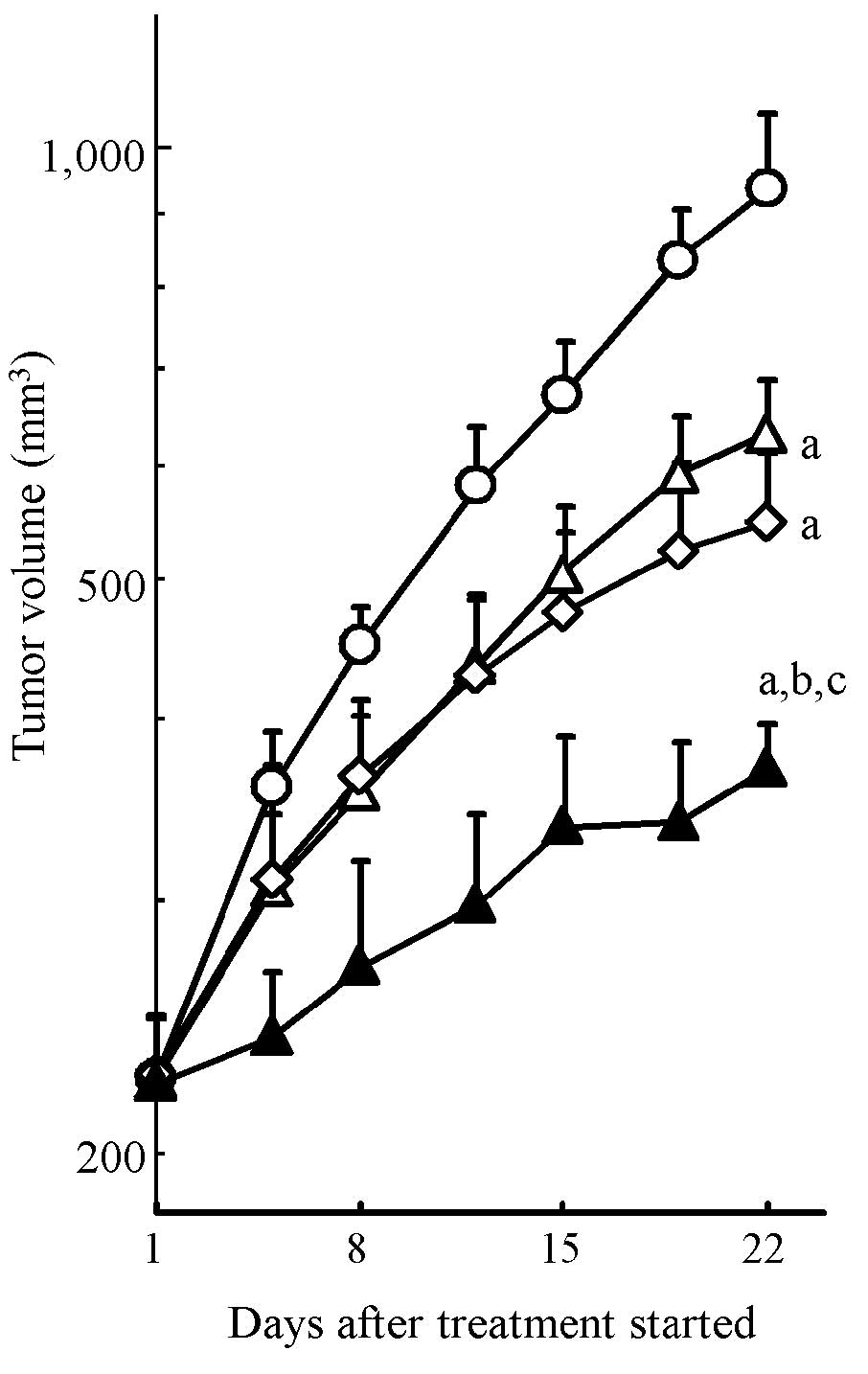
To evaluate the combination effect of erlotinib and
gemcitabine on in vivo tumor growth inhibition, we conducted
xenograft model experiments using the HPAC cell line. HPAC tumor
growth was significantly inhibited by erlotinib and gemcitabine
monotherapy (P<0.05, Fig. 3).
The combination treatment with erlotinib and gemcitabine resulted
in significantly stronger tumor growth inhibition compared to each
drug alone (P<0.05) (Fig. 3).
Tumor volume on day 22 (mean ± SD) was 371±28 mm3 in the
combination treatment group, 634±58 mm3 in the erlotinib
group, 549±66 mm3 in the gemcitabine group and 938±122
mm3 in the vehicle group. Significant body weight loss
was not observed in any treatment group throughout the experiment
(Table I). These results showed
that erlotinib enhanced the antitumor activity of gemcitabine
without additional toxicity of normal tissue in the KRAS-mutated
pancreatic cancer xenograft model.
 | Table IBody weight of mice treated with
erlotinib and/or gemcitabine in the HPAC tumor xenograft model. |
Table I
Body weight of mice treated with
erlotinib and/or gemcitabine in the HPAC tumor xenograft model.
| Group | Body weight (g) |
|---|
|
|
|---|
| Day 1 | Day 22 |
|---|
| Vehicle
(control) | 26.6±1.1 | 27.0±1.8 |
| Erlotinib (50
mg/kg) | 26.8±1.0 | 26.1±0.8 |
| Gemcitabine (20
mg/kg) | 26.5±1.1 | 26.3±1.5 |
| Erlotinib (50 mg/kg)
+ gemcitabine (20 mg/kg) | 27.0±1.0 | 26.4±1.5 |
Discussion
No clinical trial thus far, using combination agents
such as oxaliplatin, cisplatin, irinotecan, 5-fluorouracil,
marimastat (matrix metalloproteinase inhibitor) or tipifarnib
(farnesyltransferase inhibitor) with gemcitabine, has produced
significant survival improvement over gemcitabine alone (3). Erlotinib is the first agent to produce
a significant improvement in survival in combination with
gemcitabine compared with gemcitabine alone (4).
Erlotinib, an EGFR-targeting drug, has been approved
in many countries for the treatment of non-small-cell lung cancer
(NSCLC) patients who previously received chemotherapy. Recently,
the combination therapy of erlotinib with gemcitabine has been
approved for the treatment of locally advanced or metastatic
chemonaive pancreatic cancer. In a pre-clinical study, Morgan et
al demonstrated the therapeutic effect of erlotinib in
combination with gemcitabine in a mouse xenograft model using the
KRAS wild-type human pancreatic cancer cell line BxPC-3 (14). However, in the clinical setting, the
frequency of mutations in the KRAS oncogene can reach 90% in
pancreatic ductal adenocarcinomas (11). Therefore, we conducted in
vivo experiments using KRAS-mutated pancreatic cancer cell
lines. The present study showed that erlotinib significantly
enhanced the antitumor activity of gemcitabine in a xenograft model
inoculated with the KRAS-mutated pancreatic cancer cell line, HPAC.
Our results, together with those of Morgan et al, are
consistent with the results of the PA.3 clinical trial (4) and indicate the clinical benefit of a
combination therapy of erlotinib and gemcitabine for the treatment
of pancreatic cancer regardless of KRAS status.
The present in vitro and in vivo
experiments showed that erlotinib enhanced the antitumor activity
of gemcitabine against KRAS-mutated cells. However, the mechanism
involved in the effects of the combination of erlotinib and
gemcitabine remains unclear. An increase in apoptosis with a
combination treatment compared with each agent alone in head and
neck carcinoma has been reported (18). Changes in cell cycle distributions
may explain the mechanism involved in the increase of apoptosis of
cancer cells treated with a combination of an EGFR inhibitor and
gemcitabine. Gemcitabine treatment induces an accumulation of the
S-phase population, which is considered sensitive to erlotinib;
thus, the combination treatment may enhance antitumor activity.
We found that gemcitabine increased the level of
EGFR phosphorylation, which was entirely blocked by erlotinib in
the KRAS-mutated pancreatic cancer cells. EGFR expression and its
phosphorylation are one of the significant factors determining the
sensitivity of cells to erlotinib-induced growth inhibition
(19). Therefore, the increase in
EGFR phosphorylation by gemcitabine may render cancer cells more
sensitive to erlotinib. These results suggest that EGFR activation
may be a survival response in HPAC pancreatic cancer cells treated
with gemcitabine. Furthermore, the inhibition of the
gemcitabine-induced phosphorylation of EGFR by erlotinib may block
this initial survival response and promote cytotoxicity from
gemcitabine. Phosphorylation of EGFR at Y845 in response to
gemcitabine was previously shown in several pancreatic and head and
neck cancer cells (14,18). Although the mechanism involved in
the increase in phosho-EGFR from gemcitabine remains unclear, it is
plausible that the gemcitabine-mediated degradation of Cdc25A
phosphatase, which is known to directly regulate EGFR
phosphorylation levels, is involved (20). Numerous studies suggest that EGFR
activation induced by cellular stress, such as
H2O2, UV and chemotherapeutic agents
including cisplatin, 5-fluorouracil, paclitaxel, doxorubicin and
irinotecan (14), promotes an
anti-apoptotic survival response through the activation of the MAPK
or Akt signal pathways (21,22).
Therefore, gemcitabine may also be an agent of cellular stress that
induces EGFR activation.
Recently, the sequence-specific interactions of
erlotinib and chemotherapeutic drug combinations were found to
influence the efficacy of regimens in NSCLC (23). The treatment schedule of erlotinib
and gemcitabine also affects the combination effects. Gemcitabine
followed by erlotinib enhances the antitumor effects of each drug
alone, while erlotinib followed by gemcitabine has antagonistic
effects (14,18). In the present study, erlotinib and
gemcitabine were administered, not sequentially, but concurrently
in both in vitro and in vivo experiments, consistent
with the drug treatment schedule of the PA.3 clinical trial. The
difference in the effects between sequential treatment with
gemcitabine followed by erlotinib and concurrent treatment was not
determined in our experiments. However, it is plausible that
sequential treatment may be more effective compared to concurrent
treatment, since erlotinib immediately increases the G1-phase
population of the cell cycle (24)
leading to a reduction in S-phase entry, which is crucial for
gemcitabine-mediated cytotoxicity.
In conclusion, we demonstrated that the treatment of
erlotinib in combination with gemcitabine exerted antitumor
activity superior to each drug as a monotherapy in the KRAS-mutated
pancreatic cancer model. Our results confirm that the combination
of erlotinib and gemcitabine has potential therapeutic benefits
against pancreatic cancers. Our findings are useful in the
investigation of molecular agents targeted to pathways other than
EGFR signal cascade in pancreatic cancer treated with gemcitabine,
as well as in the exploration of new combination regimens including
erlotinib and gemcitabine for more efficacious therapies against
pancreatic cancer.
Acknowledgements
We thank Ms. F. Ford for the editorial
assistance.
References
|
1
|
Wanebo HJ and Vezeridis MP: Pancreatic
carcinoma in perspective. A continuing challenge. Cancer.
78:580–591. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD and von Hoff DD:
Improvements in survival and clinical benefit with gemcitabine as
first-line therapy for patients with advanced pancreas cancer: a
randomized trial. J Clin Oncol. 15:2403–2413. 1997.PubMed/NCBI
|
|
3
|
Van Cutsem E, Verslype C and Grusenmeyer
PA: Lessons learned in the management of advanced pancreatic
cancer. J Clin Oncol. 25:1949–1952. 2007.PubMed/NCBI
|
|
4
|
Moore MJ, Goldstein D, Hamm J, Figer A,
Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos
D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M and
Parulekar W: Erlotinib plus gemcitabine compared with gemcitabine
alone in patients with advanced pancreatic cancer: a Phase III
trial of the National Cancer Institute of Canada Clinical Trials
Group. J Clin Oncol. 25:1960–1966. 2007. View Article : Google Scholar
|
|
5
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dancey J and Sausville EA: Issues and
progress with protein kinase inhibitors for cancer treatment. Nat
Rev Drug Discov. 2:296–313. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ueda S, Ogata S, Tsuda H, Kawarabayashi N,
Kimura M, Sugiura Y, Tamai S, Matsubara O, Hatsuse K and Mochizuki
H: The correlation between cytoplasmic overexpression of epidermal
growth factor receptor and tumor aggressiveness: poor prognosis in
patients with pancreatic ductal adenocarcinoma. Pancreas. 29:e1–8.
2004. View Article : Google Scholar
|
|
8
|
Lee J, Jang K-T, Ki C-S, Lim T, Park YS,
Lim HY, Choi D-W, Kang WK, Park K and Park JO: Impact of epidermal
growth factor receptor (EGFR) kinase mutations, EGFR gene
amplifications and KRAS mutations on survival of pancreatic
adenocarcinoma. Cancer. 109:1561–1569. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takano T, Ohe Y, Sakamoto H, et al:
Epidermal growth factor receptor gene mutations and increased copy
numbers predict gefitinib sensitivity in patients with recurrent
non-small cell lung cancer. J Clin Oncol. 23:6829–6837. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Almoguera C, Shibata D, Forrester K,
Martin J, Arnheim N and Perucho M: Most human carcinomas of the
exocrine pancreas contain mutant c-K-ras genes. Cell. 53:549–554.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lohr M, Kloppel G, Maisonneuve P,
Lowenfels AB and Luttges J: Frequency of K-ras mutations in
pancreatic intraductal neoplasias associated with pancreatic ductal
adenocarcinoma and chronic pancreatitis: a meta-analysis.
Neoplasia. 7:17–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moore MJ, da Cunha Santos G, Kamel-Reid S,
Chin K, Tu D, Parulekar W, Ludkovski O, Squire J, Richardson F and
Tsao M: The relationship of K-ras mutations and EGFR gene copy
number to outcome in patients treated with erlotinib in the
National Cancer Institute of Canada Clinical Trials Group trial
study PA.3. Proc ASCO. 25:abs 45212007.
|
|
13
|
Dowell J, Minna JD and Kirkpatrick P:
Erlotinib hydrochloride. Nat Rev Drug Discov. 4:13–14. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morgan MA, Parsels LA, Kollar LE, Normolle
DP, Maybaum J and Lawrence TS: The combination of epidermal growth
factor receptor inhibitors with gemcitabine and radiation in
pancreatic cancer. Clin Cancer Res. 14:5142–5149. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ng SS, Tsao MS, Nicklee T and Hedley DW:
Effects of the epidermal growth factor receptor inhibitor OSI-774,
Tarceva, on downstream signaling pathways and apoptosis in human
pancreatic adenocarcinoma. Mol Cancer Ther. 1:777–783.
2002.PubMed/NCBI
|
|
16
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koizumi F, Kanzawa F, Ueda Y, Koh Y,
Tsukiyama S, Taguchi F, Tamura T, Saijo N and Nishio K: Synergistic
interaction between the EGFR tyrosine kinase inhibitor gefitinib
(‘Iressa’) and the DNA topoisomerase I inhibitor CPT-11
(irinotecan) in human colorectal cancer cells. Int J Cancer.
108:464–472. 2004.
|
|
18
|
Chun PY, Feng FY, Scheurer AM, Davis MA,
Lawrence TS and Nyati MK: Synergistic effects of gemcitabine and
gefitinib in the treatment of head and neck carcinoma. Cancer Res.
66:981–988. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mendelsohn J and Baselga J: The EGF
receptor family as targets for cancer therapy. Oncogene.
19:6550–6565. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Morgan MA, Parsels LA, Parsels JD,
Mesiwala AK, Maybaum J and Lawrence TS: Role of checkpoint kinase 1
in preventing premature mitosis in response to gemcitabine. Cancer
Res. 65:6835–6842. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, McCullough KD, Franke TF and
Holbrook NJ: Epidermal growth factor receptor-dependent Akt
activation by oxidative stress enhances cell survival. J Biol Chem.
275:14624–14631. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Miyazaki Y, Hiraoka S, Tsutsui S, Kitamura
S, Shinomura Y and Matsuzawa Y: Epidermal growth factor receptor
mediates stress-induced expression of its ligands in rat gastric
epithelial cells. Gastroenterology. 120:108–116. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mahaffey CM, Davies AM, Lara PN Jr, Pryde
B, Holland W, Mack PC, Gumerlock PH and Gandara DR:
Schedule-dependent apoptosis in K-ras mutant non-small cell lung
cancer cell lines treated with docetaxel and erlotinib: rationale
for pharmacodynamic separation. Clin Lung Cancer. 8:548–553. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ling YH, Li T, Yuan Z, Haigentz M Jr,
Weber TK and Perez-Soler R: Erlotinib, an effective epidermal
growth factor receptor tyrosine kinase inhibitor, induces p27KIP1
up-regulation and nuclear translocation in association with cell
growth inhibition and G1/S phase arrest in human non-small cell
lung cancer cell lines. Mol Pharmacol. 72:248–258. 2007. View Article : Google Scholar
|