Introduction
Nephroblastoma (Wilms’ tumor) is a common pediatric
embryonal tumor that occurs as a result of abnormal cellular
differentiation during organogenesis. Nephroblastomas typically
exhibit triphasic histological components, consisting of blastema,
epithelia and stroma (1). According
to molecular pathology, tumors are now subcategorized into 2
subclasses by Wilms’ tumor 1 gene (WT1) mutation status
(2). The WT1-mutated
subclass usually contains a concomitant β-catenin mutation while
the WT1 wild-type subclass harbors mutations of a novel
candidate gene, WTX, in varying frequencies (3,4). While
WT1 is known to be a classic tumor suppressor in the
WT1-mutated subclass, however, its role is unclear in the
WT1 wild-type subclass as it appears to possess a different
tumorigenesis pathway (2).
WT1 encodes a zinc-finger protein that
functions as a transcription factor involved in cellular growth and
differentiation (5). A
physiological role of WT1 in renal development during
embryogenesis was confirmed by a previous study (6). The gene was originally described as a
tumor suppressor gene that is involved in the tumorigenesis of
nephroblastoma. However, only 10–15% of nephroblastomas harbor
WT1 mutations, and growing evidence suggests that
nephroblastomas with mutations of WT1 possess a separate
molecular pathogenesis to nephroblastomas without mutations
(2). Thus, the role of WT1
should be re-assessed in the WT1 wild-type subclass.
WT1 plays an oncogenic role in adult renal cell carcinoma
and various other human neoplasms, particularly leukemia, breast
and prostate cancers. These factors suggest a possible oncogenic
role for WT1 in pediatric nephroblastomas.
This study aimed to observe the expression of
WT1 in pediatric nephroblastomas. The expression levels were
analyzed with regard to WT1 mutation status and compared to
other pediatric renal tumors and neuroblastomas. Furthermore,
WT1 RNA interference was performed in a primary culture
model. The negative effects thereof were also demonstrated in tumor
growth, which was related to increased apoptosis.
Materials and methods
Tissue samples and nucleic acid
extraction
Frozen tumor tissue from 24 cases of nephroblastoma
and their normal kidney counterparts, 3 cases of other pediatric
renal tumors and 10 cases of neuroblastomas were used in the
mutation and RNA-based expression studies. The ‘other renal tumor’
group consisted of 2 cases of clear cell sarcoma of the kidney and
a case of pediatric renal cell carcinoma that harbored PRCC-TFE
fusion. Sample collection and use of clinical data were performed
under the guidelines of the Institutional Research Ethics Committee
(Project 50/368-023) of the Faculty of Medicine, Prince of Songkla
University. DNA extraction from the samples was carried out using a
Genomic DNA mini kit (Geneaid, Taiwan) following the manufacturer’s
suggested protocol. Total RNA was extracted using an RNAeasy
extraction kit (Qiagen, Inc.).
For the immunohistochemical study, formalin-fixed,
paraffin-embedded archival tissue samples from the 24 cases of
nephroblastoma were used. Clinical data from the same series of
patients were published in a previous study (7). Briefly, the series included 2 patients
who presented with a partial spectrum of WAGR (Wilms’ tumor,
aniridia, genitourinary tract anomalies and retardation)
association and 2 patients with bilateral tumors. None of the
patients had a family history of renal tumors.
WT1 and β-catenin mutation study
The full coding sequence of WT1 was studied
in each nephroblastoma sample by polymerase chain reaction (PCR)
and the direct nucleotide sequencing method. The primers and PCR
conditions for WT1 were identical to a previous study
(3), with some modifications. The
mutation study of β-catenin spanned exon 3, which encodes its
phosphorylation sites. Sequencing was performed in both
directions.
WT1 and β-catenin expression study
The relative expression of WT1 and β-catenin
at the mRNA level was studied with quantitative real-time reverse
transcription PCR (qRT-PCR). In brief, cDNA was constructed from 1
μg of RNA using an Omniscript reverse transcription kit (Qiagen,
Inc.) following the manufacturer’s instructions. qRT-PCR was
conducted with an ABI7300 model real-time PCR machine (Applied
Biosystems, Inc.) and a QuantiTech Probe RT-PCR kit was used
(Qiagen, Inc.). Expression of WT1 in terms of copy numbers
was normalized with 10−5 copies of GAPDH and was
presented logarithmically. The relative expression of
β-catenin was also performed in a similar manner. Details of
the primers and Taq Man probes were previously published (8,9).
The expression and localization of WT1 at the
protein level were studied in 24 clinical nephroblastoma tumor
tissue samples using immunohistochemistry. Hematoxylin and
eosin-stained slides from the case records were selected by a
pathologist for immunohistochemical study. Sections (3-μm) were
cut, deparaffinized and rehydrated from formalin-fixed,
paraffin-embedded tissue. A WT1 monoclonal antibody (1:500; Dako,
Inc.) was used as the primary antibody. The staining followed the
manufacturer’s suggested protocol for the Dako EnVision+ System
(Dako, Inc.). Briefly, antigen retrieval was performed in a
microwave oven using Tris-EDTA buffer. Endogenous peroxidase
activity was blocked with 0.03% hydrogen peroxide containing sodium
azide. Slides were incubated with non-immune serum for 30 min and
then with the primary antibody for 120 min in a moist chamber,
followed by an additional 30-min incubation with peroxidase-labeled
polymer conjugated to goat anti-mouse immunoglobulins. Color was
developed through a liquid 3,3′-diaminobenzidine chromogen
solution. Light counterstaining was carried out with hematoxylin.
β-catenin immunostaining was performed using the same protocol
described in a previous publication (10). The immunohistological staining in
this study was performed by one technician.
Primary culture of nephroblastoma
Primary nephroblastoma cells were derived from the
ascites of a patient with recurrent nephroblastoma. The primary
tumor of this patient was in stage 3 (gross tumor rupture prior to
surgery), showed favorable histology and harbored wild-type
WT1 and β-catenin. Following centrifugation of the
ascites, the pellet was washed 3 times with phosphate-buffered
saline (PBS). The cells were resuspended with RPMI-1640
(Invitrogen, Inc.) supplemented with 20% fetal bovine serum (FBS)
(Gibco BRL), 1% penicillin/streptomycin and 0.5% glutamine. Cells
were grown in 60-mm petri dishes and incubated in a 5%
CO2 at 37°C. The first medium change was carried out on
the second day when the cells began to attach properly. The medium
was changed every 3 days until the culture reached 80% confluence.
After the two passages, an aliquot of cells was cryopreserved in
90% FBS/10% dimethyl sulfoxide and stored in liquid nitrogen. All
experiments were performed on the fourth passage. Further
subcultures showed that the cells ceased their proliferative
ability at the fourteenth passage at 4 months of continuous
culture.
WT1 RNA interference and Annexin-V
apoptosis assay
The siRNAs against WT1 (siRNAWT1)
used in this study were presented in a previous publication by our
group (11). Briefly,
siRNAWT1 was a combination of three 25-nt siRNA duplexes
targeting different non-overlapping regions of WT1 mRNA on
exons 7 and 8.
One day prior to transfection, a total of
1.5×105 cells were seeded in each well of a 24-well
plate. The cells were transfected with siRNAWT1 at a
final concentration of 100 nM using Fugene6-HD (Roche, Inc.) as a
transfection agent. The transfection agent plus a non-specific
sequence (Invitrogen, Inc.) was used as a mock control. Cells were
harvested at 48 and 72 h after transfection to check the expression
of WT1 mRNA. The expression was semi-quantitatively determined
using the RT-PCR method, using 28 cycles of reaction and the same
primer set as WT1 qRT-PCR. The cell survival assay after WT1
inhibition was performed in a 24-well plate format. The cells were
plated at a near-confluence density of 5×104 cells/well.
Following siRNAWT1 transfection, the number of cells in
each group was determined by counting under a hemocytometer using
the trypan blue exclusion technique.
The apoptosis experiment was performed in a 60-mm
disc format. Cells were plated in three discs one night prior to
transfection. siRNAWT1 and the non-specific sequence
were transfected at the same concentration as the experiment in the
6-well plate format. The cells were trypsinized and harvested at 72
h of transfection. The apoptosis assay used the double staining of
Annexin-V, the propidium iodide method and flow cytometry on the
Becton-Dickinson FACScan platform. Flow cytometric analysis was
conducted using the CellQuest program.
Results
Mutations of WT1 and β-catenin in
pediatric nephroblastoma
Among the 24 nephroblastomas examined, WT1
mutations were detected in 4 cases (Table I). Two of these cases had associated
WAGR syndrome and 1 case had bilateral tumors. Of the 4 tumors with
a WT1 mutation, concomitant mutations at the β-catenin codon
45 were detected in 3 tumors. Except for WT1 mutations
detected in the normal tissue of 2 WAGR cases, all other mutations
were somatic.
 | Table IWT1 and β-catenin genotypes and the
WT1 immunoreactivity pattern of 4 nephroblastoma cases with WT1
mutations. |
Table I
WT1 and β-catenin genotypes and the
WT1 immunoreactivity pattern of 4 nephroblastoma cases with WT1
mutations.
| Code | Clinical remarks | WT1 mutation | β-catenin
mutation | WT1
immunohistochemistry |
|---|
| WT8 | WAGR association | Exon 8 CGA413TGA
(Arg413stop) | TCT45TAT
ser45tyr | Stroma (+) |
| WT13 | Bilateral
disease | Exon 8 CGA413TGA
(Arg413stop) | - | Stroma (+) |
| WT20 | - | Exon 9 CGG445TGG
(Arg445Trp) | TCT45TGT
ser45cys | Blastema (++) |
| WT24 | WAGR association | Exon 7 151-152 (ins
32 bp) | TCT45TTT
ser45phe | Stroma (+) |
Relative overexpression of WT1 in
pediatric nephroblastoma
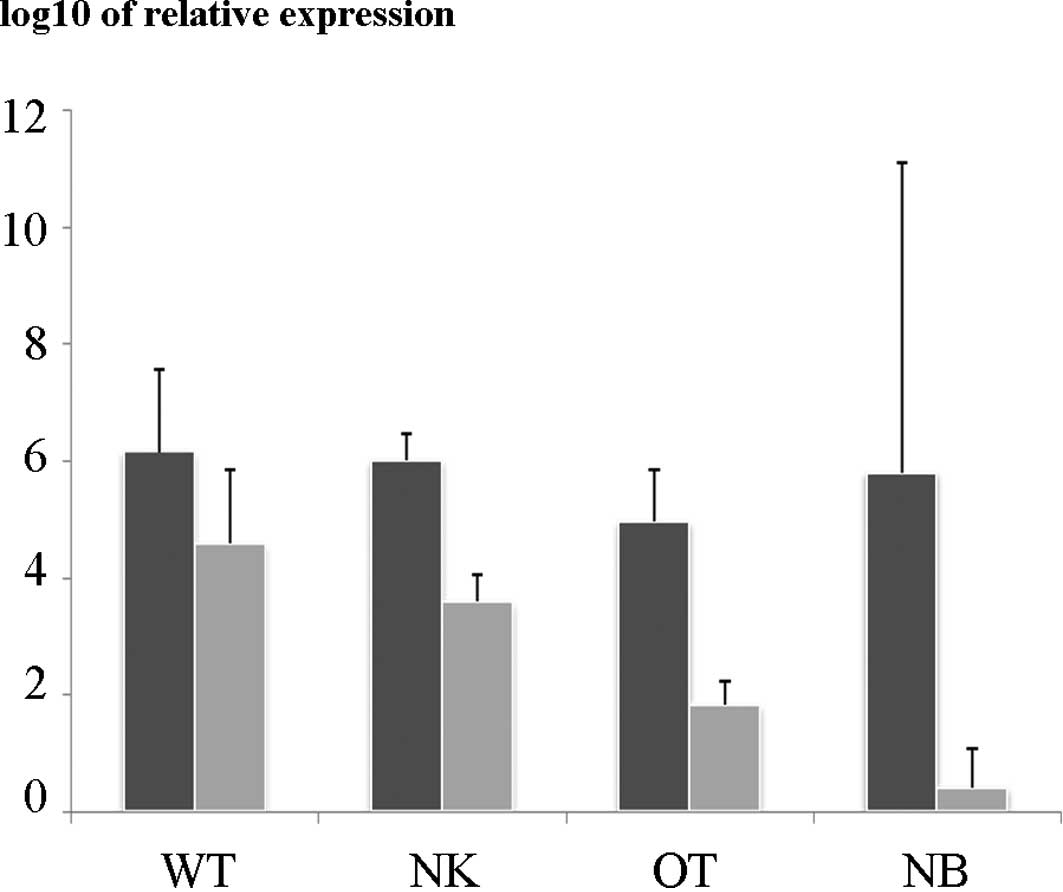
In the qRT-PCR study, the relative expression of
WT1 in the nephroblastomas was significantly higher than
that in the control renal tissue and the other renal tumors. The
expression of WT1 in the nephroblastomas and other pediatric
renal tumors was higher than that in the neuroblastoma tissues
(Fig. 1). No significant difference
was noted in the relative expression of β-catenin among the
nephroblastomas and other types of tissue studied. The relative
expression of WT1 in the nephroblastomas harboring a
WT1 mutation was not different from wild-type tumors (data
not shown).
Characteristic patterns of WT1
immunohistochemistry in nephroblastoma
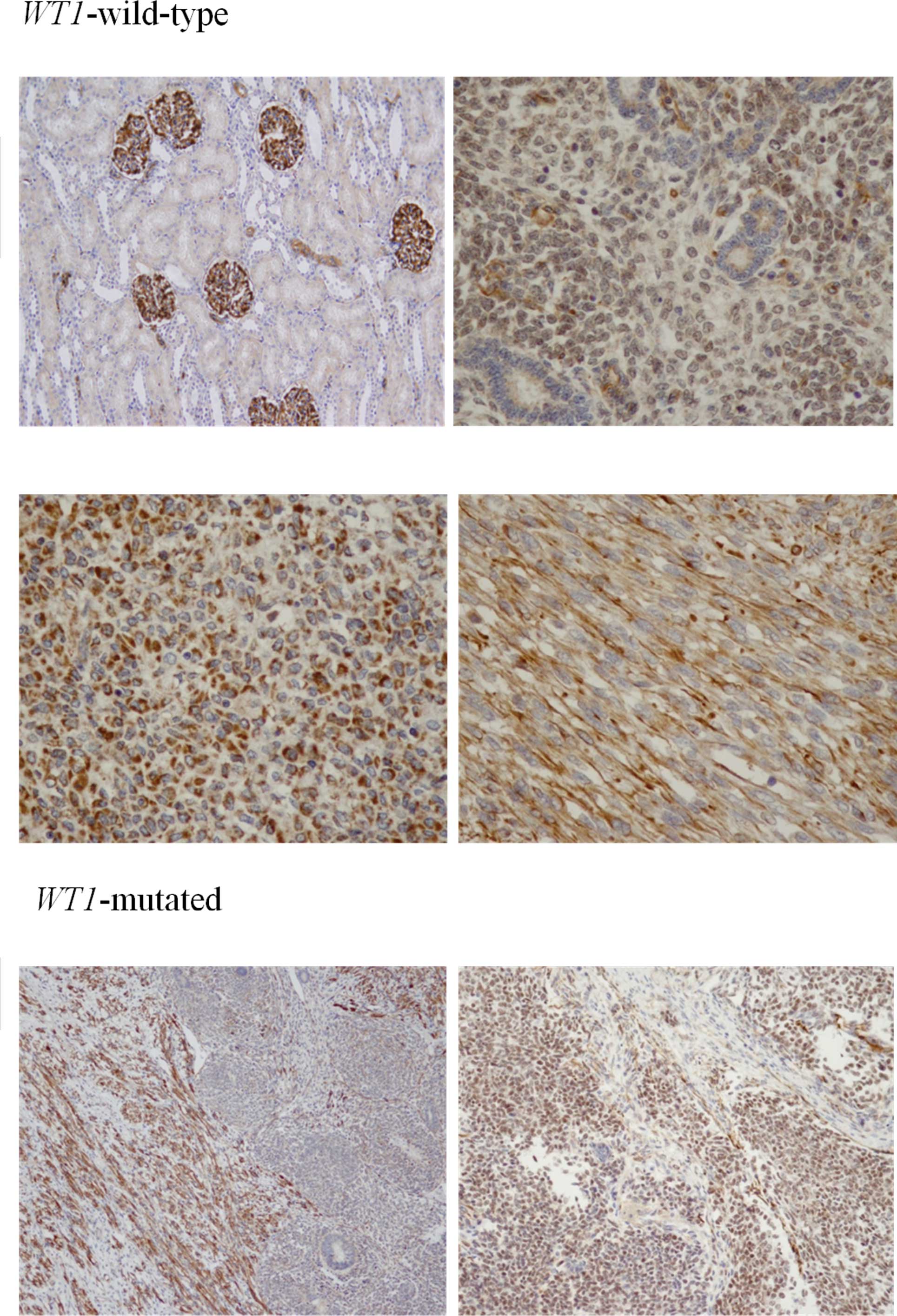
WT1 immunoreactivity was detected in varying
intensities in the nephroblastomas. Staining was limited to the
glomerular epithelium in the normal kidney tissue. Only light
positive staining was found in the CCSK and PRCC samples. In cases
with wild-type WT1, diffuse immunoreactivity was detected in
the cytoplasm of the stromal and blastemal components, sparing the
tubular epithelium. Immunoreactivity in the cases with a WT1
mutation was generally weaker and was confined to the stromal
elements in 3 cases and detectable in the blastemal component in 1
case (WT20) (Fig. 2).
The study showed nuclear accumulation of β-catenin
in the 4 cases of nephroblastoma harboring WT1 mutations,
regardless of the β-catenin mutation status (data not shown). The
positive nuclear-stained cells co-localized with WT1 immunoreactive
cells. In the case with a point mutation of WT1, for which
the WT1 immunohistochemistry was positive at the blastema,
β-catenin nuclear staining was also positive in the same component.
Membraneous staining of β-catenin was also detected in the
glomerular epithelium of adjacent non-tumor tissue.
WT1 suppression inhibited the growth of
nephroblastoma cells through increased apoptosis
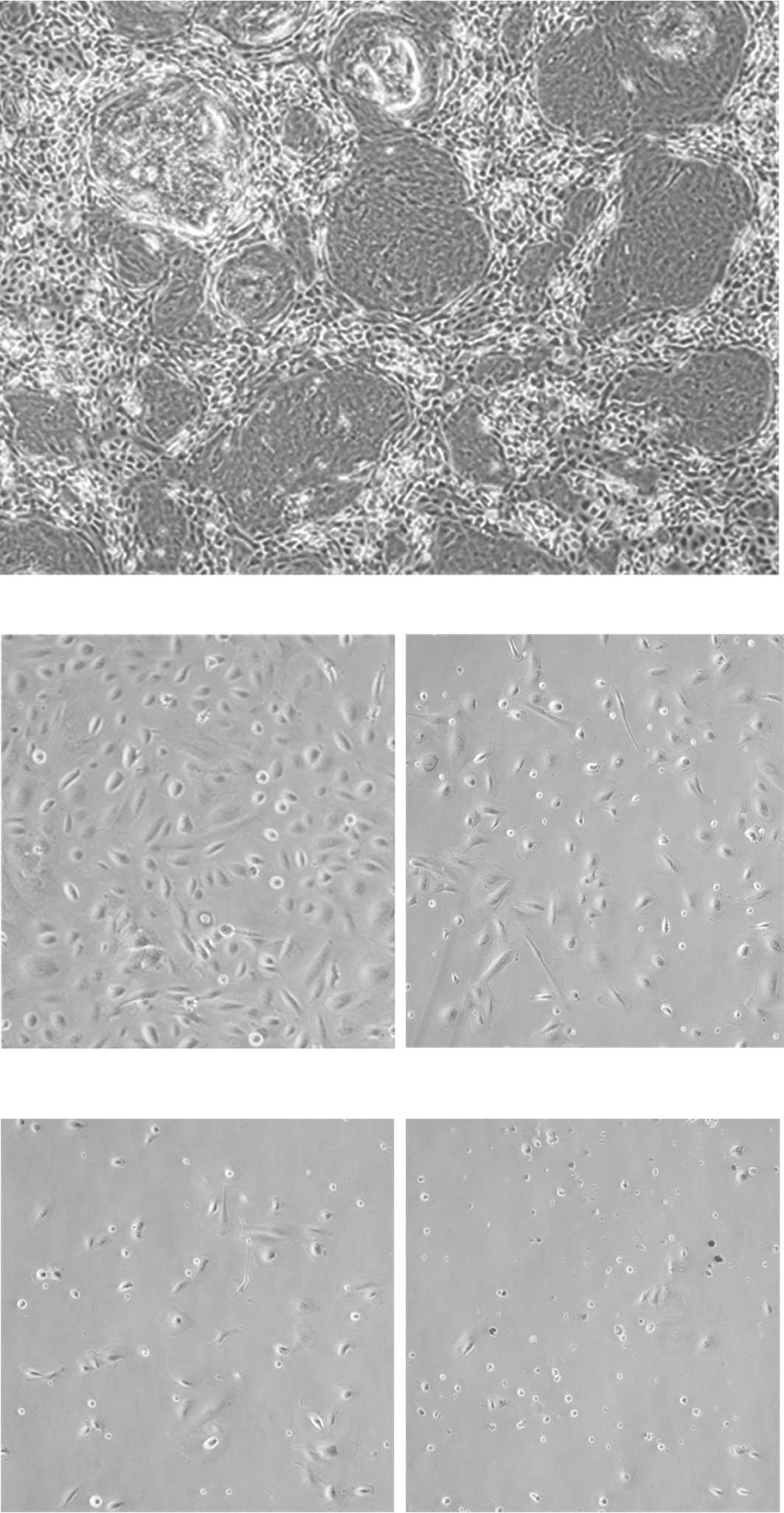
The morphology of the primary nephroblastoma culture
is shown in Fig. 3. In the
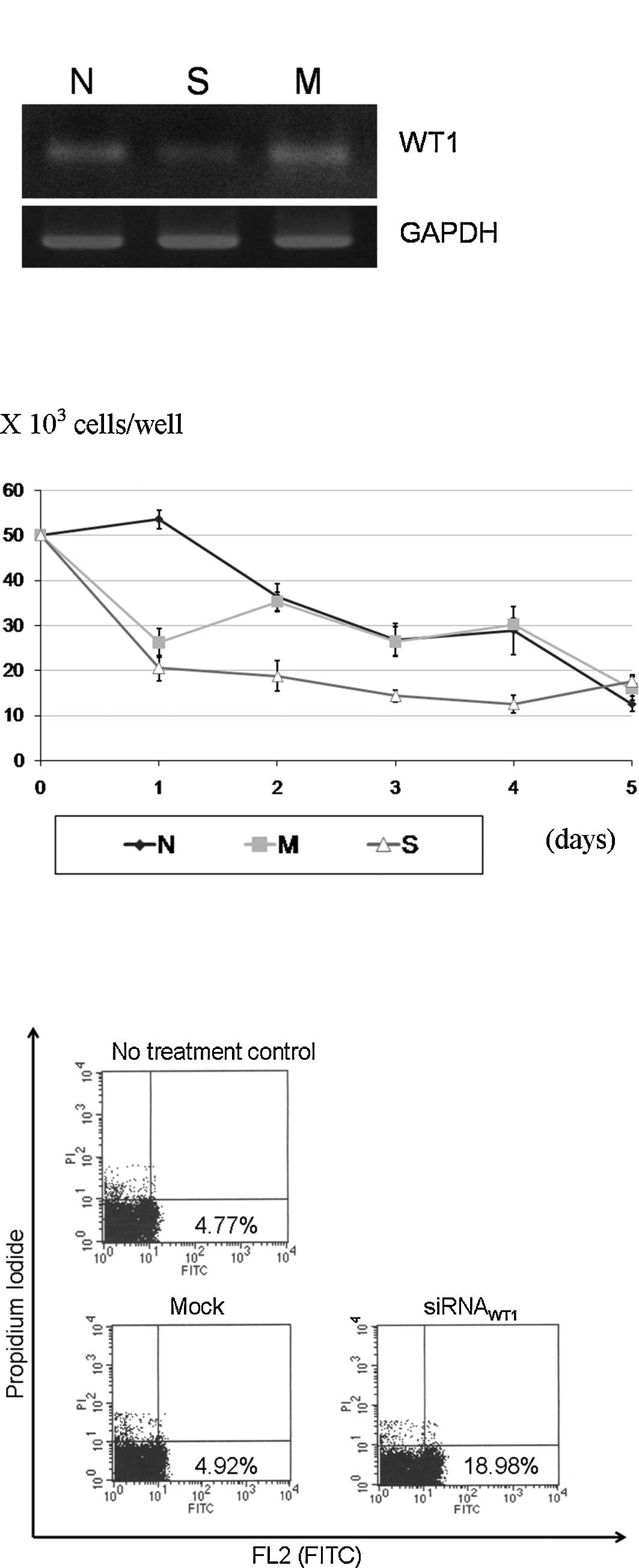
siRNAWT1 transfection experiment, the
siRNAWT1 caused a reduction in WT1 expression (Fig. 4A). The
siRNAWT1-transfected cells had a significantly slower
growth from the 2nd to the 4th day of transfection, as compared to
the mock transfection and no treatment groups (Fig. 4B). The apoptosis assay showed an
increased apoptosis in the experimental group of 3.8 times the mock
control (Fig. 4C).
Discussion
WT1 is a transcription factor that plays a
role in genitourinary organ development. In pediatric
nephroblastoma, the tumor suppressor role of WT1 is
supported by the classical evidence of mutations found in 5–10% of
tumors (5). However, the role of
the gene in the subgroup of nephroblastomas that harbors the
wild-type form of WT1 has yet to be elucidated. An oncogenic
role of WT1 in various human cancers suggests the possibility of
its involvement in the development of nephroblastomas. Data from
functional studies of WT1 in nephroblastomas are, however,
limited as the tumor has only a limited number of cell lines
available.
Our data from the qRT-PCR study showed that the
average expression of WT1 at the mRNA level in our pediatric
nephroblastomas was significantly higher than that in the
non-tumorous renal tissues. Data indicated that this overexpression
was higher than the physiologic expression in the normal kidney
tissue. When the expression level in the nephroblastoma tissues was
compared to other pediatric renal tumors and neuroblastomas, the
high level was found to be exclusive in the nephroblastomas,
suggesting that WT1 is associated with the tumorigenesis of
this tumor. We used β-catenin, a known key player in
pediatric embryonal tumors, as an external control and found a
comparable expression of this gene in various tissues studied.
Varying expression levels of β-catenin in the neuroblastomas
may be explained by variations in tumor differentiation (12).
The total levels of WT1 in WT1
wild-type and WT1-mutated tumors were not different at the
mRNA level. However, expression at the protein level in the 4
tumors with a WT1 mutation studied showed a weaker
intensity. The localization of WT1 proteins in nephroblastoma
tissues were examined and the proteins were found to express in the
blastemal and epithelial components of the tumors harboring
wild-type WT1. Additionally, the proteins were confined to
the stromal component in the WT1-mutated tumors, a
difference that suggests different roles of WT1 in the two
nephroblastoma subclasses. This speculation is consistent with
recent RNA microarray studies that found a difference in molecular
signatures between nephroblastomas with and without a WT1
mutation (4,13,14).
Fukuzawa et al described the expression
pattern of WT1 in fetal kidney tissue (15). These authors found positive WT1
nuclear immunohistochemistry mostly in glomerular epithelium and
blastema tissues. In our WT1 wild-type tumors, the
localization patterns were comparable with those reported in
developing kidneys (15). We
speculated that WT1 marks an immature renal lineage in the tumor
and may contribute to the growth of nephroblastoma tumor cells
derived from developmentally arrested renal tissue.
We further tested the consequence of suppressing WT1
expression on nephroblastoma cell growth. Using a primary
nephroblastoma culture, we created a WT1 knock-down model by
using siRNAWT1 transfection. The study found a negative
growth effect on the siRNAWT1-transfected group,
suggesting an oncogenic role of the gene in this tumor subclass.
The rapid reduction of the cells in the mock transfection and
siRNAWT1 groups on the first day was explained by
cytotoxicity caused by the transfection complexes. Additional
evaluation of apoptotic activity showed increased apoptosis in the
siRNAWT1-treated cells. These results were consistent
with previous evidence suggesting an anti-apoptotic role for
WT1 (16).
An advantage of using cells cultured from ascites
was that the culture was predominantly composed of tumor cells with
a negligible amount of contaminating fibroblasts. However, the
cells used were obtained from a patient undergoing chemotherapy.
Therefore, the cells may not have exhibited a native nephroblastoma
phenotype. The cells were from a single patient and may not have
represented the complete range of possibilities from this subclass
of WT1 wild-type nephroblastomas. Moreover, WT1 has
at least four isoforms with different functions (16). Manipulation of its expression may
also alter the isoform composition. Further studies focusing on the
role of WT1 in this nephroblastoma subclass may be
beneficial, not only for understanding the tumor biology, but also
for possibly suggesting opportunities for the use of WT1 as a
therapeutic target in this specific subset of tumor.
In conclusion, this study examined the mutation and
expression of the WT1 gene in pediatric nephroblastomas and
found a significantly high expression at the mRNA level,
irrespective of the mutation status. At the protein level,
differences in intensity and localization suggest its divergent
roles in wild-type and mutated tumors. In the WT1 wild-type
subclass of pediatric nephroblastomas, overexpression and negative
growth effects on a suppressed expression suggest an oncogenic role
of the gene in this tumor.
Acknowledgements
The authors thank Karnda Tongmitr of the Department
of Pathology for the immunohistochemical studies. The apoptosis
assays were performed at the Scientific Equipment Center, Prince of
Songkla University. Dave Patterson edited the English language in
the manuscript.
References
|
1
|
Beckwith JB, Kiviat NB and Bonadio JF:
Nephrogenic rests, nephroblastomatosis, and the pathogenesis of
Wilms’ tumor. Pediatr Pathol. 10:1–36. 1990.
|
|
2
|
Li CM, Kim CE, Margolin AA, et al: CTNNB1
mutations and overexpression of Wnt/beta-catenin target genes in
WT1-mutant Wilms’ tumors. Am J Pathol. 165:1943–1953.
2004.PubMed/NCBI
|
|
3
|
Rivera MN, Kim WJ, Wells J, et al: An X
chromosome gene, WTX, is commonly inactivated in Wilms tumor.
Science. 315:642–645. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Perotti D, Gamba B, Sardella M, et al:
Functional inactivation of the WTX gene is not a frequent event in
Wilms’ tumors. Oncogene. 27:4625–4632. 2008.PubMed/NCBI
|
|
5
|
Lee SB and Haber DA: Wilms tumor and the
WT1 gene. Exp Cell Res. 264:74–99. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moore AW, McInnes L, Kreidberg J, Hastie
ND and Schedl A: YAC complementation shows a requirement for WT1 in
the development of epicardium, adrenal gland and throughout
nephrogenesis. Development. 126:1845–1857. 1999.PubMed/NCBI
|
|
7
|
Sangkhathat S, Chotsampancharaen T,
Kayasut K, Patrapinyokul S, Chiengkriwate P, Kitichet R and Maipang
M: Outcomes of pediatric nephroblastoma in southern Thailand. Asian
Pac J Cancer Prev. 9:643–647. 2008.PubMed/NCBI
|
|
8
|
Oji Y, Yamamoto H, Nomura M, et al:
Overexpression of the Wilms’ tumor gene WT1 in colorectal
adenocarcinoma. Cancer Sci. 94:712–717. 2003.
|
|
9
|
Sangkhathat S, Kusafuka T, Miao J, et al:
In vitro RNA interference against β-catenin inhibits the
proliferation of pediatric hepatic tumors. Int J Oncol. 28:715–722.
2006.
|
|
10
|
Wanitsuwan W, Kanngurn S,
Boonpipattanapong T, Sangthong R and Sangkhathat S: Overall
expression of beta-catenin outperforms its nuclear accumulation in
predicting outcomes of colorectal cancers. World J Gastroenterol.
14:6052–6059. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Navakanit R, Graidist P, Leeanansaksiri W
and Dechsukum C: Growth inhibition of breast cancer cell line MCF-7
by siRNA silencing of Wilms tumor 1 gene. J Med Assoc Thai.
90:2416–2421. 2007.PubMed/NCBI
|
|
12
|
Sangkhathat S, Nara K, Kusafuka T, Yoneda
A and Fukuzawa M: Artificially accumulated β-catenin inhibits
proliferation and induces neurite extension of neuroblastoma cell
line NB-1 via up-regulation of trkA. Oncol Rep. 16:1197–1203.
2006.
|
|
13
|
Fukuzawa R, Anaka MR, Weeks RJ, Morison IM
and Reeve AE: Canonical WNT signaling determines lineage
specificity in Wilms tumour. Oncogene. 28:1063–1075. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fukuzawa R, Heathcott RW, More HE and
Reeve AE: Sequential WT1 and CTNNB1 mutations and alterations of
beta-catenin localisation in intralobar nephrogenic rests and
associated Wilms tumours: two case studies. J Clin Pathol.
60:1013–1016. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fukuzawa R, Heathcott RW, Sano M, Morison
IM, Yun K and Reeve AE: Myogenesis in Wilms’ tumors is associated
with mutations of the WT1 gene and activation of Bcl-2 and the Wnt
signaling pathway. Pediatr Dev Pathol. 7:125–137. 2004.
|
|
16
|
Hohenstein P and Hastie ND: The many
facets of the Wilms’ tumor gene, WT1. Hum Mol Genet. 15:R196–R201.
2006.PubMed/NCBI
|