Introduction
Colorectal cancer (CRC) is the third most common
cancer worldwide, and the second most common cause of
cancer-related death (1).
Approximately 20–40% of CRC patients that undergo curative surgery
develop local recurrence or distant metastasis, and exhibit a poor
outcome that is generally less than 2 years (2). Chemotherapy is an important strategy
in the treatment of metastatic CRC, and irinotecan (CPT-11) is one
of the major chemotherapy drugs used in metastatic CRC treatment.
Treatment with a combination of CPT-11, 5-fluorouracil (5-FU) and
leucovorin is generally approved as the standard chemotherapy for
metastatic disease and somewhat increases survival (3). However, the majority of patients
eventually succumb to the disease. Various predictive factors of
chemosensitivity were previously investigated (4). Regarding 5-FU chemotherapy, the target
enzyme thymidylate synthase and the metabolic enzyme
dihydropyrimidine dehydrogenase were suggested as predictive
factors (4). The predictive factors
underlying CPT-11 chemosensitivity have yet to be sufficiently
investigated. Thus, identification of the molecular genetic
parameters associated with response to CPT-11 is of clinical
interest.
Apoptosis or programmed cell death occurs in various
physiological and pathological situations (5). It occurs not only in normal tissues
but also in malignant tumors, and plays an important role in both
the pathogenesis of tumors and their biological behavior (6). Defects in the apoptotic pathway
contribute to cell accumulation in the colon, promoting malignancy
and subsequent metastasis, and allow tumor cells to survive in a
suspended state, thereby permitting their hematogenous or lymphatic
dissemination (7). The apoptotic
pathway may also be a final common step in the cytotoxicity exerted
by a number of anticancer drugs with various underlying mechanisms
of action (8,9). The altered expression of genes that
encode apoptotic proteins provide cells with inherent resistance to
anticancer drugs. Thus, the expression levels of apoptotic proteins
serves as a predictor of chemotherapeutic agent response.
In addition to genetic aberrations such as mutation
and deletion, increasing evidence suggests that epigenetic changes
play an important role in CRC pathogenesis (10,11).
The aberrant methylation of gene promoter regions leading to gene
silencing is currently the most widely studied epigenetic
abnormality in human malignancies, affecting multiple cellular
functions including cell growth and differentiation, cell cycle
control and DNA repair (11,12).
Given that DNA methylation affects the transcription of crucial
genes involved in the regulation of apoptosis (13,14),
it may also contribute to the evolution of resistance to
chemotherapeutic agents.
In a previous study, it was shown that the
inactivation of apoptosis-related genes due to DNA hypermethylation
may predict the response to the 5-FU-based chemotherapy treatment
of gastric cancer (15). To
identify the apoptosis-related genes involved in reduced
sensitivity to CPT-11, the genome was screened for genes responding
to the demethylating agent 5-aza-2′-deoxycytidine (DAC) using
microarray analysis. Colon cancer cells were used in the event that
DAC was able to enhance sensitivity to CPT-11. Based on our
microarray results, an apoptosis-related gene, Bcl-2/adenovirus E1B
19 kDa protein interacting protein 3 (BNIP3)was selected.
The methylation status of the gene was analyzed in primary CRCs.
Moreover, the methylation status was compared with various
clinicopathological variables and BNIP3 methylation was
investigated as a predictor of prognosis and response to CPT-11
treatment in CRC patients.
Materials and methods
Reagents
SN-38, an active metabolite of CPT-11, was provided
by Yakult (Tokyo, Japan). SN-38 was dissolved in dimethyl sulfoxide
at a concentration of 1 μM and stored at −20°C, before further
dilution in phosphate-buffered saline (PBS) and filter
sterilization immediately prior to use. DAC was obtained from Sigma
(St. Louis, MO, USA), dissolved in PBS and filter-sterilized.
Cell lines
The human colon adenocarcinoma cell lines HCT-15 and
HT29 were provided by the Cell Resource Center for Biomedical
Research, Tohoku University (Miyagi, Japan) and the American Type
Culture Collection (Manassas, VA, USA). HCT-15 cells were
maintained in RPMI-1640 (Sigma) containing 10% heat-inactivated
fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml
streptomycin, 10 mM HEPES (Gibco-BRL, Gaithersburg, MD, USA) and
1.0 mM sodium pyruvate (Gibco). HT29 cells were maintained in Mac
Coy’s medium containing 10% heat-inactivated FBS, 100 U/ml
penicillin and 100 μg/ml streptomycin. The cell lines were cultured
at 37°C in 5% CO2. Cells (5×103 per well)
were plated in 24-well plates on day 1 of the culture. On day 3,
the medium was removed and new medium containing DAC was added. The
cells were treated with 0.5 μM DAC (or PBS as a control) for 72 h.
On day 6, the cells were rinsed twice with FBS-free medium, and
fresh medium containing 0.0015 μM SN-38 (or PBS) was added. The
dose of each drug was administered on the basis of its
pharmacological dose as previously reported (14), and our preliminary experiments (data
not shown). The cells were harvested following incubation with
trypsin-EDTA on day 9, stained with trypan blue and counted. Total
mRNA or genomic DNA was extracted on day 6 and utilized in the
analysis of mRNA expression or methylation status following DAC
treatment.
Microarray analysis
Total RNA was extracted from cultured cells using
the RNeasy kit (Qiagen, Hilden, Germany). The integrity of the RNA
was assessed using the Agilent 2100 BioAnalyzer (Agilent
Technologies, Palo Alto, CA, USA), and the samples were confirmed
to have an RNA Integrity Number >5.0 prior to gene expression
analysis. Contaminant DNA was removed via digestion with RNase-free
DNase (Qiagen). Using 2 μg total RNA, cRNA was prepared using the
one-cycle target labeling and control reagents kit (Affymetrix,
Santa Clara, CA, USA). Hybridization and signal detection of
HG-U133 plus 2.0 arrays (Affymetrix) were performed following the
manufacturer’s instructions. The 4 microarray datasets obtained
were normalized using the robust multi-array average (RMA) method
(16) and R 2.4.1 statistical
software (17) together with a
BioConductor package. Normalized gene expression levels were
log2-transformed, and 62 control probe sets were removed for
further analysis. For each of the 54,613 probe sets, a fold change
analysis was performed using each pair of treated and untreated
HCT15 and HT29 cells. Probe sets that showed >1.5-fold increase
or decrease were identified as differentially expressed genes.
Real-time polymerase chain reaction
(RT-PCR)
Total cellular RNA was extracted from the treated
cells using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and
incubated with DNase as previously reported (14). cDNA was then synthesized from 10 ng
extracted RNA using the High-Capacity cDNA reverse transcription
kit (Applied Biosystems). PCR was performed as previously described
(14), using the conditions:
initial denaturation for 96°C for 2 min, followed by 26 cycles of
denaturation for 1 min at 96°C, annealing for 1 min at 57–60°C and
extension for 1 min at 72°C. As an internal control for RT-PCR
analysis, glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
transcripts were amplified from the same cDNA samples. Primer
sequences were: GAPDH, 5′-CAACAGCCTCAAGATCATCAGC-3′ (forward) and
5′-TCCTAGACGGCAGGTCAGGTC-3′ (reverse); BNIP3,
5′-GGATGCAGGAGGAGAGCCT-3′ (forward) and 5′-CGAG GTGGGCTGTCACAGT-3′
(reverse); and SOCS3, 5′-GACCA GCGCCACTTCTT-CAC-3′ (forward) and
5′-ACTGGATGCG CAGGTTCTTG-3′ (reverse). Following amplification, PCR
products were separated on 2% agarose gels, stained with ethidium
bromide and visualized under ultraviolet illumination.
DNA extraction
Tumor or normal mucosal tissue was cut into 10-μm
sections from tissue blocks, with a change of blade and cleaning of
the microtome between specimens. The specimens were deparaffinized
and washed, and tumor tissue was manually dissected using a razor
blade, using a hematoxylin and eosin (H&E)-stained slide as a
guide. Dissected tissues or cultured cell lines were then incubated
overnight with proteinase-K in digestion buffer and the genomic DNA
was extracted using a standard phenol chloroform method.
Methylight analysis
Sodium bisulfite conversion and DNA recovery were
performed using EpiTect Bisulfite (Qiagen). Following sodium
bisulfite conversion, the genomic DNA was analyzed using the
Methylight technique, a fluorescence-based RT-PCR (Q-PCR) assay
(18) and the ABI Prism 7300 R-T
PCR System (Taq Man; Applied Biosystems, Foster City, CA, USA). Two
sets of primers and probes, designed specifically for
bisulfite-converted DNA were used. One set detected methylation in
the gene of interest and the second set served as a reference set
for β-actin (ACTB) to normalize for input DNA. The reference
primers and probes were designed in a region of the ACTB gene that
lacks CpG dinucleotides, allowing for equal amplification
regardless of the methylation levels. Primer and probe sequences
were: ACTB, 5′-AGGTTGGGGAAGTTT GTTTTTG-3′ (forward),
5′-CCACCACCCAACACACAATA-3′ (reverse) and
5′-TGGGGTGGTGATGGAGGAGGTT-3′ (probe); BNIP3,
5′-TTCGGTCGGAGGAATTTATAGG-3′ (forward), 5′-CCCCAATCGCGACCAA-3′
(reverse) and 5′-ACGACGCGACCGCAAAT-3′ (probe); as well as SOCS3
5′-TCGCGTTTTTTT-TTTCGTAGTTT-3′ (forward), 5′-CGC GACCTCCGCACA-3′
(reverse) and 5′-CGACCGCTACCG CATCCCGA-3′ (probe).
SssI-treated HCT-15 DNA was used as a fully methylated
positive control (100% methylation ratio). Parallel Taq Man PCR
reactions were performed with specific primers for the
bisulfite-converted methylated sequence for a particular locus and
with the ACTB reference primers. In each case, triplicate threshold
cycle (Ct) values were obtained and averaged, and expression levels
were then evaluated by the 2-ΔΔCt method (19). As an internal standard, each
individual sample was normalized to its ACTB content and compared
to the gene expression level of SssI-treated HCT-15 DNA as
positive controls (calibration sample) as follows:
2ΔΔCt, where ΔΔCt = (Ct-target-Ct-reference)-treated
sample – (Ct-target-Ct-reference) calibrator sample. The percentage
of fully-methylated reference (PMR) was defined as 2ΔΔCt
× 100%.
Patients and tissue samples
Patients entered into this study had undergone
surgical resection for primary sporadic colorectal cancer at the
Department of Surgical Oncology, Tokyo Medical and Dental
University (Tokyo, Japan). This study was pre-approved by the
institutional review board of Tokyo Medical and Dental University,
and written informed consent was obtained from all 112 participants
prior to the commencement of the study. Following surgery for both
colon and rectal cancer, patients exhibiting stage III cancer
received oral or intravenous 5-FU-based adjuvant chemotherapy, and
those with stage IV tumor or following recurrence received
5-FU-based systemic chemotherapy. The resected specimens were fixed
in 10% neutral buffered formalin and embedded in paraffin. For all
cases, archival H&E slides of the primary tumors were retrieved
and reviewed to confirm pathological features. In addition to the
International Union Against Cancer Tumor-Node-Metastasis (TNM)
classification (20),
clinicopathological factors such as age, degree of histological
differentiation, pathological tumor class and lymph node metastasis
at the time of diagnosis in stage I–III patients during the
follow-up period were studied. The response rate to chemotherapy
including CPT-11 treatment was assessed by Response Evaluation
Criteria In Solid Tumors (21).
Statistical analysis
Statistical analysis was carried out using StatView
Software (version 5.0). To estimate the differences between groups,
the Chi-square test, Welch’s t-test and log-rank test were used
where appropriate. The Kaplan-Meier method was used to estimate
survival. Survival was calculated from the date of surgery.
Prognostic factors were examined by univariate and multivariate
analysis using the Cox proportional hazards model. P<0.05 was
considered to be statistically significant.
Results
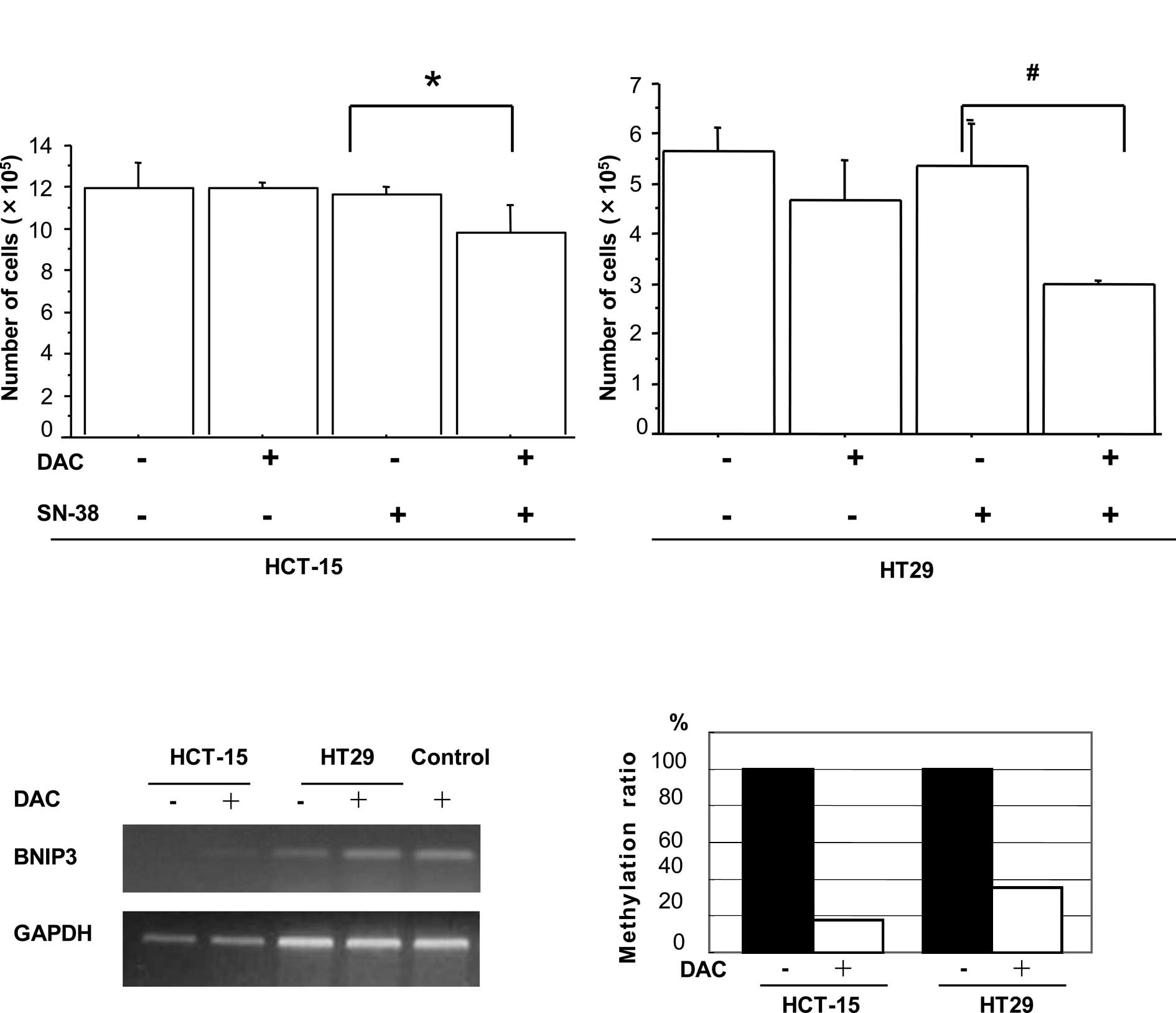
DAC increased sensitivity to SN-38 in
colorectal cancer cells
Pre-incubation with the demethylating agent DAC was
examined to investigate whether it influenced the sensitivity of
the colon cancer cell lines HCT-15 and HT29 to SN-38. On day 9, the
total number of HCT-15 cells that had been pre-incubated with DAC
prior to exposure to SN-38 was found to be significantly decreased
in comparison to cells treated with SN-38 alone (p=0.0049, Fig. 1A). A similar reduction was observed
in the HT29 cells (p=0.0038, Fig.
1A). Treatment with DAC or SN-38 alone resulted in no
significant effect on the rate of cell proliferation in the two
cell lines.
Identification of pro-apoptotic genes
induced by DAC in colon cancer cells
Since inactivation of the apoptotic pathway is
associated with chemoresistance (8), identification of the pro-apoptotic
genes that may be reactivated by DAC was attempted. Using
oligonucleotide microarrays, the global changes in gene expression
following the treatment of HCT-15 and HT29 with DAC were analyzed,
and the expression changes in treated and untreated cells were
compared. Expression levels of 943 genes were found to be increased
by >1.5-fold following DAC treatment in HCT-15 cells, and 2,720
genes were increased in the HT29 cells. A total of 234 of these
genes were found to be similarly altered in the two cell lines.
Among the 234 genes, 10 were identified as having known
pro-apoptotic activities (Table I).
As the expression levels of BNIP3 and SOCS3 are
frequently reduced due to promoter methylation in gastrointestinal
cancers (22,23), up-regulation of these genes
following DAC treatment was analyzed to determine whether it was
caused by DNA demethylation. The BNIP3 mRNA expression was
up-regulated and its DNA promoter was demethylated in both the
HCT-15 and HT29 cell lines following treatment with DAC (Fig. 1B and C). In contrast, although
SOCS3 mRNA expression was enhanced after DAC treatment,
primers and probes specific for methylated SOCS3 failed to
amplify any product in the two cell lines by Methylight analysis
(data not shown). The results indicated that SOCS3
expression may be induced by DAC treatment independently of the
methylation status of the DNA promoter.
 | Table IApoptosis-related genes up-regulated
by DAC treatment in colon cancer cell lines. |
Table I
Apoptosis-related genes up-regulated
by DAC treatment in colon cancer cell lines.
| | Fold change |
|---|
| |
|
|---|
| Symbol | Name | HCT-15 | HT29 |
|---|
| UNC5B | Unc-5 homolog B
(C. elegans) | 5.21 | 11.8 |
| NUPR1 | Nuclear protein
1 | 2.26 | 5.68 |
| ATF5 | Activating
transcription factor 5 | 4.76 | 3.00 |
| IFI6 | Interferon,
α-inducible protein 6 | 2.92 | 3.28 |
| MX1 | Myxovirus
(influenza virus) resistance 1, interferon-inducible protein p78
(mouse) | 2.07 | 3.34 |
| SCIN | Scinderin | 6.55 | 2.34 |
| STAT1 | Signal transducer
and activator of transcription 1, 91 kDa | 2.03 | 1.80 |
| SOCS3 | Suppressor of
cytokine signaling 3 | 3.48 | 1.61 |
| BNIP3 | BCL2/adenovirus E
1B 19 kDa interacting protein 3 | 22.1 | 1.54 |
| EEF1A2 | Eukaryotic
translation elongation factor α2 | 1.51 | 1.67 |
Relationship between clinicopathological
factors and BNIP3 methylation in CRC patients
The median PMR value for BNIP3 methylation in
16 randomly selected patients with normal colon epithelial tissue
was 0. The positive methylation was defined as PMR>0. The
BNIP3 methylation status was then investigated in 112
patients who had undergone surgical resection for primary CRC
between March 2000 and April 2003. Patients were prospectively
followed over a median post-operative duration of 42 months and
methylation of BNIP3 was detected in 58% of the 112
patients. No significant correlations were found between
BNIP3 methylation and clinicopathological factors such as
age, gender, tumor depth, vessel invasion, lymphatic invasion,
lymph node metastasis and stage (Table
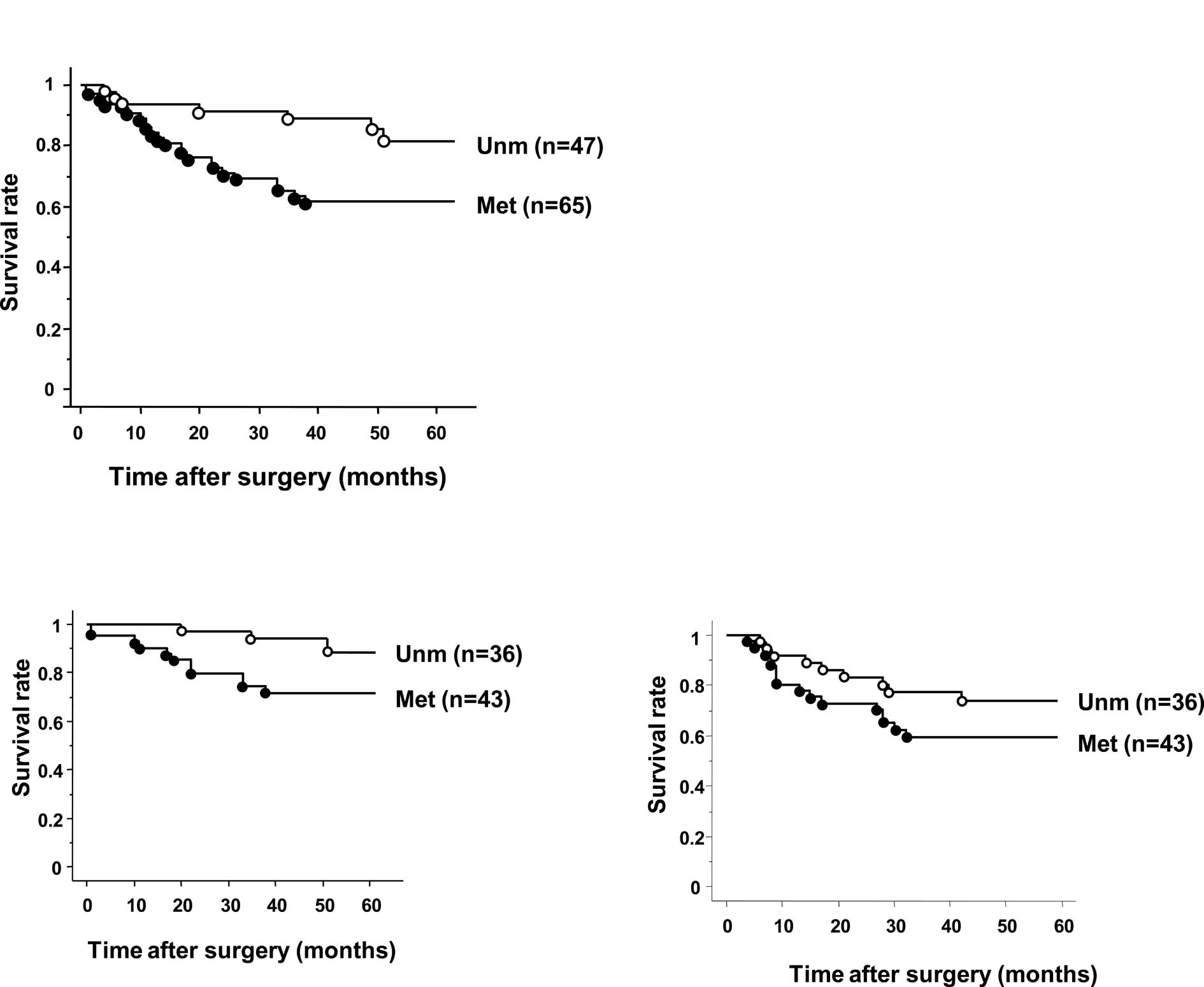
II). Patients with BNIP3 methylation also exhibited a
significantly shorter overall survival time (OS) compared to those
without methylation (p=0.012, Fig.
2A). In stage II/III tumors, patients with BNIP3
methylation also exhibited a significantly shorter OS than those
without methylation (p=0.039, Fig
2B). No difference in relapse-free survival (RFS) was observed
between patients with or without methylation (p=0.16, Fig. 2C). Regarding stage II/III patients,
investigation of the univariate analysis of survival using the Cox
proportional hazards model showed that BNIP3 methylation
(p=0.043) and lymph node metastasis (p=0.0086) were significantly
associated with OS. All of the variables with a significance level
at p<0.35 in the univariate analyses were subsequently used for
multivariate analyses. Multivariate analysis of the variables
failed to show BNIP3 methylation to be an independent
prognostic factor (Table
III).
| Table IIRelationship between
clinicopathological factors and BNIP3 methylation. |
Table II
Relationship between
clinicopathological factors and BNIP3 methylation.
| BNIP3 |
|---|
|
|
|---|
| Met (n=65) | Unm (n=47) | p-value |
|---|
| Age | | | 0.88 |
| <64 | 30 | 21 | |
| ≥64 | 35 | 26 | |
| Gender | | | 0.48 |
| Male | 43 | 28 | |
| Female | 22 | 19 | |
| Location | | | 0.88 |
| Right | 24 | 18 | |
| Left | 41 | 29 | |
| Lymphatic
invasion | | | 0.25 |
| Negative | 12 | 13 | |
| Positive | 53 | 34 | |
| Vessel
invasion | | | 0.88 |
| Negative | 5 | 4 | |
| Positive | 60 | 43 | |
| Depth of
invasion | | | 0.23 |
| T1, T2 | 14 | 6 | |
| T3, T4 | 51 | 41 | |
| Lymph node
metastasis | | | 0.24 |
| Negative | 30 | 27 | |
| Positive | 35 | 20 | |
| Stage | | | 0.19 |
| I | 9 | 4 | |
| II | 17 | 21 | |
| III | 25 | 16 | |
| IV | 14 | 6 | |
| Table IIIAnalysis of overall survival in stage
II/III patients using the Cox proportional hazard model. |
Table III
Analysis of overall survival in stage
II/III patients using the Cox proportional hazard model.
| Stage II/III
patients (n=79) |
|---|
|
|
|---|
| Prognosis
factor | Hazard ratio | 95% CI | p-value |
|---|
| Univariate
analysis |
| BNIP3
methylation | 3.744 | 1.044–13.432 | 0.043 |
| Age | 1.647 | 0.579–4.819 | 0.34 |
| Gender | 2.457 | 0.684–8.850 | 0.17 |
| Tumor
differentiation | 0.555 | 0.155–1.993 | 0.37 |
| Tumor
location | 0.614 | 0.215–1.752 | 0.36 |
| Depth of
invasion | 1.170 | 0.153–8.929 | 0.88 |
| Lymph node
metastasis | 4.739 | 1.484–15.152 | 0.0086 |
| Lymphatic
invasion | 2.762 | 0.361–21.277 | 0.33 |
| Multivariate
analysis |
| BNIP3
methylation | 3.260 | 0.887–11.984 | 0.075 |
| Age | 1.366 | 0.468–3.985 | 0.57 |
| Gender | 2.105 | 0.574–7.752 | 0.26 |
| Lymph node
metastasis | 3.040 | 0.670–13.889 | 0.15 |
| Lymphatic
invasion | 1.481 | 0.130–16.928 | 0.75 |
Prognostic value of BNIP3 methylation in
CRC patients who received treatment with chemotherapeutic agents
containing CPT-11
BNIP3 methylation was examined to determine
whether it was a prognostic marker in primary CRC patients treated
with CPT-11. Of the patients who had undergone surgical resection
for primary CRC between May 1998 and September 2007, 30 patients
(including 5 from 112 patients between 2000 and 2003) who
demonstrated recurrence after radical resection or incomplete
resection due to distant metastasis, received first-line
chemotherapy with CPT-11. Patients were subsequently observed over
a median duration of 15 months following the initial administration
of CPT-11. One patient had complete response (CR), 10 partial
response (PR), 7 stable disease (SD) for >6 months, and 12
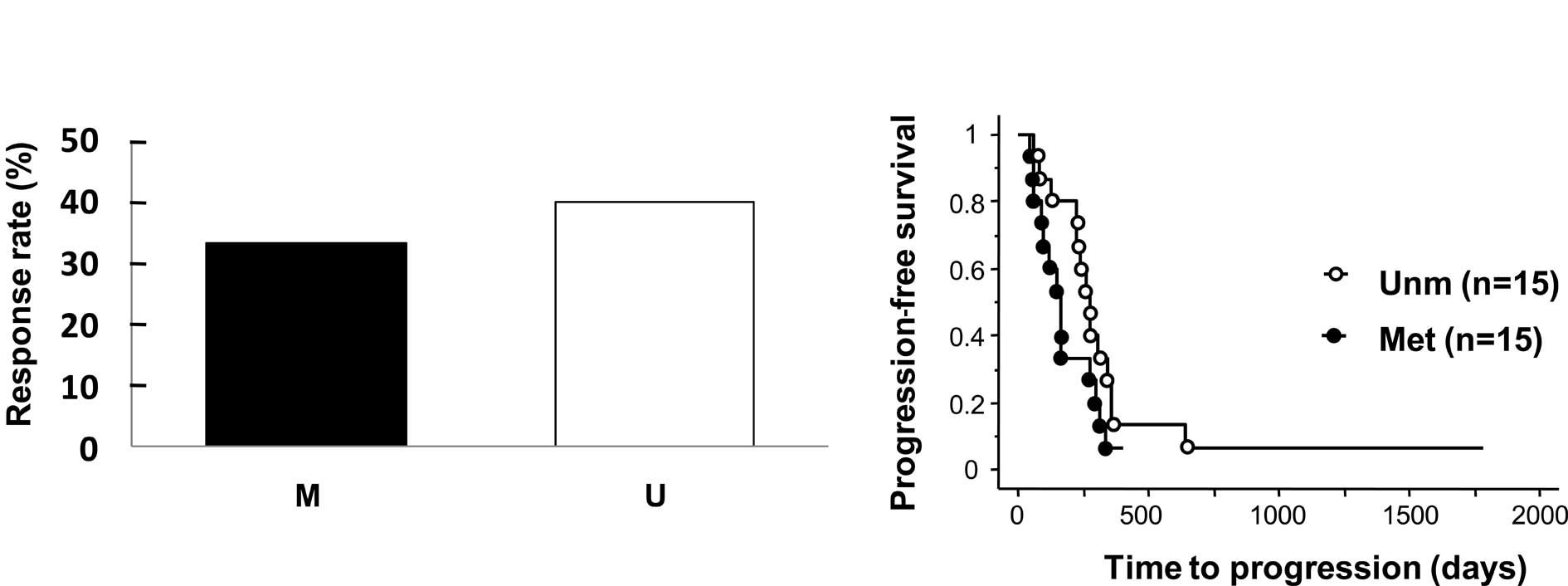
progression of disease (PD). When CR and PR patients were defined
as responders, the observed response rate was lower in patients
with BNIP3 methylation than those without (33.3 vs. 40%),
but this difference was not statistically significant (p=0.70,
Fig. 3A). Time to progression (TTP)
in patients with BNIP3 methylation was slightly shorter than
that in patients without methylation, but this difference was not
statistically significant (median 196.5 vs. 237 days) (p=0.10,
Fig. 3B).
Discussion
In this study, 2 colon cancer cell lines were
utilized in which DAC treatment enhanced sensitivity to SN-38, and
identified the apoptosis-related genes that were up-regulated
following treatment with DAC by microarray analysis. Among these
genes, we confirmed methylation of the BNIP3 promoter in
colon cancer cell lines, and subsequently detected its methylation
in more than half of the cases of primary CRC. Our results showed
that BNIP3 methylation is associated with poor clinical
outcome and chemoresistance.
Hypoxia is a common phenomenon observed in solid
tumors and is known to stabilize hypoxia-inducible factor-1
(HIF-1), which is involved in the pathways underlying angiogenesis,
glucose metabolism and cell proliferation (24–26).
HIF-1 transactivates a large number of genes that may either
promote or inhibit the growth and survival of individual tumor
cells (27). Thus, selection of an
individual tumor cell may maintain the expression of beneficial
HIF-1 transcriptional targets, while silencing pro-death
hypoxia-induced signals may result in lethal malignancy (28). BNIP3 is a pro-apoptotic
member of the Bcl-2 family whose role is mediated by HIF-1
(29–31). BNIP3 is also known to play an
important role in the regulation of apoptosis (32–34).
An increased BNIP3 expression induces cell death through
mitochondrial dysfunction, membrane depolarization, mitochondrial
permeability transition pore opening and increased production of
reactive oxygen species (35). A
reduced BNIP3 expression has been identified in a wide range
of cancer cells and primary malignancies (23,36,37). A
number of recent studies showed that methylation of the
BNIP3 promoter may play an important role in silencing
expression in a range of tumor types. These studies also showed
that BNIP3 methylation is detected in various primary
cancers (23,36,37).
In this study, BNIP3 methylation was present in almost 60%
of the primary CRC cases. Taken together, silencing BNIP3 by
methylation may be important in tumorigenesis processes in the
colon and rectum. Given the potential ability of HIF-1 to evoke
apoptosis through its target gene, the down-regulation of
BNIP3 by methylation may disrupt the HIF-1-BINP3 apoptotic
pathway and permit cancer cells with high malignant potential to
survive, since CRC patients with BNIP3 methylation had a
poorer outcome than those with without. It was reported that
BNIP3 silencing induces metastatic growth of breast cancer
in the liver, lung and bone (28).
In this study, 70% (14/20) of patients with distant metastasis
exhibited BNIP3 methylation in the 112 CRC cases examined
(Table II), suggesting that
BHIP3 silencing may contribute to the acquisition of
metastatic potential in cancer cells.
CPT-11 is a topoisomerase I (topo-I) inhibitor that
forms stable topo-I DNA-cleavable complexes and inhibits the
progression of the replication fork. However, the relationship
between the expression levels of topo-I mRNA and chemosensitivity
of CPT-11 is unclear (38,39). In a previous study, we showed that
DAC increases the growth inhibitory effects of CPT-11 on the colon
cancer cell line HCT-15, despite showing no effect on topo-I
expression levels (14).
Furthermore, our microarray experiment did not identify changes in
topo-I expression following treatment with DAC. Thus, topo-I may
not be involved in the enhanced sensitivity to CPT-11 following DAC
treatment.
Apoptosis is a significant mechanism through which
chemotherapeutic agents exert their cytotoxic effects on cancer
cells. However, cancer cells acquire resistance to apoptosis due to
an altered expression or mutation of apoptosis-related genes during
the tumorigenesis processes (40).
Numerous studies have shown a relationship between disruption of
the apoptosis pathway and chemoresistance in various tumors
(8,9). In addition, the susceptibility of
cancer cells to cytotoxic drugs appears to be at least partially
due to a dependence on the balance between pro-and anti-apoptotic
members of the Bcl-2 family (41).
Given that BNIP3 is a member of the Bcl-2 family of
pro-apoptotic proteins, and that it appears to antagonize the
activity of pro-survival proteins, including Bcl-2 and Bcl-xL
(35), it may also contribute to
chemosensitivity. Previously, BNIP3 expression was found to
be down-regulated in clones with acquired resistance to 5-FU
compared to their parental CRC cell lines (33). Additionally, BNIP3 expression
was found to be associated with paclitaxel response in an ovarian
cancer model (42). BNIP3
down-regulation results from the addition of small interfering RNA
enhanced chemoresistance in pancreatic cancer cells (43,44).
By contrast, the overexpression of BNIP3 in rat fibroblast
cells increased sensitivity to apoptosis induced by topo-I and -II
inhibitors (45). Our previous
study showed that the inhibitory effects of SN-38 on tumor tissue
are increased in a CRC xenograft model when BNIP3 expression
is restored via promoter demethylation following treatment with DAC
(14). In this study, BNIP3
was up-regulated following treatment with DAC in SN-38-resistant
CRC cells when DAC increased sensitivity to SN-38. Moreover, CRC
patients with BNIP3 methylation exhibited a shorter TTP for
CPT-11 treatment. Thus, BNIP3 may play an important role in
the reduced response to CPT-11 treatment in CRC patients.
In conclusion, the relatively high frequency of
BNIP3 methylation in primary CRC suggests that this gene is
involved in carcinogenesis of the colon and rectum. Moreover, since
BNIP3 methylation is associated with poor OS and decreased
sensitivity to CPT-11, the methylation status of this gene may be a
predictive factor for prognosis and responsiveness to CPT-11 in CRC
patients. Since methylation appears to be reversed by a chemical
agent, BNIP3 reactivation via a demethylating agent may be a
novel target for the treatment of CRC.
References
|
1
|
Ricchi P, Zarrilli R, Di Palma A and
Acquaviva AM: Nonsteroidal anti-inflammatory drugs in CRC: from
prevention to therapy. Br J Cancer. 88:803–807. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kobayashi H, Mochizuki H, Sugihara K, et
al: Characteristics of recurrence and surveillance tools after
curative resection for colorectal cancer, a multicenter study.
Surgery. 141:67–75. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tournigand C, André T, Achille E, Lledo G,
Flesh M, Mery-Mignard D, Quinaux E, Couteau C, Buyse M, Ganem G,
Landi B, Colin P, Louvet C and de Gramont A: FOLFFIRI followed by
FOLFOX6 or the reverse sequence in advanced colorectal cancer: a
randomized GERCOR study. J Clin Oncol. 22:229–237. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ichikawa W, Uetake H, Shirota Y, Yamada H,
Nishi N, Nihei Z, Sugihara K and Hirayama R: Combination of
dihydropyrimidine dehydrogenase and thymidilate synthase gene
expressions in primary tumors as predictive parameters for the
efficacy of fluoropyrimidine-based chemotherapy for metastatic
colorectal cancer. Clin Cancer Res. 9:86–91. 2003.
|
|
5
|
Hengarter MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar
|
|
6
|
Wyllie AH: Apoptosis. Br J Cancer.
67:205–208. 1993.
|
|
7
|
Compagni A and Chiristofori G: Recent
advances in research on multistage tumorigenesis. Br J Cancer.
83:1–5. 2000.PubMed/NCBI
|
|
8
|
Makin G and Dive C: Apoptosis and cancer
chemotherapy. Trends Cell Biol. 11:S22–S26. 2001. View Article : Google Scholar
|
|
9
|
Fulda S and Debatin KM: Extrinsic vs.
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Suzuki H, Gabrielson E, Chen W, Anbazhagan
R, van Engeland M, Weijenberg MP, Herman JG and Baylin SB: A
genomic screen for genes upregulated by demethylation and histone
deacetylase inhibition in human colorectal cancer. Nat Genet.
31:141–149. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kondo Y and Issa JP: Epigenetic changes in
colorectal cancer. Cancer Metastasis Rev. 23:29–39. 2004.
View Article : Google Scholar
|
|
12
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
13
|
Teodoridis JM, Strathdee G and Brown R:
Epigenetic silencing mediated by CpG island methylation: potential
as a therapeutic target and as a biomarker. Drug Resist Updat.
7:267–278. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ishiguro M, Iida S, Uetake H, Morita S,
Makino H, Kato K, Takagi Y, Enomoto M and Sugihara K: Effect of
combined therapy with low-dose 5-aza-2′-deoxycytidine and
irinotecan on colon cancer cell line HCT-15. Ann Surg Oncol.
14:1752–1762. 2007.
|
|
15
|
Kato K, Iida S, Uetake H, Takagi Y,
Yamashita T, Inokuchi M, Yamada H, Kojima K and Sugihara K:
Methylated TMS1 and DAPK genes predict prognosis and response to
chemotherapy in gastric cancer. Int J Cancer. 122:603–608. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
R, Development Core Team. R, A language
and environment for statistical computing. R, Foundation for
Statistical Computing; Vienna, Austria: ISBN: 3-900051-07-0URL
http://www.R-project.org.
2006
|
|
17
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar
|
|
18
|
Eads CA, Danenberg KD, Kawakami K, Saltz
LB, Blake C, Shibata D, Danenberg PV and Laird PW: MethyLight: a
high-throughput assay to measure DNA methylation. Nucleic Acids
Res. 28:E322000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shirota Y, Stoehlmacher J, Brabender J,
Xiong YP, Uetake H, Danenberg KD, Groshen S, Tsao-Wei DD, Danenberg
PV and Lenz HJ: ERCC1 and thymidylate synthase mRNA levels predict
survival for colorectal cancer patients receiving combination
oxaliplatin and fluorouracil chemotherapy. J Clin Oncol.
19:4298–4304. 2001.
|
|
20
|
Sobin LH and Wittekind C: TNM
Classification of Malignant Tumours. 6th edition. New York: Wiley;
2002
|
|
21
|
Therasse P, Arbuck SG, Eisenhauer EA,
Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van
Oosterom AT, Christian MC and Gwyther SG: New guidelines to
evaluate the response to treatment in solid tumors. J Natl Cancer
Inst. 92:205–216. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tischoff I, Hengge UR, Vieth M, Ell C,
Stolte M, Weber A, Schmidt WE and Tannapfel A: Methylation of
SOCS-3 and SOCS-1 in the carcinogenesis of Barrett’s
adenocarcinoma. Gut. 56:1047–1053. 2007.PubMed/NCBI
|
|
23
|
Murai M, Toyota M, Suzuki H, et al:
Aberrant methylation and silencing of the BNIP3 gene in colorectal
and gastric cancer. Clin Cancer Res. 11:1021–1027. 2005.PubMed/NCBI
|
|
24
|
Hocker M, Schlenger K, Hockel S and Vaupel
P: Hypoxic cervical cancers with low apoptotic index are highly
aggressive. Cancer Res. 59:4525–4528. 1999.PubMed/NCBI
|
|
25
|
Hocker M and Vaupel P: Tumor hypoxia:
definitions and current clinical, biologic, and molecular aspects.
J Natl Cancer Inst. 93:266–276. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zagzag D, Zhong H, Scalziti JM, Laughner
E, Simons JW and Semenza GL: Expression of hypoxia-inducible factor
1α in brain tumors: association with angiogenesis, invasion, and
progression. Cancer. 88:2606–2618. 2000.
|
|
27
|
Blagosklonny MV: Antiangiogenic therapy
and tumor progression. Cancer Cell. 5:13–17. 2004. View Article : Google Scholar
|
|
28
|
Manka D, Spicer Z and Millhorn DE:
Bcl-2/Adenovirus E1B 19 kDa interacting protein-3 knockdown enables
growth of breast cancer metastases in the lung, liver, and bone.
Cancer Res. 65:11689–11693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bruick RK: Expression of the gene encoding
the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad
Sci USA. 97:9082–9087. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo K, Searfoss G, Krolikowski D, Pagnoni
M, Franks C, Clark K, Yu KT, Jaye M and Ivashchenko Y: Hypoxia
induces the expression of the pro-apoptotic gene BNIP3. Cell Death
Differ. 8:367–376. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sowter HM, Ratcliffe PJ, Watson P,
Greenberg AH and Harris AL: HIF-1-dependent regulation of hypoxic
induction of the cell death factors BNIP3 and NIX in human tumors.
Cancer Res. 61:6669–6673. 2001.PubMed/NCBI
|
|
32
|
Bacon AL, Fox S, Turley H and Harris AL:
Selective silencing of the hypoxia-inducible factor 1 target gene
BNIP3 by histone deacetylation and methylation in colorectal
cancer. Oncogene. 26:132–141. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
De Angelis PM, Fjell B, Kravik KL, Haug T,
Tunheim SH, Reichelt W, Beigi M, Clausen OP, Galteland E and Stokke
T: Molecular characterizations of derivatives of HCT116 colorectal
cancer cells that are resistant to the chemotherapeutic agent
5-fluorouracil. Int J Oncol. 24:1279–1288. 2004.PubMed/NCBI
|
|
34
|
Kothari S, Cizeau J, McMillan-Ward E,
Israels SJ, Bailes M, Ens K, Kirshenbaum LA and Gibson SB: BNIP3
plays a role in hypoxic cell death in human epithelial cells that
is inhibited by growth factors EGF and IGF. Oncogene. 22:4734–4744.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vande Velde C, Cizeau J, Dubik D, Alimonti
J, Brown T, Israels S, Hakem R and Greenberg AH: BNIP3 and genetic
control of necrosis-like cell death through the mitochondrial
permeability transition pore. Mol Cell Biol. 20:5454–5468.
2000.PubMed/NCBI
|
|
36
|
Okami J, Simeone DM and Logsdon CD:
Silencing of the hypoxia-inducible cell death protein BNIP3 in
pancreatic cancer. Cancer Res. 64:5338–5346. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Murai M, Toyota M, Satoh A, Suzuki H,
Akino K, Mita H, Sasaki Y, Ishida T, Shen L, Garcia-Manero G, Issa
JP, Hinoda Y, Tokino T and Imai K: Aberrant DNA methylation
associated with silencing BNIP3 gene expression in haematopoietic
tumors. Br J Cancer. 92:1165–1172. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tan KB, Mattern MR, Eng WK, McCabe FL and
Johnson RK: Nonproductive rearrangement of DNA Topoisomerase I and
II genes: correlation with resistance to topoisomerase inhibitors.
J Natl Cancer Inst. 81:1732–1735. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bras-Gonçalves RA, Rosty C, Laurent-Puig
P, Soulié P, Dutrillaux B and Poupon MF: Sensitivity to CPT-11 of
xenografted human colorectal cancers as a function of
microsatellite instability and p53 status. Br J Cancer. 82:913–923.
2000.PubMed/NCBI
|
|
40
|
Schmaltz C, Harrigan PH, Wells A and
Fisher DE: Regulation of proliferation-survival decisions during
tumor cell hypoxia. Mol Cell Biol. 18:2845–2854. 1998.PubMed/NCBI
|
|
41
|
Oizumi S, Isobe H, Ogura S, Ishida T,
Yamazaki K, Nishimura M, Kawakami Y and Dosaka-Akita H:
Topoisomerase inhibitor-induced apoptosis accompanied by down
regulation of Bcl-2 in human lung cancer cells. Anticancer Res.
22:4029–4037. 2002.PubMed/NCBI
|
|
42
|
Bani MR, Nicoletti MI, Alkharouf NW,
Ghilardi C, Petersen D, Erba E, Sausville EA, Liu ET and Giavazzi
R: Gene expression correlating with response to paclitaxel in
ovarian carcinoma. Mol Cancer Ther. 3:111–121. 2004.PubMed/NCBI
|
|
43
|
Erkan M, Kleeff J, Esposito I, Giese T,
Ketterer K, Büchler MW, Giese NA and Friess H: Loss of BNIP3
expression is a late event in pancreatic cancer contributing to
chemoresistance and worsened prognosis. Oncogene. 24:4421–4432.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Akada M, Crnogorac-Jurcevic T, Lattimore
S, Mahon P, Lopes R, Sunamura M, Matsuno S and Lemoine NR:
Intrinsic chemoresistance to gemcitabine is associated with
decreased expression of BNIP3 in pancreatic cancer. Clin Cancer
Res. 11:3094–3101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen G, Ray R, Dubik D, Shi L, Cizeau J,
Bleackley RC, Saxena S, Gietz RD and Greenberg AH: The E1B
19K/bcl-2-binding proteins Nip3 is a dimeric mitochondrial protein
that activates apoptosis. J Exp Med. 186:1975–1983. 1997.
View Article : Google Scholar : PubMed/NCBI
|