Introduction
TRAIL (tumor necrosis factor (TNF)-related
apoptosis-inducing ligand) belongs to the TNF superfamily, which
can induce apoptosis in a wide variety tumor cells but not normal
cells (1). Because of its ability,
TRAIL is showing promise as a cancer therapeutic agent. TRAIL
induces apoptosis through interacting with death receptor 4 (DR4;
TRAIL-R1) and death receptor 5 (DR5; TRAIL-R2) leading to the
formation of the death-inducing signal complex (DISC) with binding
of caspase-8, leading to apoptosis (extrinsic or death receptor
pathway) (1,2). In addition, TRAIL induces apoptosis
via the disruption of the mitochondria membrane permeability,
release of cytochrome c into the cytoplasm and activation of
caspase-9 (intrinsic or mitochondria pathway) (3). Despite the beneficial effect of TRAIL
to selectively kill tumor cells, many cancer cells appear to show
resistance to TRAIL (2). The
mechanism of TRAIL resistance is not clearly, but several studies
has been reported that TRAIL resistance is intimately associated
with overexpression of anti-apoptosis including FADD-like apoptosis
regulator (c-FLIP), anti-apoptotic Bcl-2 family proteins (e.g.,
Bcl-2 and Bcl-xL) and inhibitor of apoptosis proteins (IAPs)
(2). However, single treatment with
TRAIL may not be sufficient for the treatment of various malignant
tumor cells, TRAIL-resistant cancer cells can be sensitized by
TRAIL sensitizer such as chemotherapeutic drugs and biochemical
inhibitors that suppress the expression of
anti-apoptosis-associated proteins including Bcl-2, c-FLIP or XIAP,
indicating that combination therapy may be a possibility.
Therefore, understanding the molecular mechanisms of TRAIL
resistance and ways to sensitize these cells to undergo apoptosis
by TRAIL are important issues for effective cancer therapy.
Dioscin, a plant glucoside saponin extracted from
the roots of Polygonatum zanlanscianense, has
anti-inflammatory, lipid-lowering, anticancer and hepatoprotective
effects (4–7). Several mechanisms have been proposed
for the anticancer activity of dioscin, including induction of
apoptosis and arrest of cell cycle (8,9).
Dioscin-induced apoptosis were mediated by activation of caspase-9
and -3, together with downregulation of anti-apoptotic Bcl-2
protein (8,10) or by the elevated oxidative stress
mediated by downregulation of peroxiredoxins as well as through
mitochondria dysfunction (5,11).
The aim of this study is to evaluate dioscin as a
sensitizer of TRAIL and to understand the mechanism of the synergy
between dioscin and TRAIL against human renal cancer cells. Dioscin
treatment rendered human renal cancer cells more sensitive to
TRAIL. These results suggest that this combined treatment with
dioscin and TRAIL may provide a safe and effective therapeutic
strategy against malignant cancer that are resistant to various
conventional treatments. Furthermore, we provide novel evidence
that the prominent sensitizing effect of dioscin on TRAIL-induced
apoptosis is due to ROS generation which causes downregulation of
c-FLIP.
Materials and methods
Cells and materials
The Caki cells were obtained from the American Type
Culture Collection (ATCC, Rockville, MD, USA). The culture medium
used throughout these experiments was Dulbecco’s modified Eagle’s
medium (DMEM), containing 10% fetal calf serum (FCS), 20 mM HEPES
buffer and 100 μg/ml of gentamycin. Anti-Bcl-2, anti-PARP,
anti-pro-caspase-3, anti-Mcl-1, and anti-actin antibodies were
purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA).
Anti-c-FLIP antibody was purchased from Alexis Corp. (San Diego,
CA, USA). Dioscin was isolated from Polygonatum
zanlanscianse PAMP) and were directly added to cell cultures at
the indicated concentrations. N-acetyl-L-cysteine (NAC) and
pan-caspase inhibitor (Z-VAD-FMK) were purchased from Calbiochem
(San Diego, CA, USA).
Purification and identification of
dioscin
The root of Dioscorea nipponica Makino was
obtained from the Uiseong Medicinal Farm (Uiseong, Korea). Three
kilograms of the roots were extracted three times with 5 liters of
methanol each time. After filtration, the extract was evaporated in
vacuo to give 115 g of dry sample. The following procedures of
purification of dioscin based on silica-gel chromatography were the
same as previously reported (12).
The purified compound was identified as dioscin by analyses of IR
spectroscopy (Perkin-Elmer, Shelton, CT, USA) and 1H- and 13C-NMR
spectroscopy (Bruker AMX 300, Rheinsten, Germany).
HPLC analysis of dioscin
The purity of dioscin was confirmed by HPLC analysis
as was previously reported (12).
Dioscin and its derivatives, such as prosapogenin A and
prosapogenin C were determined by HPLC system comprising an SCL-10A
system controller, LC-10AD pump and SPD-10A UV detector (Shimadzu,
Japan). The analytical column was a Mightysil RP-C18 GP-250 (Kanto
Chemical Co., USA). The mobile phase for HPLC consisted of 75%
acetonitrile (v/v) with a flow rate of 0.7 ml/min. The column
temperature was maintained at 30˚C. A 10 μl of the sample dissolved
in methanol (1 mg/ml) was injected into the HPLC system, and the UV
absorption at 215 nm was recorded. The retension time of dioscin
was 3.25 min and the purity of dioscin was identified as above
98.5%.
Western blotting
Cellular lysates were prepared by suspending
6×105 cells in 100 μl of lysis buffer (137 mM NaCl, 15
mM EGTA, 0.1 mM sodium orthovanadate, 15 mM MgCl2, 0.1%
Triton X-100, 25 mM Mops, 100 μM phenylmethlsulfonyl fluoride, and
20 μM leupeptin, adjusted to pH 7.2). The cells were disrupted by
sonication and extracted at 4˚C for 30 min. Lysates containing
proteins were quantified using BCA protein assay kit (Pierce,
Rockford, IL, USA). The proteins were electrotransferred to
Immobilon-P membranes (Millipore Corp., Bedford, MA, USA).
Detection of specific proteins was carried out with an ECL western
blotting kit (Millipore) according to the manufacturer’s
instructions.
Cell count and flow cytometry
analysis
Cell counts were performed using a hemocytometer.
Approximately 1×106 Caki cells were suspended in 100 μl
of PBS, and 200 μl of 95% ethanol were added while vortexing. The
cells were incubated at 4˚C for 1 h, washed with PBS, and
resuspended in 250 μl of 1.12% sodium citrate buffer (pH 8.4)
together with 12.5 μg of RNase. Incubation was continued at 37˚C
for 30 min. The cellular DNA was then stained by applying 250 μl of
propidium iodide (50 μg/ml) for 30 min at room temperature. The
stained cells were analyzed by fluorescent activated cell sorting
(FACS) on a FACScanto flow cytometer for relative DNA content based
on red fluorescence.
RNA isolation and reverse
transcriptase-polymerase chain reaction (RT-PCR)
Total cellular RNA was extracted from cells using
the Easy-blue Total RNA Extraction kit (iNtRon, Sungnam, Korea). A
cDNA was synthesized from 5 μg of total RNA using M-MLV reverse
transcriptase (Promega, Madison, WI, USA). The cDNAs for c-FLIP,
Bcl-2 and actin were amplified by PCR with specific primers. The
sequence of the sense primer for c-FLIPL was 5′-CGG ACT ATA GAG TGC
TGA TGG-3′ and the antisense primers were 5′-GAT TAT CAG GCA GAT
TCC TAG-3′. PCR products were analyzed by agarose gel
electrophoresis and visualized by ethidium bromide.
Statistical analysis
Three or more separate experiments were performed.
Statistical analysis was done by paired Student’s t-test or ANOVA.
A P-value <0.05 was considered to have pronounced difference
between experimental and control groups.
Results
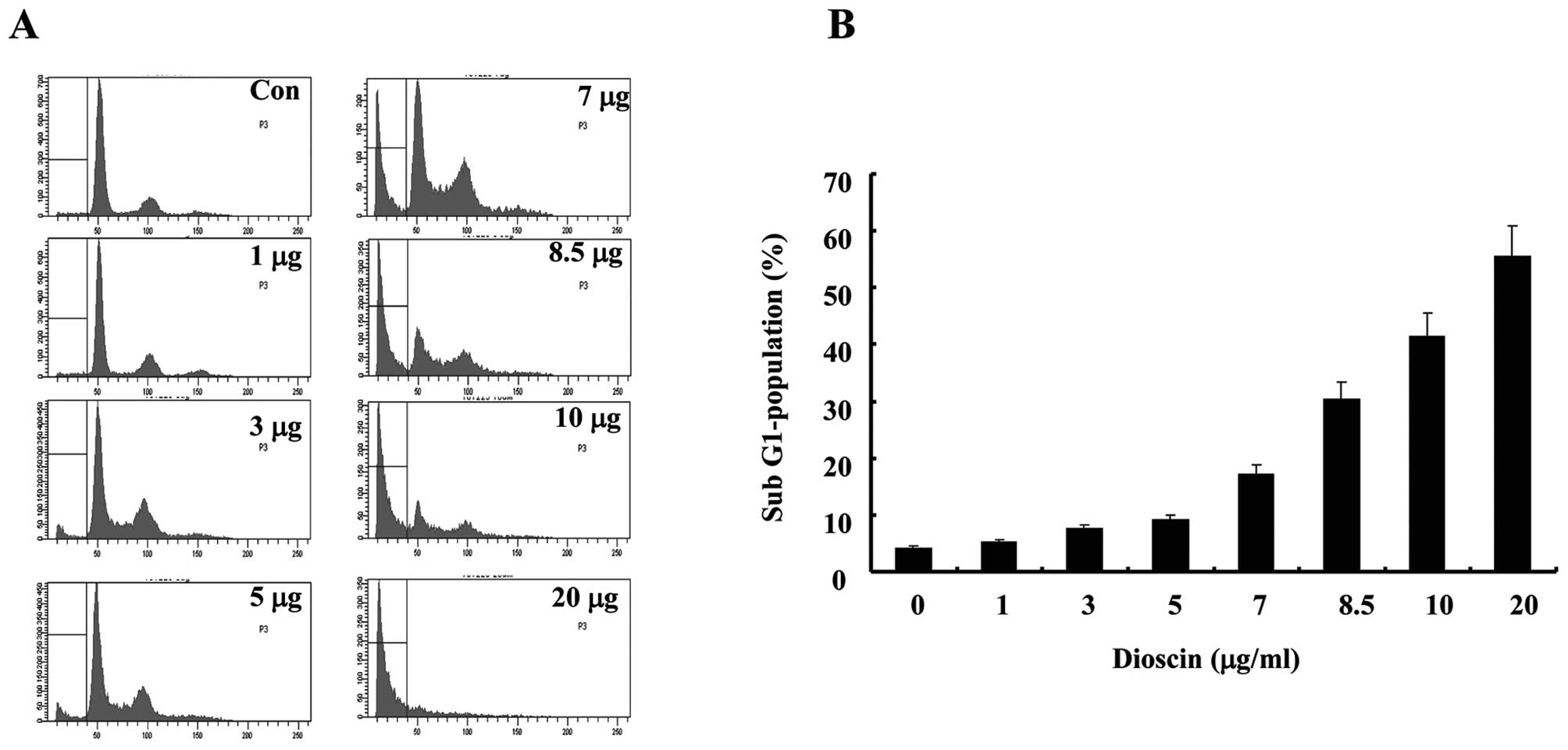
Dioscin treatment induces apoptosis in a
dose-dependent manner in Caki cells
To investigate the effect of dioscin-induced
apoptosis, human renal carcinoma Caki cells were treated with
various concentrations of dioscin. Two established criteria were
subsequently used to assess apoptosis in this study. Apoptosis was
determined in Caki cells using flow cytometry analysis
demonstrating hypo-diploid DNA. Fig.
1A shows treatment with dioscin in Caki cells resulted in a
markedly increased accumulation of sub-G1 phase in a dose-dependent
manner of dioscin. Because cells undergoing apoptosis executed the
death program by activating caspases and cleavage of PARP, we
analyzed expression levels of pro-caspase-3, and cleavage of PARP.
As demonstrated in Fig. 1B,
exposure to dioscin led to a reduction of the 32-kDa precursor,
accompanied by a concomitant revealed cleavage of PARP. Next, we
analyzed nuclear condensation, which is another hallmark of
apoptosis. Combinatory treatment with dioscin plus TRAIL induced
nuclear condensation in Caki cells. In contrast, nuclear
condensation in Caki cells treated with TRAIL alone or dioscin
alone was barely detected.
Dioscin sensitizes renal cancer cells to
TRAIL-mediated apoptosis
In an attempt to search for novel strategies to
overcome TRAIL resistance in cancer cells, we investigated the
effect of the combined treatment with dioscin and TRAIL in Caki
cells. Co-treatment of Caki cells with dioscin and TRAIL resulted
in a markedly increased accumulation of sub-G1 phase cells,
compared with Caki cells treated with dioscin or TRAIL alone
(Fig. 2A). In addition, combinatory
treatment of Caki cells with dioscin and TRAIL strongly stimulated
reduction of the protein levels of pro-caspases 3, Bcl-2, Mcl-1,
and c-FLIPL (Fig.
2B).
Dioscin downregulates Bcl-2, Mcl-1 and
c-FLIP protein expressions
To investigate the underlying mechanisms involved in
dioscin enhanced TRAIL-induced apoptosis, we analyzed the changes
in the expression levels of various apoptosis-regulating proteins.
Bcl-2, Mcl-1 and c-FLIPL protein expressions were
decreased by the indicated concentrations of dioscin-treated Caki
cells in a dose-dependent manner. To further elucidate the
mechanism responsible for the changes in amounts of proteins level,
we determined the levels of Bcl-2, Mcl-1 and c-FLIPL
mRNAs by RT-PCR. c-FLIPL and Mcl-1 mRNA levels remain
constant through the dioscin treatment at different doses in Caki
cells. We found that dioscin treatment of Caki cells
dose-dependently decreased the mRNA levels of Bcl-2 from RT-PCR
analysis, suggesting that dioscin modulates Bcl-2 expression at the
transcriptional level and c-FLIPL and Mcl-1 at the
post-transcriptional level (Fig.
3).
Dioscin plus TRAIL-induced apoptosis was
mediated via caspase-dependent pathway
We next examined whether activation of caspase
pathway plays a critical role in dioscin plus TRAIL-induced
apoptosis. As shown in Fig. 4A,
dioscin plus TRAIL-induced apoptosis was completely prevented by
pre-treatment with a general and potent inhibitor of caspases,
z-VAD-fmk, as determined by FACS analysis. These results suggest
that the combined treatment with dioscin and TRAIL-induced
apoptosis was mediated by caspase-dependent apoptosis pathways. We
also found that z-VAD-fmk prevented all these caspase-related
events such as cleavage of pro-caspase-3 and PARP (Fig. 4B). Pretreatment with z-VAD–fmk
recovered Mcl-1 protein which were downregulated by combination
treatment with dioscon plus TRAIL to basal level, but z-VAD-fmk
partly blocked dioscin plus TRAIL-induced downregulation of
c-FLIPL protein, indicating that the decreased
c-FLIPL protein level was partly caused by caspase
activation. These results suggested the possibilities that the
decreased c-FLIPL protein was partly caused by
caspase-independent pathways (Fig.
4B).
To further clarify the underlying mechanisms of the
decreased c-FLIPL protein level in dioscin-treated
cells, we performed c-FLIPL protein stability test. Caki
cells were treated with cycloheximide (CHX) and dioscin for
different doses. We found that the degradation of
c-FLIPL protein was facilitated by dioscin treatment
(Fig. 4C), implying that dioscin
treatment caused reduction of c-FLIP protein stability.
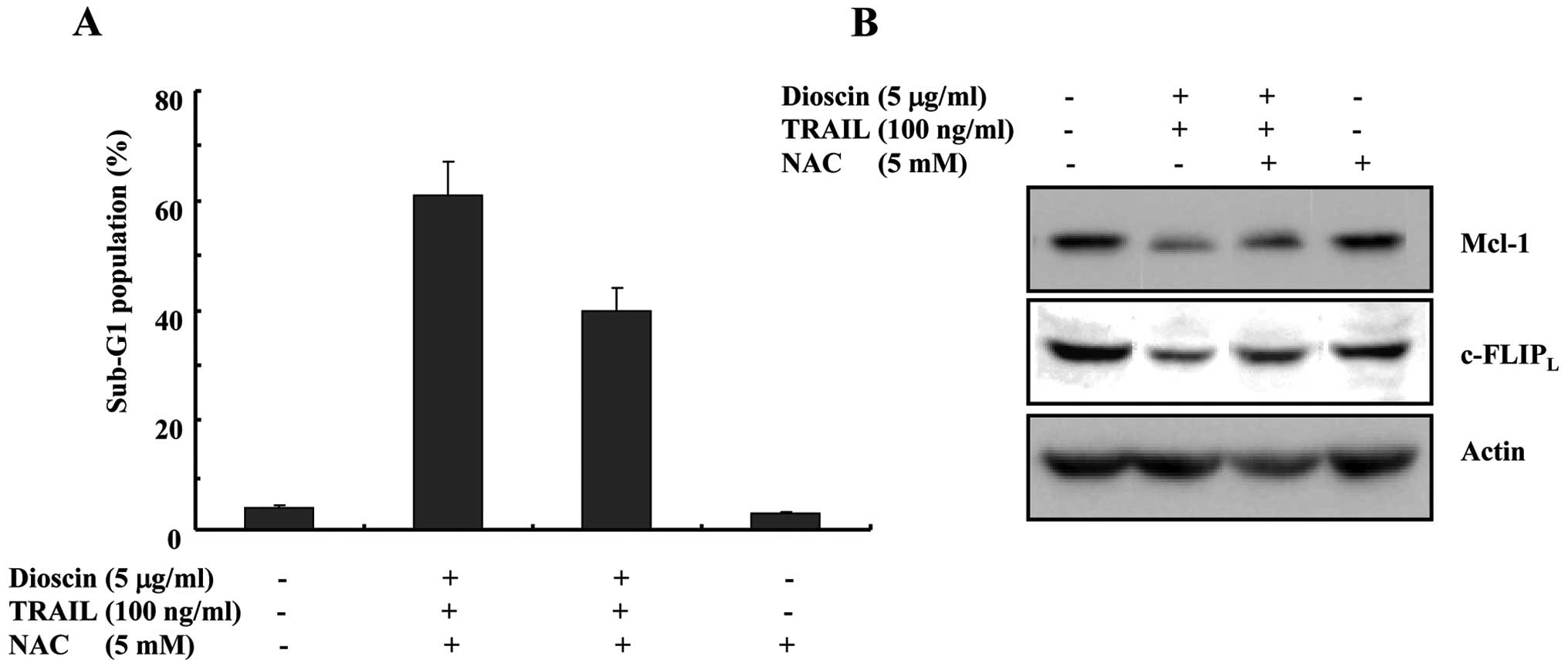
Dioscin-stimulated TRAIL-induced
apoptosis appears to be partially dependent on the formation of
reactive oxygen species (ROS) via downregulation of
c-FLIPL and Bcl-2
Numerous investigations have documented that ROS may
play an important role during apoptosis induction (13,14).
It has been reported that dioscin increases ROS production in
various cancer cells (5,10). Therefore, we investigated whether
ROS generation is directly associated with dioscin plus
TRAIL-induced apoptosis. As shown in Fig. 5A, dioscin plus TRAIL-induced
apoptosis was completely prevented by pretreatment with NAC, as
determined by FACS analysis. As shown in Fig. 5B, pretreatment with NAC decreased
the increased expression levels of c-FLIPL and Bcl-2 by
dioscin treatment to basal levels, dioscon-induced downregulation
of c-FLIPL protein was partly blocked by NAC treatment.
Downregulations of c-FLIPL
contribute to dioscin-stimulated TRAIL-induced apoptosis
We examined whether dowregulation of
c-FLIPL by dioscon is critical to stimulate
TRAIL-induced apoptosis. Overexpression of c-FLIPL in
Caki cells significantly attenuated dioscin-facilitated
TRAIL-induced apoptosis, whereas co-treatment with dioscin plus
TRAIL induced significant apoptosis in Caki/vector cells (Fig. 6). This result suggests that
c-FLIPL downregulation also contributes to
dioscin-facilitated TRAIL-induced apoptosis.
Discussion
In this study, we demonstrated for the first time
that combination treatment with dioscin plus TRAIL on renal cancer
cells synergistically induced apoptosis. Dioscin-mediated
dowregulation of Bcl-2 is controlled at the transcriptional level
in a dose-dependent manner. In contrast, dioscin-induced
downregulation of c-FLIPL is caused by facilitating
degradation of c-FLIPL protein. In addition, we also
found that production of ROS by dioscin treatment seemed to
partially take part in c-FLIPL downregulation.
Several reagents such as compound C, rosiglitazone,
LBH589, and silibinin can induce downregulation of c-FLIP and
subsequent sensitization to TRAIL-induced apoptosis in different
cancer cells (15–18). It is generally recognized that
c-FLIPL protein levels can be regulated by
ubiquitin/proteasome mediated degradation (19,20) or
by their transcriptional control through the NF-κB or c-Fos pathway
(21,22). In this study, dioscin promotes
ubiquitin/proteasome-mediated degradation of c-FLIPL,
leading to downregulation of c-FLIP, but not by transcriptional
control. However, further work is needed for the mechanistic study
to elucidate dioscin-induced activation of the proteasomal
signaling pathway.
It has been suggested that cells can regulate
proteasome function in response to increased ROS level both by
altering the total number of proteasomes and by altering the
subunit components of the ubiquitin-proteasome (23). Dioscin sensitizing HL-60 cells to
apoptosis through a ROS-dependent mechanism is supported by direct
measurement of ROS generation (5).
Recently, several studies have shown that ROS downregulates c-FLIP
levels and increases the sensitivity to apoptotic stimuli (24,25).
Therefore, we investigated whether downregulations of
c-FLIPL was actually mediated by ROS signaling pathway.
In the presence of NAC, the decreased levels of c-FLIPL
caused by dioscin were partly restored. Taken together,
dioscin-stimulated TRAIL-induced apoptosis appears to be dependent
on the formation of ROS for downregulations of
c-FLIPL.
Recently, it has been suggested that cytotoxicity of
dioscin was mediated by activating death receptor through
upregulation of Fas, FasL (Fas ligand), TNF-α, TNF receptor-1, and
TNF receptor-associated factor 1 as well as by activating
mitochondrial pathways through downregulation of Bcl-2 and in human
gastric carcinoma cells (26).
However, we found that the expression of Bcl-2 was downregulated by
dioscin treatment at transcriptional level in our study, the
expression level of TRAIL death receptor (DR5) was not altered by
dioscin treatment.
In summary, we suggest that the use of dioscin is a
potentially important therapeutic approach for enhancing
sensitivity to TRAIL via downregulation of proteins related to the
inhibition of the apoptotic processes such as Bcl-2 and c-FLIP.
Acknowledgements
This research was supported by the Yeungnam
University research grants in 2009.
References
|
1
|
Tan ML, Ooi JP, Ismail N, Moad AI and
Muhammad TS: Programmed cell death pathways and current antitumor
targets. Pharm Res. 26:1547–1560. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu GS: TRAIL as a target in anti-cancer
therapy. Cancer Lett. 285:1–5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deng Y, Lin Y and Wu X: TRAIL-induced
apoptosis requires Bax-dependent mitochondrial release of
Smac/DIABLO. Genes Dev. 16:33–45. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sautour M, Mitaine-Offer AC, Miyamoto T,
Dongmo A and Lacaille-Dubois MA: A new steroidal saponin from
Dioscorea cayenensis. Chem Pharm Bull. 52:1353–1355. 2004.
View Article : Google Scholar
|
|
5
|
Wang Y, Che CM, Chiu JF and He QY: Dioscin
(saponin)-induced generation of reactive oxygen species through
mitochondria dysfunction: a proteomic-based study. J Proteome Res.
6:4703–4710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaskiw MJ, Tassotto ML, Mok M, Tokar SL,
Pycko R, Th’ng J and Jiang ZH: Structural analogues of diosgenyl
saponins: synthesis and anticancer activity. Bioorg Med Chem.
17:7670–7679. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu B, Yin L, Xu L and Peng J: Application
of proteomic and bioinformatic techniques for studying the
hepatoprotective effect of dioscin against CCl4-induced
liver damage in mice. Planta Med. 77:407–415. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cai J, Liu M, Wang Z and Ju Y: Apoptosis
induced by dioscin in HeLa cells. Biol Pharm Bull. 25:193–196.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gao LL, Li FR, Jiao P, et al: Paris
chinensis dioscin induces G2/M cell cycle arrest and apoptosis in
human gastric cancer SGC-7901 cells. World J Gastroenterol.
17:4389–4395. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang Z, Zhou J, Ju Y, Zhang H, Liu M and
Li X: Effects of two saponins extracted from the polygonatum
Zanlanscianense pamp on the human leukemia (HL-60) cells. Biol
Pharm Bull. 24:159–162. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhiyu W, Yue C, Neng W, et al: Dioscin
induces cancer cell apoptosis through elevated oxidative stress
mediated by downregulation of peroxiredoxins. Cancer Biol Ther.
13:138–147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kwon CS, Sohn HY, Kim SH, et al:
Anti-obesity effect of Dioscorea nipponica Makino with
lipase-inhibitory activity in rodents. Biosci Biotechnol Biochem.
67:1451–1456. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sheng-Tanner X, Bump EA and Hedley DW: An
oxidative stress-mediated death pathway in irradiated human
leukemia cells mapped using multilaser flow cytometry. Radiat Res.
150:636–647. 1998. View
Article : Google Scholar
|
|
14
|
Choi YK, Seo HS, Choi HS, Choi HS, Kim SR,
Shin YC and Ko SG: Induction of Fas-mediated extrinsic apoptosis,
p21WAF1-related G2/M cell cycle arrest and ROS generation by
costunolide in estrogen receptor-negative breast cancer cells,
MDA-MB-231. Mol Cell Biochem. 363:119–128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Son YG, Kim EH, Kim JY, et al: Silibinin
sensitizes human glioma cells to TRAIL-mediated apoptosis via DR5
up-regulation and down-regulation of c-FLIP and survivin. Cancer
Res. 67:8274–8284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim YH, Jung EM, Lee TJ, et al:
Rosiglitazone promotes tumor necrosis factor-related
apoptosis-inducing ligand-induced apoptosis by reactive oxygen
species-mediated up-regulation of death receptor 5 and
down-regulation of c-FLIP. Free Radic Biol Med. 44:1055–1068. 2008.
View Article : Google Scholar
|
|
17
|
Kauh J, Fan S, Xia M, Yue P, Yang L, Khuri
FR and Sun SY: c-FLIP degradation mediates sensitization of
pancreatic cancer cells to TRAIL-induced apoptosis by the histone
deacetylase inhibitor LBH589. PLoS One. 5:e103762010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jang JH, Lee TJ, Yang ES, et al: Compound
C sensitizes Caki renal cancer cells to TRAIL-induced apoptosis
through reactive oxygen species-mediated down-regulation of c-FLIPL
and Mcl-1. Exp Cell Res. 316:2194–2203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Poukkula M, Kaunisto A, Hietakangas V, et
al: Rapid turnover of c-FLIPshort is determined by its unique
C-terminal tail. J Biol Chem. 280:27345–27355. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kaunisto A, Kochin V, Asaoka T, Mikhailov
A, Poukkula M, Meinander A and Eriksson JE: PKC-mediated
phosphorylation regulates c-FLIP ubiquitylation and stability. Cell
Death Differ. 16:1215–1226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li W, Zhang X and Olumi AF: MG-132
sensitizes TRAIL-resistant prostate cancer cells by activating
c-Fos/c-Jun heterodimers and repressing c-FLIP(L). Cancer Res.
67:2247–2255. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Benayoun B, Baghdiguian S, Lajmanovich A,
et al: NF-kappaB-dependent expression of the antiapoptotic factor
c-FLIP is regulated by calpain 3, the protein involved in
limb-girdle muscular dystrophy type 2A. FASEB J. 22:1521–1529.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Glickman MH and Raveh D: Proteasome
plasticity. FEBS Lett. 579:3214–3223. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nitobe J, Yamaguchi S, Okuyama M, et al:
Reactive oxygen species regulate FLICE inhibitory protein (FLIP)
and susceptibility to Fas-mediated apoptosis in cardiac myocytes.
Cardiovasc Res. 57:119–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kanayama A and Miyamoto Y: Apoptosis
triggered by phagocytosis-related oxidative stress through FLIPS
down-regulation and JNK activation. J Leukoc Biol. 82:1344–1352.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu M, Xu L, Yin L, et al: Cytotoxicity of
dioscin in human gastric carcinoma cells through death receptor and
mitochondrial pathways. J Appl Toxicol. Feb 14–2012.(Epub ahead of
print).
|