Introduction
Hepatocellular carcinoma (HCC) is the most common
type of human liver cancer, leading to increasing mortality rates
worldwide. Currently available chemotherapies cannot achieve good
prognosis for patients with unresectable HCC (1,2).
Therefore, it is necessary to investigate the molecular mechanisms
responsible for HCC development in order to identify new targets
for early diagnosis and novel treatments. Proteases have been found
to be important factors in pathophysiology of tumor dieases.
Besides their contribution to cancer progression by degrading
extracellular matrix proteins, certain proteases serve as signaling
molecules through binding to specific membrane receptors, termed
the protease-activated receptors (PARs). PARs are
seven-transmembrane G-protein-coupled receptors, which are
activated by a unique proteolytic mechanism that involves
N-terminal domain cleavage by specific serine proteases. The
N-terminal cleavage in turn generates a new tethered ligand for
PARs binding and receptor activation (3). PAR-1, PAR-3 and PAR-4 are cleaved and
activated by thrombin. PAR-2 is activated by trypsin and
trypsin-like proteases, including some coagulation factors
(4). PAR-2 can also be activated
in vitro by synthetic peptides that resemble the new
sequence (SLIGKV) produced after receptor cleavage (5).
Importance of trypsin, a major serine protease, has
been evidenced recently in many cancers, including digestive tract
tumors. Extra-pancreatic production of trypsin was found in ovarian
(6), lung (7), gastric (8), and colonic tumors (9) and colon cancer cell lines as well
(10,11). In addition, overexpression of
exogenous trypsinogen cDNA in human gastric cancer cells increases
their tumorigenicity in nude mice (8).
Recently, it has been shown that trypsin targeted to
PAR-2 is a very robust growth factor for human colon cancer cells
(12). However, expression and
functional consequences of activation of PAR-2 in hepatocellular
carcinoma have not yet been reported. This study is to determine
the PAR-2 expression in HepG2 cells, the effect on hepatocellular
carcinoma proliferation of PAR-2 activated by trypsin or agonist
peptide, and the signaling pathway molecules downstream of PAR-2
that may contribute to colon cancer cell proliferation. By using
the human hepatoma cell line HepG2, we showed that upregulation of
c-fos and PCNA (two downstream molecules of ERK1/2) results from
activation of PAR-2 by trypsin or the synthetic activating peptide
SLIGKV-NH2. These data provide the first piece of
evidence that PAR-2 is expressed in human hepatoma cells, and PAR-2
activation plays an essential role in growth of hepatoma cells,
partially through the ERK/AP-1 pathway.
Materials and methods
Reagents
The PAR-2 activating peptide AP2
(SLIGKV-NH2) and its reverse sequence RP
(VKGILS-NH2) were obtained from Xi’an Lianmei
Biotechnology Co., Ltd. (China). Trypsin was purchased from
Sigma-Aldrich (St. Louis, MO, USA). The MEK inhibitor PD98059 was
purchased from Promega (USA). The goat polyclonal anti-PAR-2
antibody was purchased from Santa Cruz Biotechnology, Inc. The
rabbit polyclonal anti-c-fos antibody and mouse monoclonal
anti-PCNA antibody were obtained from NeoMarkers for Lab Vision
Corp., USA.
Cell culture
HepG2 cells were cultured in high-glucose (4.5 g/l)
Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 12%
fetal calf serum (FCS, Gibco), and 100 μg/ml
penicillin/streptomycin. The cells were incubated at 37°C, 5 %
CO2 in a humidified atmosphere.
Immunocytochemical staining
The cells were fixed with cold methanol:acetone
(1:1) at −20°C for 10 min, and washed three times with PBS. The
fixed cells were treated with 3% hydrogen peroxide for 10 min to
eliminate endogenous peroxidase. After blocking with rabbit serum
for 10 min, the cells were incubated with goat polyclonal antibody
to PAR-2 (1:100, final dilution) for 2 h at 37°C. Control cells
were incubated with non-immune goat IgG2, and the concentration of
which was adjusted to that of the primary antibody to verify the
specificity of the labeling. The cells were then incubated with
biotinylated rabbit anti-goat IgG, followed by avidin peroxidase.
Samples were washed and the color was developed using the Dako 3,
3-diaminobenzidine tetrahydrochloride (DAB) substrate-chromogen
system. After further washes, the samples were counterstained with
hematoxylin, dehydrated, and coverslipped.
Immunofluorescence staining
Immunofluorescence staining was performed on HepG2
cells grown on glass coverslips. The cells were treated with 0.2%
Triton X-100 for 20 min at room temperature. After blocked with
rabbit serum for 10 min, the cells were incubated with goat
polyclonal antibody to PAR-2 (1:100, final dilution) overnight at
4°C. The control slides were not treated with the primary antibody.
After three washes with PBS, the cells were incubated with
FITC-labelled rabbit anti-goat IgG (1:100, final dilution) for 40
min at 37°C. After three washes with PBS, confocal fluorescence
images were taken with Zeiss LSM510 METANLO (Germany).
Flow cytometry
HepG2 cells were seeded at the density of
8×104/ml and allowed to attach for 24 h. The medium was
removed and the attached cells were rinsed twice with serum-free
medium. The cells were replenished with serum-free medium and
starved in serum-free media for 24 h to maintain quiescence. Then
the cells were incubated with or without PAR-2 agonists (50 μM
SLIGKV-NH2 or 25 nM trypsin) or 50 μM
VKGILS-NH2. After 24 h of culture, the cells were
harvested, washed twice with cold PBS, and fixed in 75% ethanol for
at least 24 h at 4°C. The cells were then washed with PBS
containing 1% BSA and incubated with 100 μg/ml RNase A (Sigma, USA)
and 50 μg/ml propidium iodide (Sigma) for 2 h at room temperature.
Finally, the stained cells were analyzed on a FACSCalibur flow
cytometer. Proliferation index (PI) was calculated according to the
following equation: PI =
(S+G2M)/(G0G1+S+G2M) ×
100%.
Reverse-transcription polymerase chain
reaction (RT-PCR)
To evaluate the mRNA level of PAR-2 expression in
HepG2 cells, and the effects of PAR-2 agonists
(SLIGKV-NH2 or trypsin) and VKGILS-NH2 on
PAR-2 expression, the culture flasks were replenished with
serum-free medium and the cells were incubated for 24 h to maintain
quiescence. Then the cells were incubated with or without PAR-2
agonists (50 μM SLIGKV-NH2 or 25 nM trypsin) or 50 μM
VKGILS-NH2. After 24 h of culture, total RNA was
extracted from the cells with TRIzol Reagent (Invitrogen Corp.,
Carlsbad, CA, USA). First-strand cDNA was synthesized by reverse
transcription of the RNA with the Superscript Preamplification
system according to the manufacturer’s instructions. PCR
amplification was performed with 2.5 U of Taq DNA polymerase on 5
μg of cDNA. The reaction was allowed to proceed for 35 cycles at
94°C for 45 sec, 51°C for 45 sec, and 72°C for 90 sec. Control PCRs
were performed by substituting water for cDNA and omitting RT
during the DNA synthesis. To evaluate the effects of PAR-2 agonists
and VKGILS-NH2 on c-fos and PCNA mRNA expression, the
wells were replenished with serum-free medium and the cells were
incubated for 24 h to maintain them quiescent. Then the cells were
incubated with or without 50 μM SLIGKV-NH2, 25 nM
trypsin or 50 μM VKGILS-NH2. In some experiments, HepG2
cells were preincubated for 1 h with 50 μM PD98059 before
stimulated with PAR-2 agonists. After 50 min and 24 h of culture,
total RNA was extracted from the cells to detect c-fos or PCNA mRNA
expression, respectively. The reaction was allowed to proceed for
35 cycles at 94°C for 45 sec, 55°C for 1 min, and 72°C for 45 sec.
The quality of the PCR product was checked by 1.5% agarose gel
electrophoresis at 90 V for 45 min and visualized by 0.5 μg/ml
ethidium bromide. Primers used in this study are given in Table I.
 | Table IOligonucleotides used in this
study. |
Table I
Oligonucleotides used in this
study.
| Human PAR-2-F |
5′-AGAAGCCTTATTGGTAAGGTT-3′ |
| Human PAR-2-R |
5′-AACATCATGACAGGTCGTGAT-3′ |
| c-fos-F |
5′-AGAATCCGAAGGGAAAGGAA-3′ |
| c-fos-R |
5′-CTTCTCCTTCAGCAGGTTGG-3′ |
| PCNA-F |
5′-TTTCTAGGTCTCAGCCGGTC-3′ |
| PCNA-R |
5′-GCAAATTCACCAGAAGGCAT-3′ |
| β-actin-F |
5′-TGTTTGAGACCTTCAACACCC-3′ |
| β-actin-R |
5′-AGCACTGTGTTGGCGTACAG-3′ |
Proliferation assay
HepG2 cells were seeded in 96-well culture plates at
5000 cells/well and allowed to attach for 24 h. The medium was
removed and attached cells were rinsed twice with serum-free
medium. They were then grown in 100 μl of culture medium without
FCS for 24 h to maintain quiescence. Then 200 μl of a fresh
serum-free medium, with or without PAR-2 agonists (50 μM
SLIGKV-NH2 or 25 nM trypsin) or 50 μM
VKGILS-NH2, were added. In some experiments, HepG2 cells
were preincubated for 1 h with 50 μM PD98059 before stimulated with
PAR-2 agonists. After 24 h of culture, cell proliferation was
measured using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
(Sigma-Aldrich). Finally, the treatment medium was removed and DMSO
was added to each plate, and absorbance of each well was measured
at 590 nm using a 96-well-microplate reader.
Western blot analysis
For the changes of c-fos and PCNA protein
expression, HepG2 cells were grown to 50% confluence and
serum-deprived for 24 h before the addition of PAR-2 agonists (50
μM SLIGKV-NH2 or 25 nM trypsin) or 50 μM
VKGILS-NH2. In some experiments, HepG2 cells were
preincubated for 1 h with 50 μM PD98059 before their stimulation
with 50 μM PAR-2 AP or 25 nM trypsin. After 2 and 24 h of culture,
HepG2 cells were lysed in lysis buffer containing leupeptin and
phenylmethylsulfonyl fluoride to detect c-fos and PCNA protein
expression, respectively. Electorophoresis was performed through a
10% sodium dodecyl sulfate (SDS) polyacrylamide gel and transferred
to polyvinylidene difluoride (PVDF) membranes. Membranes were
blocked in 5% non-fat milk and washed in Tris-buffered saline (TBS)
containing 0.1% Tween-20 for 1 h at room temperature followed by
the addition of anti-c-fos or anti-PCNA antibody (1:200) overnight
at 4°C. Then, the membrane was washed with TBS/Tween-20 for 1 h and
horseradish peroxidase-labeled anti-mouse or anti-rabbit antibody
(1:5000) was used as the secondary antibody. Blots were developed
with a chemiluminescence detection system (BeyoECL Plus, Beyotime
Institute of Biotechnology).
Statistical analysis
Results are expressed as means ± SD. The P-value of
differences between single subgroups was calculated with the Least
Significance Difference post hoc test (LSD test).
Results
PAR-2 expression in human hepatoma cell
line HepG2
To confirm the expression and localization of PAR-2
at protein level, immunohistochemical and immunofluorescence
analysis were carried out on human hepatoma cell line HepG2. As
shown in Fig. 1, immunocytochemical
and immunofluorescence staining for PAR-2 in HepG2 cells revealed
its expression in the cytoplasm. No immunocytochemical or
immunofluorescence signal was observed in the negative-control
groups. In order to confirm whether HepG2 cells express PAR-2 at
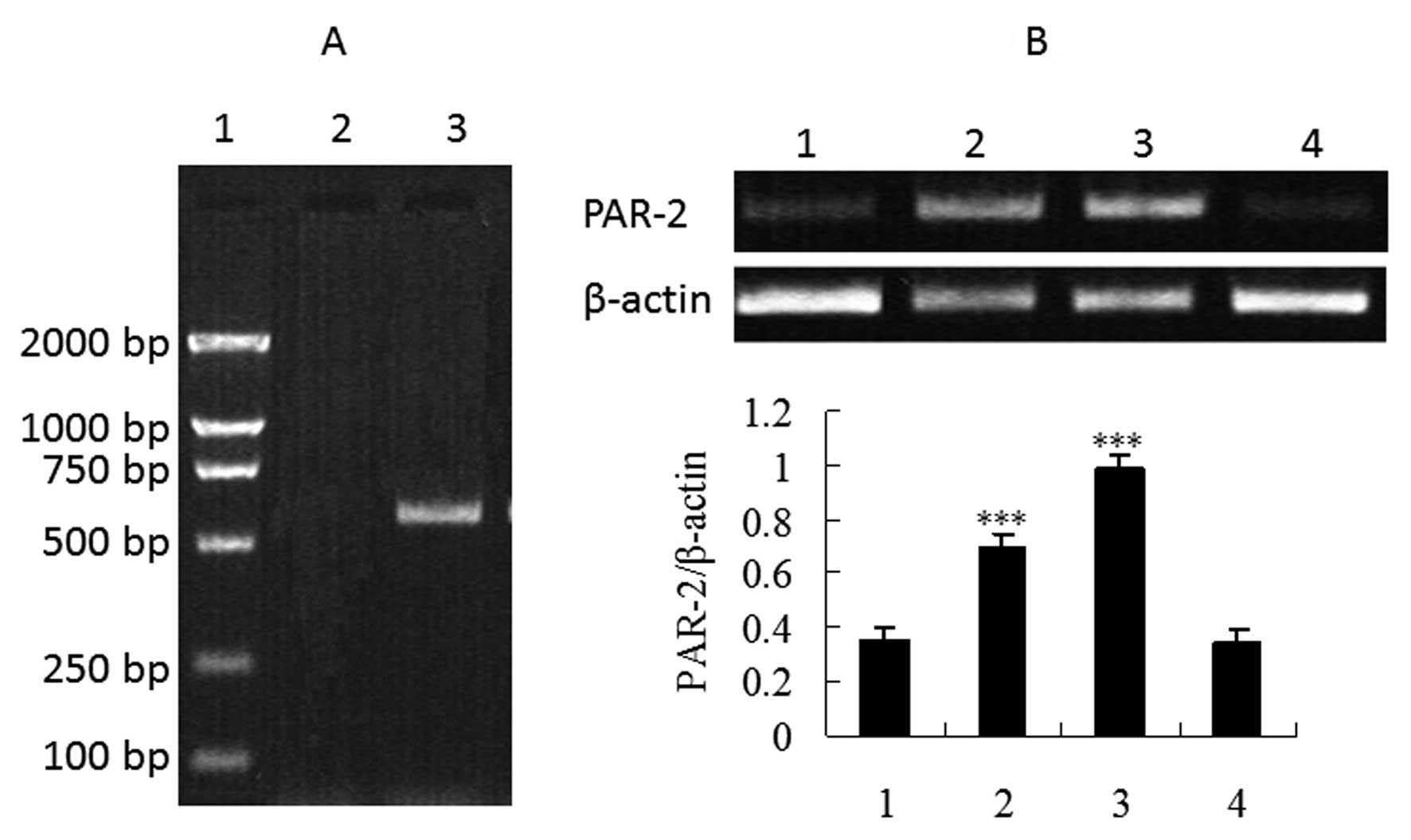
the mRNA level, we investigated the presence of specific PAR-2 mRNA
transcripts in total RNA extracted from HepG2 cells. Since PAR-2
has been shown to be activated by trypsin or PAR-2-activating
peptide AP2 (13), we also
performed RT-PCR studies following incubation of HepG2 cells with
the two activators. Fig. 2 shows
that mRNA expression dramatically increased in the cells treated
with trypsin or SLIGKV-NH2 compared to the control group
and the reverse PAR-2 AP group (P<0.001), further demonstrating
that PAR-2 expression has been upregulated by trypsin or
PAR-2-activating peptide AP2.
Trypsin and PAR-2 AP promote HepG2 cell
growth by accelerating the hepatoma cell cycle progression
To identify the phase of cell cycle affected by
trypsin, SLIGKV-NH2 or VKGILS-NH2, cell cycle
distribution was assayed by flow cytometry. Cells were synchronized
with 24-h serum starvation and then induced to re-enter the cell
cycle by treatment with trypsin, SLIGKV-NH2 or
VKGILS-NH2. Flow cytometric analysis was performed after
propidium iodide staining. After 1-day treatment, as shown in
Table II, trypsin or
SLIGKV-NH2 treatment significantly increased the
percentage of cells in the S phase, G2/M phase and the
proliferation index (PI) of HepG2 cells (P<0.001). Consequently,
the percentage of cells in the G0/G1 phase
was reduced (P<0.001). But there was no statistical significance
of the difference between the reverse PAR-2 agonists and control
group. These results suggest that trypsin or SLIGKV-NH2
promotes cell cycle progression and stimulate the growth of HepG2
cells (Fig. 3).
| Table IIEffects of trypsin, SLIGKV or VKGILS
on cell cycle distribution in HepG2 cells. |
Table II
Effects of trypsin, SLIGKV or VKGILS
on cell cycle distribution in HepG2 cells.
| Cell cycle (%) | |
|---|
|
| |
|---|
| Group |
G0/G1 |
G2/M | S | PI |
|---|
| Control group | 79.12±0.67 | 9.54±0.34 | 11.34±0.55 | 20.88±0.67 |
| 50 μM SLIGKV | 57.85±0.46a | 13.20±0.15a | 28.95±0.54a | 42.15±0.46a |
| 25 nM trypsin | 56.11±0.85a | 13.49±0.44a | 30.41±0.44a | 43.88±0.86a |
| 50 μM VKGILS | 79.27±0.85 | 9.51±0.47 | 11.23±0.54 | 20.73±0.85 |
Trypsin and PAR-2 AP stimulate cell
proliferation in HepG2 cells via ERK phosphorylation
To evaluate the role of PAR-2 on hepatoma cell
proliferation, serum-starved HepG2 cells were cultured in the
presence or absence of trypsin, SLIGKV-NH2 or
VKGILS-NH2, and the proliferation rate of HepG2 cells
was evaluated after 24 h of culture. In some experiments, HepG2
cells were preincubated for 1 h with 50 μM PD98059 before cell
stimulation with PAR-2 agonists. Because ERK1/2 has been shown to
play a pivotal role in the pathway leading to growth
factor-regulated proliferation (14), we used the pharmacological inhibitor
PD98059 to determine the involvement of ERK1/2 in the
growth-stimulating effect of PAR-2. The results from MTT assays
showed that, the proliferation rate of HepG2 cells treated with
trypsin or SLIGKV-NH2 was significantly increased
(P<0.001), and pretreatment of HepG2 cells with PD98059 resulted
in significant decrease of cell proliferation induced by SLIGKV or
trypsin (P<0.001). There was no statistical significance of the
difference between VKGILS and control group. These data clearly
show that PAR-2 agonists act as growth factors for HepG2 cells
through the MEK/ERK1/2 pathway.
Blockade of ERK1/2 phosphorylation
inhibits PAR-2 mediated enhancement of transcription and protein
expression of c-fos in HepG2 cells
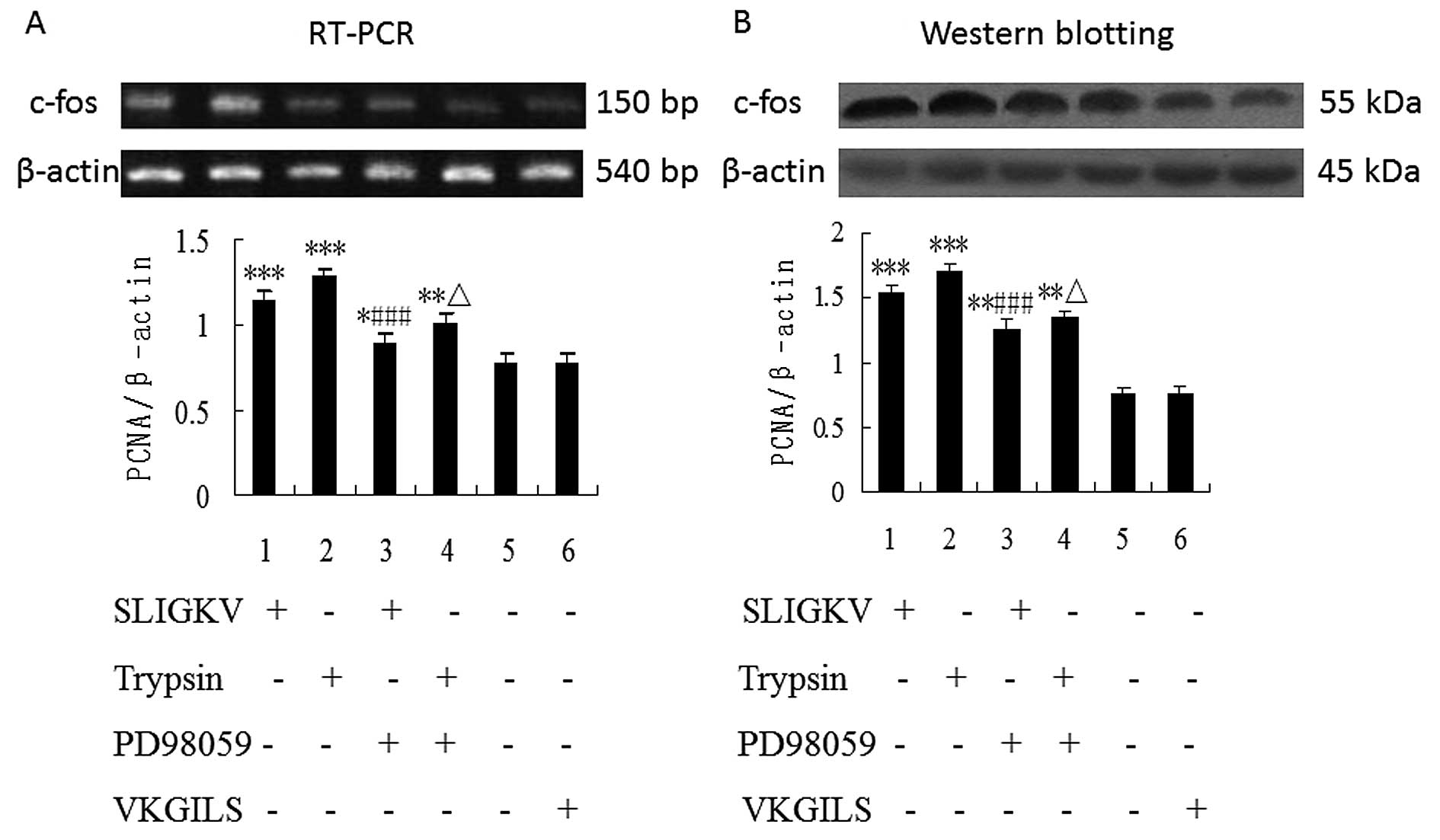
Because PAR-2-mediated HepG2 cell proliferation
could be influenced by inhibitor of MEK, we sought to identify
whether PD98059 inhibits PAR-2-mediated enhancement of
transcription and protein expression of the immediate-early gene
c-fos in HepG2 cells. As shown in Fig.
4, the mRNA and protein expression of c-fos were upregulated in
HepG2 cells stimulated by PAR-2 agonists (50 μM
SLIGKV-NH2 or 25 nM trypsin) (P<0.001) and the
upregulation of c-fos by PAR-2 activators was significantly blocked
by PD98059 (P<0.001). There was no statistical significance of
the difference between VKGILS and control group. Densitometric
analyses showed significant differences for each of the
experiments.
PD98059 inhibits PAR-2 agonist-induced
PCNA mRNA and protein upregulation
As shown by RT-PCR and western blot analysis
(Fig. 5), the mRNA and protein
expression of PCNA were unregulated in HepG2 cells stimulated by
PAR-2 agonists (50 μM SLIGKV-NH2 or 25 nM trypsin), 24 h
of treatment (P<0.001). Furthermore, the upregulation of PCNA
expression induced by PAR-2 activators in HepG2 cells was
significantly blocked by pretreatment with the inhibitor PD98059
(P<0.001). In contrast, no change of PCNA expression was
observed in HepG2 cells with VKGILS-NH2 treatment.
Discussion
Tumor micro-environment is rich in active molecules
such as growth factors, inflammatory mediators, cytokines and
proteinases (e.g. metalloproteases and serine proteases).
Proteolytic enzymes regulate the proliferation, angiogenesis,
invasion and metastasis in cells (15). Trypsin not only degrades
extracellular matrix proteins but also modulates cellular functions
through PAR-2 activation (16). New
insights into tumor biology research have revealed a key role of
trypsin in stomach, colorectal and pancreatic tumor progression
(17). Recently, it has been shown
that downregulation of trypsin (11) or serine protease inhibitors suppress
carcinogenesis in many in vivo and in vitro assays
(18). Thus, we speculate that
trypsin, and possibly other serine proteases targeting PAR-2, can
be used as new important signaling proteins in the control of HCC
growth.
In this study, the expression of PAR-2 and role of
PAR-2 in human hepatoma cell line were reported for the first time.
The human hepatoma cell line HepG2 was used as a model cell line
and our result showed that PAR-2 was expressed mainly in cell
membrane and cytoplasm. RT-PCR studies showed PAR-2 mRNA was
expressed in HepG2 cells, and the mRNA expression dramatically
increased in the cells treated with trypsin or
SLIGKV-NH2 compared to the control group or the reverse
PAR-2 AP treated group. The upregulation of PAR-2 mRNA may be due
to the appearance of desensitization and hydrolization induced by
PAR-2 activation, resulting in the feedback upregulation of PAR-2
mRNA. PAR-2 activation by trypsin plays a key role in hepatoma
cells and we found activation of PAR-2 in hepatoma cells by a
specific, synthetic, peptide SLIGKV resulted in enhancement of cell
proliferation, mimicking the trypsin effect. In contrast, the
reverse peptide was devoid of any mitogenic effect. Based on the
above, it is conceivable that the use of selective and potent PAR-2
antiagonists or trypsin inhibitors may be useful for HCC treatment.
As previously reported by others (19), compared to trypsin, AP2 is an
agonist with lower potency. The differences in the potency of AP2
are probably due to the non-sufficient presentation of AP2 to the
binding domain of PAR-2. To clarify the role of PAR-2 activation by
trypsin or PAR-2 AP in HepG2 cell cycle progression, the percentage
of G1 phase, S phase, G2/M phase and
proliferation index (PI) of HepG2 cells were investigated. We found
that trypsin and SLIGKV-NH2 significantly reduced the
ratio of G0/G1 phase, but increased the
percentage of S phase, G2/M phase and proliferation
index (PI) in HepG2 cells, compared to the control group and
VKGILS-NH2 group. These data suggest that PAR-2 agonists
accelerate the progress of cell cycle from
G0/G1-phase to S and G2/M phase,
and promote the synthesis of DNA of hepatoma cells. Previous
studies by others have demonstrated that cyclin D1 promoted the
G1-S phase of the cell cycle and was frequently
overexpressed in many human cancers including HCC (20,21),
and downregulation of cyclin D1 inhibited HCC growth in animal
models (22). Therefore, we
speculate that one mechanism by which PAR-2 activation induced HCC
proliferation is through the control of cyclin D1 expression.
Further research is ongoing to demonstrate this phenomenon.
The efficient and potent mitogenic action of trypsin
on hepatoma cells raises the question of the endogenous source(s)
of trypsin or other serine proteases that can activate PAR-2 in
situ in HCC. The importance of locally secreted trypsin at the
vicity of HCC should be emphasized: i) normal epithelia cells
surrounding hepatoma cells are likely a source of active trypsin
(23); ii) blood vessels
surrounding tumors also express trypsin (24); iii) it has been suggested from
studies in pancreas that trypsin-like enzymes secreted by tumor
cells could directly regulate growth of pancreatic cells in an
autocrine manner by interacting with PAR-2 (20). In gastric carcinoma cells, it has
recently been reported that trypsinogen secreted by tumor cells,
when activated to trypsin, can stimulate the growth and
adhesiveness of the producer cells in an autocrine manner (25). Thus, we speculate that some hepatoma
cell lines may produce and secrete trypsinogens as well (4). Trypsin is also present in serum at
nanomolar concentrations and can diffuse from blood to tumor cells.
Elevated trypsin levels were reported in the serum of patients with
HCC (26), suggesting that serum
may be an important source of trypsin in cancer patients. These
data suggest the possibility of autocrine/paracrine regulation of
PAR-2 activity by trypsin in hepatoma cells.
To elucidate the mechanism by which PAR-2 induces
hepatoma cell proliferation, the cell signaling pathway leading to
cell proliferation after activaton of PAR-2 was analyzed.
PAR-mediated signaling is known to be involved in the activation of
MAPK cascades in a number of cells (27). The extracellular signal-regulated
kinase 1/2 (ERK1/2) pathway typically transduces growth factor
signals that lead to cell differentiation or proliferation. Our
data demonstrated that the MEK (upstream activator of ERK1/2)
inhibitor PD98059 strongly decreased hepatoma cell proliferation
stimulated by PAR-2 agonists. Thus, the relationship between ERK1/2
and cell proliferation is well demonstrated. Although the
intracellular pathways responsible for PAR-2-mediated stimulation
of ERK1/2 phosphorylation by PAR-2 agonists needs further research,
it is worth mentioning that PAR-2 agonists transactivate the EGF-R
through a pathway that includes matrix metalloproteinase-dependent
cleavage and release of TGF-α, which in turn activates the EGF-R
and downstream MAPK cascade, leading to cell proliferation
(28). Moreover, PAR-2 couples
Gαq/11 and phospholipase Cβ, leading to hydrolysis of
phosphatidylinositol bisphosphate, Ca2+ mobilization,
and activation of protein kinase C (PKC) and ERK1/2 (29). These data suggest that PAR-2
agonists may stimulate the proliferation of hepatoma cells by
EGFR-ERK1/2 or Ca2+-ERK1/2 pathway.
The activator protein-1 (AP-1) transcription factor
is a family of transcription factors composed of homodimers and
heterodimers which are members of Jun, Fos, and ATF subfamilies
that bind to a common DNA site, the AP-1 binding site (30). MAPKs are upstream activators of AP-1
(31). ERK, p38, and JNK,
subfamilies of the MAPK pathway, induce Fos and Jun production by
activating different transcription factors such as Elk-1 and ATF.
It has been demonstrated that AP-1 plays a central role in
tumorigenesis (32). ERK1/2 pathway
is responsible for the phosphorylation and activation of AP-1
protein. Fos protein differ significantly in both of their DNA
binding sites and trans-activation potential as well as their
target gene regulation. Overexpression of c-fos can efficiently
transform cells and lead to tumor formation. Proliferating cell
nuclear antigen (PCNA) is a 36-kDa nuclear marker of cell
proliferation since its expression and distribution are correlated
with the rate of cell proliferation and DNA synthesis in various
tumors (33). PCNA is an auxiliary
protein of DNA polymerase-δ that functions during the cell cycle
(34). The distribution of PCNA
increases during G1 phase, peaks at the G1S
interphase and decreases during G2 phase (35). The PCNA gene contains AP-1 sites in
promoter region and its expression is regulated by AP-1 activity
(36). In this study, we have shown
significant upregulation of c-fos and PCNA in response to trypsin
or PAR-2 AP, and the effect was strongly decreased by PD98059. The
results suggest that PAR-2 agonists may elevate the rate of cell
proliferation and DNA synthesis in hepatoma cells by ERK1/2-AP-1
pathway. Additional studies are needed to explore other
transcription factors and their target genes in the progression of
hepatoma induced by activation of PAR-2. Previous studies have
demonstrated that the activation of PAR-2 stimulated proliferation
of ESC through p38MAPK, p42/44MAPK, and
SAPK/JNK pathways (37). It has
also been demonstrated that nuclear factor kappa B (NF-κB)
signaling mediated by PAR-2 is regulated by intracellular
Ca2+ in skin epithelial cell line NCTC2544, independent
of ERK and p38MAPK pathways (38). NF-κB transcription factors play a
key role in many physiological processes such as cell
proliferation, cell death, and inflammation (39). Thus, we speculate that the
proliferation of HepG2 induced by PAR-2 agonists may also be
related to other pathways, such as the p38MAPK,
SAPK/JNK, Ca2+/NF-κB pathways.
In conclusion, our data demonstrated that PAR-2
played an important role in proliferation of hepatoma cells, and
PAR-2 activation promoted the proliferation of hepatoma cells
partially via the ERK/AP-1 pathway. Further studies are needed to
clarify other mechanisms of PAR-2-induced signaling pathways
leading to hepatoma cell proliferation.
Acknowledgements
We thank instructor Zhong-wei Xu and Xia Mai
(Department of Cell Biology, Logistics College of the Chinese
People’s Armed Police Forces) for assistance with western blotting
technique; Dr Yu-xian Yan and instructor Jing-tian Han (Centralab,
Logistics College of the Chinese People’s Armed Police Forces) for
help with immunocytochemical and immunofluorescence analysis; and
Dr Xue-jun Cui (Department of English, Medical College of the
Chinese People’s Armed Police Forces) for careful revision of the
manuscript.
References
|
1
|
Bosch FX, Ribes J, Díaz M and Cléries R:
Primary liver cancer: worldwide incidence and trends.
Gastroenterology. 127:S5–S16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bruix J, Boix L, Sala M and Llovet J:
Focus on hepatocellular carcinoma. Cancer Cell. 5:215–219. 2004.
View Article : Google Scholar
|
|
3
|
Hollenberg MD and Compton SJ:
International Union of Pharmacology. XXVIII: protease-activated
receptors. Pharmacol Rev. 54:203–219. 2002. View Article : Google Scholar
|
|
4
|
Uusitalo-Jarvinen H, Kurokawa T, Mueller
BM, Andrade-Gordon P, Friedlander M and Ruf W: Role of protease
activated receptor 1 and 2 signaling in hypoxia induced
angiogenesis. arterioscler Thromb Vasc Biol. 27:1456–1462. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kirkland JG, Cottrell GS, Bunnett NW and
Corvera CU: Agonists of protease-activated receptors 1 and 2
stimulate electrolyte secretion from mouse gallbladder. Am J
Physiol Gastrointest Liver Physiol. 293:335–346. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hirahara F, Miyagi Y, Miyagi E, et al:
Trypsinogen expression in human ovarian carcinomas. Int J Cancer.
63:176–181. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kawano N, Osawa H, Ito T, et al:
Expression of gelatinase A, tissue inhibitor of
metalloproteinases-2, matrilysin, and trypsin (ogen) in lung
neoplasms: an immunohistochemical study. Hum Pathol. 28:613–622.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miyata S, Miyagi Y, Koshikawa N, et al:
Stimulation of cellular growth and adhesion to fibronectin and
vitronectin in culture and tumorigenicity in nude mice by
overexpression of trypsinogen in human gastric cancer cells. Clin
Exp Metastasis. 16:613–622. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Williams SJ, Gotley DC and Antalis TM:
Human trypsinogen in colorectal cancer. Int J Cancer. 93:67–73.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miyata S, Koshikawa N, Higashi S, et al:
Expression of trypsin in human cancer cell lines and cancer tissues
and its tight binding tosoluble form of Alzheimer amyloidprecursor
protein in culture. J Biochem. 125:1067–1076. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bernard-Perrone F, Carrere J, Renaud W, et
al: Pancreatic trypsinogen I expression during cell growth and
differentiation of two human colon carcinoma cells. Am J Physiol.
274:G1077–G1086. 1998.PubMed/NCBI
|
|
12
|
Darmoul D, Marie JC, Devaud H, Gratio V
and Laburthe M: Initiation of human colon cancer cell proliferation
by trypsin acting at protease-activated receptor-2. Br J Cancer.
85:772–779. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hansen KK, Oikonomopoulou K, Li Y and
Hollenberg MD: Proteinases, proteinase-activated receptors (PARs)
and the pathophysiology of cancer and diseases of the
cardiovascular, musculoskeletal, nervous and gastrointestinal
systems. Naunyn Schmiedebergs Arch Pharmacol. 377:377–392. 2008.
View Article : Google Scholar
|
|
14
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Roles of the Raf/MEK/ERK pathway in cell growth, malignant
transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
van Kempen LC, de Visser KE and Coussens
LM: Inflammation, proteases and cancer. Eur J Cancer. 42:728–734.
2006.PubMed/NCBI
|
|
16
|
Sánchez-Hernández PE, Ramirez-Dueñas MG,
Albarran-Somoza B, García-Iglesias T, del Toro-Arreola A,
Franco-Topete R and Daneri-Navarro A: Protease-activated receptor-2
(PAR-2) in cervical cancer proliferation. Gynecol Oncol. 108:19–26.
2008.PubMed/NCBI
|
|
17
|
Nyberg P, Ylipalosaari M, Sorsa T and Salo
T: Trypsins and their role in carcinoma growth. Exp Cell Res.
312:1219–1228. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kennedy AR, Billings PC, Wan XS and
Newberne PM: Effects of Bowman-Birk Inhibitor on Rat Colon
Carcinogenesis. Nutr Cancer. 43:174–186. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vergnolle N: Proteinase-activated
receptors - novel signals for gastrointestinal pathophysiology.
Aliment Pharmacol Ther. 14:257–266. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
O’Brien PJ, Molino M, Kahn M and Brass LF:
Protease activated receptors: theme and variations. Oncogene.
20:1570–1581. 2001.PubMed/NCBI
|
|
21
|
Masaki T, Shiratori Y, Rengifo W, et al:
Cyclins and cyclin-dependent kinases: comparative study of
hepatocellular carcinoma versus cirrhosis. Hepatology. 37:534–543.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Uto H, Ido A, Moriuchi A, et al:
Transduction of antisense cyclin D1using two-step gene transfer
inhibits the growth of rat hepatoma cells. Cancer Res.
61:4779–4783. 2001.PubMed/NCBI
|
|
23
|
Koshikawa N, Hasegawa S, Nagashima Y, et
al: Expression of trypsin by epithelial cells of various tissues,
leukocytes, and neurons in human and mouse. Am J Pathol.
153:937–944. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koshikawa N, Nagashima Y, Miyagi Y,
Mizushima H, Yanoma S, Yasumitsu H and Miyazaki K: Expression of
trypsin in vascular endothelial cells. FEBS Lett. 409:442–448.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miyata S, Koshikawa N, Yasumitsu H and
Miyazaki KJ: Trypsin stimulates integrin α(5)β(1)-dependent
adhesion to fibronectin and proliferation of human gastric
carcinoma cells through activation of proteinaseactivated
receptor-2. J Biol Chem. 275:4592–4598. 2000.
|
|
26
|
Hendstrom J, Haglund C, Haapiainen C and
Stenman UH: Serum trypsinogen-2 and trypsin-2-α1-antitrypsin
complex in malignant and benign digestive-tract disease.
Preferential elevation in patients with cholangiocarcinoma. Int J
Cancer. 66:326–331. 1996.
|
|
27
|
Sabri A, Guo J, Elouardighi H, Darrow AL,
Andrade-Gordon P and Steinberg SF: Mechanisms of protease-activated
receptor-4 actions in cardiomyocytes. Role of Src tyrosine kinase.
J Biol Chem. 278:11714–11720. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Caruso R, Pallone F, Fina D, et al:
Protease-activated receptor-2 activation in gastric cancer cells
promotes epidermal growth factor receptor trans-activation and
proliferation. Am J Pathol. 169:268–278. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ossovskaya VS and Bunnett NW:
Protease-activated receptors: contribution to physiology and
disease. Physiol Rev. 84:579–621. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Whitmarsh AJ and Davis RJ: Transcription
factor AP-1 regulation by mitogen-activated protein kinase signal
transduction pathway. J Mol Med. 74:589–607. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Karin M, Liu Z and Zandi E: AP-1 function
and regulation. Curr Opin Biol. 9:240–246. 1997. View Article : Google Scholar
|
|
32
|
Sun Y and Oberley LW: Redox regulation of
transcriptional activators. Free Radic Biol Med. 21:335–348. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu CC and Filipe MI: Update on
proliferation-associated antibodies applicable to formalin-fixed
paraffinembedded tissue and their clinical apprications. Histochem
J. 25:843–853. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Williams GJ, Johnson K, Rudolf J, et al:
Structure of the heterotrimeric PCNA from Sulfolobus
solfataricus. Acta Crystal1ograph Sect F Struct Biol Cryst
Commun. 62:944–948. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sourisseau T, Georgiadis A, Tsapara S, Ali
R, Pestell R, Matter K and Balda MS: Regulation of PCNA and cyclin
D1 expression and epithelial morphogenesis by the ZO-1-regulated
transcription factor ZONAB/DbpA. Mol Cell Biol. 26:2387–2398. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gillardon F, Moll I and Uhlmann E:
Inhibition of c-Fos expression in the UV-irradiated epidermis by
topical application of antisense oligodeoxynucleotides suppresses
activation of proliferating cell nuclear antigen. Carcinogenesis.
16:1853–1856. 1995. View Article : Google Scholar
|
|
37
|
Hirota Y, Osuga Y, Hirata T, et al:
Activation of protease-activated receptor 2 stimulates
proliferation and interleukin (IL)-6 and IL-8 secretion of
endometriotic stromal cells. Hum Reprod. 20:3547–3553. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Macfarlane SR, Sloss CM, Cameron P, Kanke
T, McKenzie RC and Plevin R: The role of intracellular
Ca2+ in the regulation of proteinase-activated
receptor-2 mediated nuclear factor kappa B signalling in
keratinocytes. Br J Pharmacol. 45:535–544. 2005.
|
|
39
|
Baud V and Karin M: Is NF-kappaB a good
target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov.
8:33–40. 2009. View Article : Google Scholar : PubMed/NCBI
|