Introduction
Cancer affects a significant proportion of the
population worldwide. Although numerous immune effector cells and
molecules are involved in the recognition and destruction of cancer
cells by a process known as cancer immunosurveillance, the cancer
cells can escape such monitoring through the production of poorly
immunogenic tumour cell variants and the subversion of the immune
system. Therefore, a method that can induce the immunogenic
transformation of tumour cells and restore the body’s immune
response against tumour cells could represent an effective pathway
for tumour prevention and therapy (1).
Whole-cell tumour vaccines have theoretical
advantages over epitope-specific vaccines in that the former can
present multiple and unknown tumour antigens to the immune system.
However, since most types of tumour cells are poorly immunogenic in
practise (2), improving the
immunogenicity of the tumour vaccine is important for
tumour-vaccine mediated immunotherapy.
Calreticulin (CRT) is a 46-kDa
Ca2+-binding protein that is mainly located in the
endoplasmic reticulum (ER). Previous studies have indicated that
CRT is a multifunctional protein involved in a wide variety of
cellular processes, including cell adhesion (3), the folding of newly synthesised
glycoproteins (4–6), gene expression (4), Ca2+ homeostasis (7) and lectin-like chaperone activity
(6). In addition to the ER, CRT has
been found to be localised in various subcellular compartments,
such as the cytosol, nucleus and cell surface membrane.
The CRT on the cell surface has multiple functions,
including the modulation of cell adhesion and migration (8–11), and
recent studies have found that the CRT on the cell surface also
plays an important role in mediating the phagocytosis of apoptotic
tumour cells by professional and non-professional phagocytes.
During the process of tumour cell apoptosis, CRT is rapidly
translocated from the ER to the cell surface and serves as an ‘eat
me’ signal that can be recognised by the phagocytes within hours
after the initiation of immunogenic cell death; at the same time,
the tumour cells undergoing apoptosis down-regulate the expression
of ‘do not eat me’ signals such as surface CD47 to facilitate the
recognition and engulfment of the tumour cells by phagocytes and
then to induce antitumour immune response (12). It has been reported that tumour
cells killed by UVC, γ irradiation (13) and some chemotherapeutics (14–17)
could induce this type of tumour-specific immune response.
Since the CRT translocation from the ER to the cell
surface is not an inherent feature of apoptosis in tumour cells but
is only an event induced by specific stimuli in some tumour cell
lines, a method that can easily coat CRT onto the surface of any
tumour cell would aid in providing a potent research tool for
antitumour immunotherapy. In our previous studies, we used a B16-F1
mouse melanoma cell line coated with mCRT-vGPCR as a whole-cell
tumour vaccine to immunise experimental animals and found that this
whole-cell vaccine could induce a strong antitumour effect against
the homologous tumour (18). In
this study, we further evaluated the immune responses induced by
this mCRT-vGPCR-coated whole-cell vaccine both in vivo and
in vitro.
Materials and methods
Mice and cell line
Female C57BL mice 5–6 weeks of age were purchased
from the animal centre of Wuhan University. The animals were housed
under specific pathogen-free conditions and the experiments were
conducted in accordance with the ethical guidelines for the care
and use of laboratory animals at Wuhan University. The mouse
melanoma cell line B16-F1 was purchased from the Cell Bank of China
(Wuhan). The cells were maintained at 37°C in 5% CO2 and
RPMI-1640 medium, which contains 10% heat-inactivated foetal bovine
serum, 100 μg/ml streptomycin and 100 U/ml penicillin. The B16-F1
cell line with the mCRT-vGPCR high expression was constructed as
previously reported (18).
Materials
Lipofectamine 2000™ transfection reagent and TRIzol
reagent were obtained from Invitrogen (San Diego, CA, USA). Rabbit
anti-human CRT polyclonal antibody was purchased from Stressgen
(Victoria, BC, Canada). Rhodamine-labeled goat anti-rabbit IgG
antibodies, mouse IFN-γ ELISPOT kit and anti-mouse CD11c PE were
purchase from eBioscience (San Diego, CA, USA). Recombinant murine
IL-4 and GM-CSF were purchased from Peprotech (Rocky Hill, NJ,
USA). CytoTox 96® Non-Radioactive Cytotoxicity assay was
purchased from Promega (Madison, WI, USA). The RPMI-1640 cell
culture medium and the other chemicals were from Sigma (St. Louis,
MO, USA). BENS (Bis-ethyl-norspermine) was kindly provided by
Professor Robert A. Casero at Johns Hopkins University. All primer
synthesis and DNA sequencing were performed by Sangon Biologic
Engineering Technology & Services Co. (Shanghai, China).
Detection of mCRT-vGPCR mRNA by
RT-PCR
When the mCRT-vGPCR-containing or empty plasmids
were transfected into the B16-F1 cells, the total RNA was extracted
from the stably transfected cells using the TRIzol reagent and then
used to amplify mCRT-vGPCR or vGPCR mRNA by RT-PCR. The following
were the PCR primers used in this experiment: for mCRT-vGPCR,
upstream 5′-GATGGATGGAGAGTGGGAACC-3′; downstream
5′-TCGATCTAGACTACCGCGATCGGTGCTTGCAAAA-3′. For vGPCR, upstream
5′-ATATCTCGAGATGGCGGCCGAGGATTTCCTAACC-3′; downstream
5′-TCGATCTAGACTACCGCGATCGGTGCTTGCAAAA-3′. For GAPDH (the internal
control), upstream 5′-CAAGGTCATCCATGACAACTTTG-3′; downstream
5′-GTCCACCACCCTGTTGCTGTAG-3′. The two primers for mCRT-vGPCR were
located within the CRT and vGPCR portions, respectively. The length
of the PCR product was 728 bp for mCRT-vGPCR, 241 bp for vGPCR and
496 bp for GAPDH. The PCR products were separated on a 1.5% (w/v)
agarose gel, and the DNA was visualised by ethidium bromide
staining.
Detection of mCRT-vGPCR fusion protein by
western blotting
The stably transfected cell lines were washed with
PBS and lysed using Cymal-5 solution. The proteins in the lysate
were separated by 10% SDS-PAGE and transferred onto a PVDF
membrane. The membrane was blocked with fat-free milk solution (5%,
w/v) overnight and then incubated with the rabbit anti-CRT
polyclonal antibody (1:1000) at 4°C for 12 h. After washing 3
times, the membrane was incubated with an HRP-conjugated
anti-rabbit IgG antibody (1:4000) at room temperature for 1 h and
developed using ECL.
Detection of mCRT-vGPCR expression by
cell immunofluorescence (CIF)
The stably transfected cells were seeded in 24-well
plates and cultured for 12 h. After washing with PBS, the cells
were fixed with 2% (m/v) paraformaldehyde (PFA) and incubated with
10% (v/v) goat serum to block non-specific interactions. The cells
were then incubated with a rabbit anti-CRT polyclonal antibody
(1:200) at 4°C overnight. After washing 3 times with PBS, the cells
were incubated with a rhodamine-conjugated goat anti-rabbit IgG
polyclonal antibody (1:500) at room temperature for 1 h (in the
dark). After washing 3 times with PBS, the cells were incubated
with 300 nM 4,6-diamidino-2-phenylindole (DAPI) for 10 min at room
temperature (in the dark) to stain the nuclei. The samples were
analysed using fluorescence microscopy (Nikon TE2000, Japan) with
NIS-Elements BR 3.1 software (Nikon, Japan) to identify the
mCRT-vGPCR fusion protein.
In vitro phagocytosis assay
Bone marrow (BM) cells were collected from the
tibias and femurs of the C57BL mice using culture medium. Following
centrifugation, the BM cells were resuspended in red-cell lysis
solution (0.15 M NH4Cl, 0.01 M KHCO3, and 1
mM EDTA) for 1 min to remove the red blood cells. The BM cells were
collected by centrifugation and cultured in a medium supplemented
with 10 ng/ml recombinant mouse GM-CSF and 5 ng/ml recombinant
mouse IL-4 in 6-well plates (1×106 cells/well). After 7
days, the non-adherent and loosely adherent cells were harvested as
the dendritic cells (DCs) and used as the effector cells for the
phagocytosis assay. The stably transfected B16-F1 cells incubated
with BENS for 48 h were labelled with the green dye CFDA-SE (1 μM)
for 20 min and used as the target cells. The effector and target
cells were co-cultured at 37°C for 2 h at a 1:1 E/T
(effector/target) ratio. After 3 washes, anti-mouse CD11c PE was
added to the cell mixture for 30 min at room temperature (in the
dark) to label the effector cells. The cells were washed with PBS
and analysed by flow cytometry (FCM). The phagocytotic efficiency
was represented by the cell ratio of the double-positive cell
number over total cell number.
Whole-cell vaccine immunisation
The 7- to 8-week-old female C57BL mice were randomly
divided into four groups: i) B16-mCRT-vGPCR (n=10); ii) B16-vGPCR
(n=10); iii) B16 (n=10), and iv) PBS (n=10). In groups 1–3, the
B16-F1 cells were treated by 10 μM BENS for 48 h to induce cell
apoptosis and then used as the whole-cell vaccine, subcutaneously
inoculated into the back of each mouse. Instead of B16-F1 cells,
PBS was used to immune the mice in group 4. The whole-cell vaccine
or PBS was injected 3 times at Days 0, 10 and 20, and the mice were
sacrificed 10 days after the last injection. The spleens from the
vaccinated mice were collected for analysis.
Determination of specific cytotoxic T
lymphocyte (CTL) activity
The splenocytes from the immunised mice were
suspended in complete RPMI-1640 medium with 10% fetal calf serum
and used as the effector cells to assay their specific cytotoxic T
lymphocyte (CTL) activity. B16-F1 cells were used as the target
cells in this experiment. The effector cells
(5×104–2×105 cells/well) were stimulated with
IL-2 before use and incubated with the target cells
(1×104 cells/well) at E:T ratios of 20:1, 10:1, and 5:1
in a total volume of 200 μl RPMI-1640 medium. The released lactate
dehydrogenase (LDH) was measured according to the manufacturer’s
instructions after 4 h of incubation at 37°C in 5% CO2.
The percentage of specific killing was calculated as follows:
specific killing % = (experimental release − spontaneous
release)/(total release − spontaneous release).
Enzyme-linked immunospot (ELISPOT)
assay
The number of splenocytes that could produce
interferon-γ (IFN-γ) was quantified by the cytokine-specific
enzyme-linked immunospot (ELISPOT) assay. Plates (96 wells) were
coated with 100 μg of anti-mouse IFN-γ McAb overnight at 4°C,
washed with PBS and blocked for 1 h with 5% BSA. The splenocytes
were seeded into each well (2×105 cells/well) and
cultured for 24 h in RPMI-1640 alone (negative control) or with the
apoptotic B16-F1 cells induced by BENS or 10 μg/ml concanavalin A
(positive control). After washing with PBS, followed by PBS-0.05%
Tween-20, the biotinylated rabbit anti-IFN-γ antibody was added and
incubated for 1 h at room temperature. The plates were washed in
PBS-0.05% Tween-20, and then avidin-HRP solution was added and
incubated for 45 min at room temperature. After rewashing, the
cells were treated with 100 μl of freshly prepared AEC substrate
solution and incubated at room temperature in the dark to monitor
the development of spots. The reaction was stopped by washing 3
times with 200 μl/well distilled water. The plate was air-dried,
and the spots were counted using an automated ELISPOT plate
reader.
Statistical analysis
Student’s t-tests were used for the comparison of
the results between the different groups. The tests were performed
using SPSS software. P<0.05 was considered to indicate
statistically significant differences.
Results
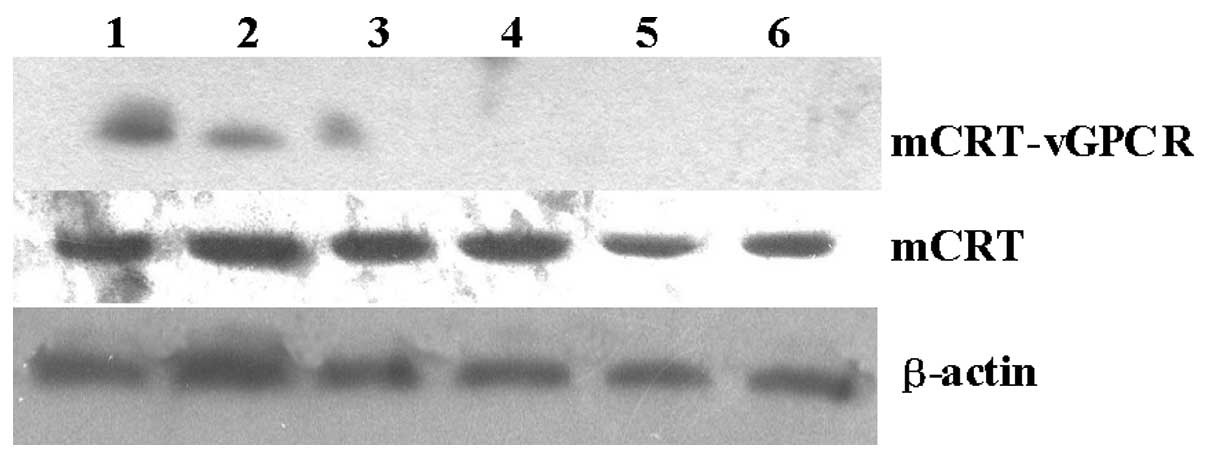
Expression of mCRT-vGPCR in B16-F1
cells
When the mCRT-vGPCR-containing or empty plasmids
were transfected into the B16-F1 cells, a high level of mCRT-vGPCR
mRNA was observed only in the mCRT-vGPCR-transfected cell line by
RT-PCR (Fig. 1). As the two primers
used for mCRT-vGPCR in the PCR reactions were located in the mCRT
and vGPCR regions, respectively, only the mRNA from the fusion gene
could be used as the template for the PCR. Using western blot
analysis with an anti-CRT antibody, the mCRT-vGPCR fusion protein
was also detected only in the mCRT-vGPCR- transfected cells. As
shown in Fig. 2, the mCRT-vGPCR
fusion protein has a higher molecular weight (110 kDa) than the
native CRT protein (55 kDa).
A CIF analysis was used to further determine whether
the mCRT-vGPCR fusion protein could be localised to the cell
surface. The two B16-F1 cell lines positively transfected with
pcDNA3.1(+)-mCRT-vGPCR showed significant membrane expression of
mCRT-vGPCR (Fig. 3), indicating
that the mCRT-vGPCR fusion protein was efficiently expressed on the
cell surface of the B16-F1 tumour cells.
mCRT-vGPCR on the cell surface enhances
the phagocytosis of B16-F1 cells by DCs in vitro
In view of the established role of CRT as an ‘eat
me’ signal, we further investigated the possible effect of
mCRT-vGPCR on the phagocytosis of BENS-treated B16-mCRT-vGPCR cells
by DCs, which are among the most efficient antigen-presenting
cells. The labelled-DCs from C57BL mice (the effector cells) and
transfected B16-F1 cells (the target cells) were co-cultured for 2
h in a 1:1 effector/target ratio, and FCM was used for the
analysis. The results showed that, compared to the B16-F1 cells
transfected with the empty vector, >5-fold higher phagocytotic
efficiency was observed when the B16-F1 cells transfected with
pcDNA3.1(+)-mCRT-vGPCR were used as the target cells, indicating
that the mCRT-vGPCR on the cell surface enhanced the phagocytosis
of the tumour cells by the DCs (Fig.
4).
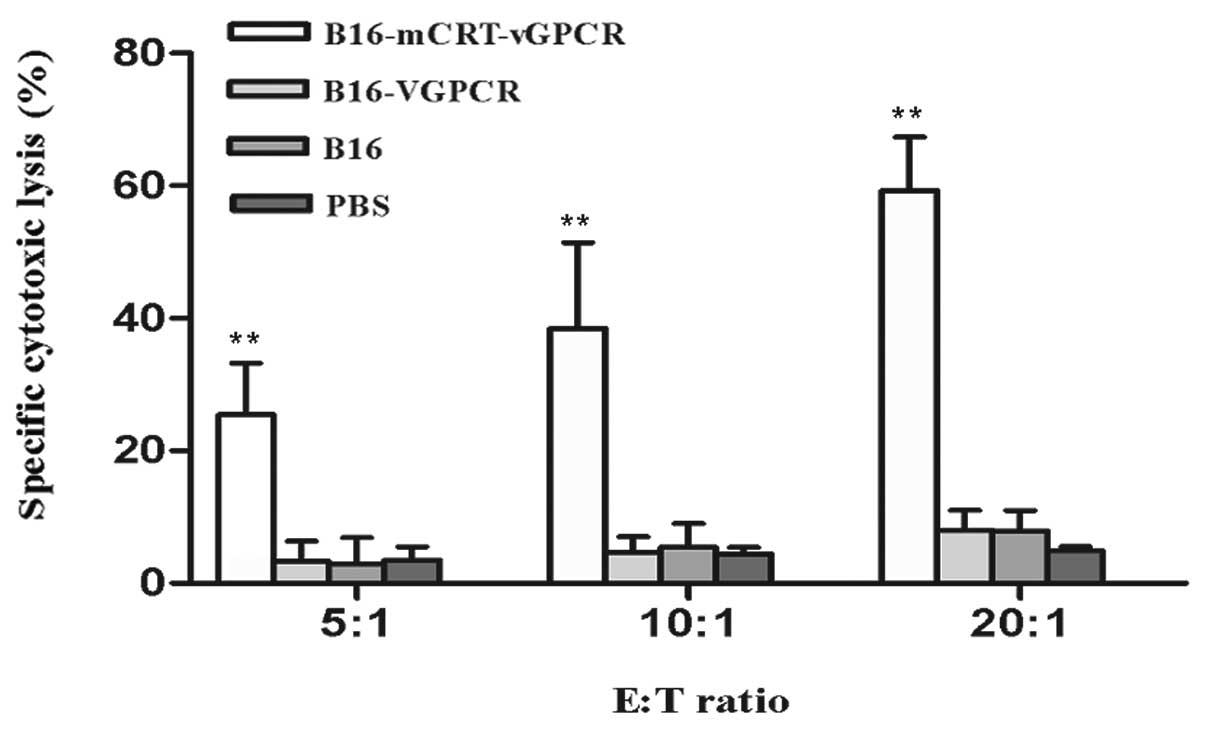
The mCRT-vGPCR-coated whole-cell vaccine
significantly induces specific CTL activity in mice
The splenocytes from the immunised mice were used as
the expanded effector (E) cells, and the B16-F1 cells were used as
the target (T) cells in this experiment. The effector cells were
stimulated by the BENS-treated B16-F1 cells in vitro before
use and then incubated with the target cells at E:T ratios of 20:1,
10:1, and 5:1 for 4 h. The specific CTL activities were determined
using the LDH assay. The results showed that the CTL activities
were enhanced in the mice immunised with the BENS-treated
B16-mCRT-vGPCR cells compared to the other groups (P<0.01)
(Fig. 5).
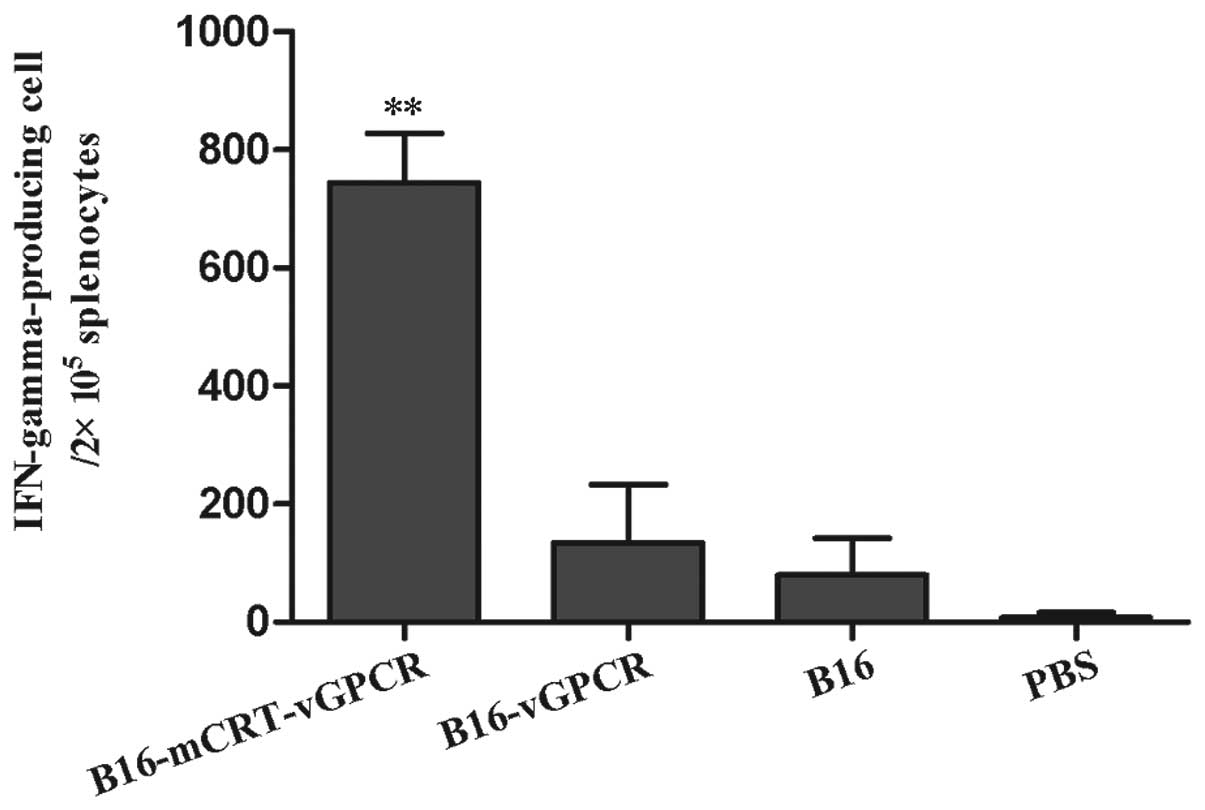
Cytokine secretion assay
To examine whether Th1 or Th2 responses occurred in
each immunised group, the IFN-γ-producing cells in the splenocytes
from the immunised-mice groups were examined using the ELISPOT
technique. The results showed that the immunisation with the
BENS-treated B16-mCRT-vGPCR cell vaccine elicited a significantly
higher number of IFN-γ-producing cells in the splenocytes (Fig. 6). Since IFN-γ is an indicator of the
Th1-based response, our data suggested that the mCRT-vGPCR- coated
cell vaccine mainly induced Th1-based immune responses.
Discussion
Whole-cell tumour vaccines have been investigated
for more than 20 years in both preclinical models and clinical
trials in humans (19–21). The advantage of whole-cell
vaccinations over other types of immunotherapy that target specific
antigens is that multiple and unknown tumour antigens may be
targeted by both the innate and adaptive immune systems. However,
the whole-cell vaccines have not resulted in significant long-term
therapeutic benefits (22,23). One possible explanation is that only
a small proportion of the molecules expressed on the cell surface
are specific for cancer cells, whereas the vast majority of cell
surface components are derived from housekeeping genes,
carbohydrates, lipids and other molecules that are ubiquitously
expressed by normal cells (24). As
a result, the immune response induced by whole-tumour cell vaccine
is insufficient to eliminate the cancer cells in vivo.
Therefore, further improvements are necessary to increase the
immunogenicity of cancer cell-based vaccines.
Recent studies have identified a new CRT-mediated
mechanism for tumour cell immunogenic death (12,13,25).
CRT is an ER resident chaperone protein with multiple physiological
functions and can rapidly translocate to the cell surface of tumour
cells during the apoptosis induced by specific stimuli, such as
anthracycline treatment. When these apoptotic tumour cells with CRT
coated on the surface were used as the vaccines to immune mice, the
specific antitumour immune response was induced.
As the translocation of CRT from the ER to the cell
surface is not an inherent feature of apoptosis in tumour cells, a
method that can easily coat CRT onto the surface of tumour cells
would provide a potent research tool for antitumour immunotherapy.
In our previous studies, an mCRT-vGPCR recombinant gene was
constructed and stably transfected into mouse melanoma B16-F1
cells. vGPCR is a membrane protein that contains seven
transmembrane domains, with the N-terminus protruding
extracellularly (26). Therefore,
the mCRT-vGPCR fusion protein was efficiently expressed on the cell
surface via the ability of vGPCR to localise to the membrane. When
this B16-F1 cell line coated with mCRT-vGPCR as a whole-cell
vaccine to immunise the mice, we found that this whole-cell vaccine
could strongly inhibit the growth of homologous tumour. In this
study, we further evaluated the immune responses induced by this
mCRT-vGPCR-coated whole-cell vaccine both in vivo and in
vitro.
DCs play important roles in processing and
presenting antigens, thus the recognition and phagocytosis of a
tumour cell vaccine by DCs is a key event in whole-cell tumour
vaccine-mediated immunotherapy. In this study, we found that,
similar to the native CRT, the mCRT-vGPCR on the tumour cell
surface could also mediate the phagocytosis of the tumour cells by
DCs. Compared with the B16-F1 cells transfected with the empty
vector, more than 5-fold higher phagocytotic efficiency was
observed when the B16-F1 cells transfected with pcDNA3.1(+)-
mCRT-vGPCR were used as the target cells.
Subsequently, these mCRT-vGPCR-coated B16-F1 cells
were treated by polyamine analogue BENS to induce cell apoptosis
and then used as the whole-cell vaccine to immunise C57BL mice. We
chose BENS as an apoptosis-inducing agent as it has no effect on
the subcellular localisation of CRT (27). The results indicated that the
mCRT-vGPCR-coated whole-cell vaccine could significantly induce
specific CTL activity and IFN-γ secretion of the splenocytes, both
of which are important indicators of the specific immune responses
stimulated by the vaccine in the model animals.
In conclusion, the mCRT-vGPCR-coated whole-cell
vaccine can induce specific antitumour immunity and these results
may provide an experimental basis for the development of new tumour
vaccines.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (no. 30973445), the
Fundamental Research Funds for the Central Universities, the
Research Foundation of Outstanding Young Innovative Team Projects
of Hubei Provence (T201203).
References
|
1
|
Zitvogel L, Tesniere A and Kroemer G:
Cancer despite immunosurveillance: immunoselection and
immunosubversion. Nat Rev Immunol. 6:715–727. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang X, Ye D and Thorpe PE: Enhancing the
potency of a whole-cell breast cancer vaccine in mice with an
antibody-IL-2 immunocytokine that targets exposed
phosphatidylserine. Vaccine. 29:4785–4793. 2011. View Article : Google Scholar
|
|
3
|
Johnson S, Michalak M, Opas M and Eggleton
P: The ins and outs of calreticulin: from the ER lumen to the
extracellular space. Trends Cell Biol. 11:122–129. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Michalak M, Corbett EF, Mesaeli N, et al:
Calreticulin: one protein, one gene, many functions. Biochem J.
344:281–292. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Trombetta ES and Parodi AJ: Quality
control and protein folding in the secretory pathway. Annu Rev Cell
Dev Biol. 19:649–676. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Trombetta ES: The contribution of
N-glycans and their processing in the endoplasmic reticulum
to glycoprotein biosynthesis. Glycobiology. 13:R77–R91. 2003.
|
|
7
|
Michalak M, Robert Parker JM and Opas M:
Ca2+ signaling and calcium binding chaperones of the
endoplasmic reticulum. Cell Calcium. 32:269–278. 2002.
|
|
8
|
Zhu Q, Zelinka P, White T and Tanzer ML:
Calreticulin-integrin bidirectional signaling complex. Biochem
Biophys Res Commun. 232:354–358. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kwon MS, Park CS, Choi K, et al:
Calreticulin couples calcium release and calcium influx in
integrin-mediated calcium signaling. Mol Biol Cell. 11:1433–1443.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gray AJ, Park PW, Broekelmann TJ, et al:
The mitogenic effects of the B-chain of fibrinogen are mediated
through cell surface calreticulin. J Biol Chem. 270:26602–26606.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
White TK, Zhu Q and Tanzer ML: Cell
surface calreticulin is a putative mannoside lectin which triggers
mouse melanoma cell spreading. J Biol Chem. 270:15926–15929. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Obeid M, Tesniere A, Ghiringhelli F, et
al: Calreticulin exposure dictates the immunogenicity of cancer
cell death. Nat Med. 13:54–61. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Obeid M, Panaretakis T, Joza N, et al:
Calreticulin exposure is required for the immunogenicity of gamma
irradiation and UVC light-induced apoptosis. Cell Death Differ.
14:1848–1850. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Spisek R and Dhodapkar MV: Towards a
better way to die with chemotherapy: role of heat shock protein
exposure on dying tumor cells. Cell Cycle. 6:1962–1965. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Spisek R, Charalambous A, Mazumder A,
Vesole DH, Jagannath S and Dhodapkar MV: Bortezomib enhances
dendritic cell (DC)-mediated induction of immunity to human myeloma
via exposure of cell surface heat shock protein 90 on dying tumor
cells: therapeutic implications. Blood. 109:4839–4845. 2007.
View Article : Google Scholar
|
|
16
|
Tesniere A, Panaretakis T, Kepp O, et al:
Molecular characteristics of immunogenic cancer cell death. Cell
Death Differ. 15:3–12. 2008. View Article : Google Scholar
|
|
17
|
Panaretakis T, Joza N, Modjtahedi N, et
al: The co-translocation of ERp57 and calreticulin determines the
immunogenicity of cell death. Cell Death Differ. 15:1499–1509.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qin Y, Han Y, Cao C, Ren Y, Li C and Wang
Y: Melanoma B16-F1 cells coated with fusion protein of mouse
calreticulin and virus G-protein coupled receptor induced the
antitumor immune response in Balb/C mice. Cancer Biol Ther.
11:574–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Copier J and Dalgleish A: Overview of
tumor cell-based vaccines. Int Rev Immunol. 25:297–319. 2006.
View Article : Google Scholar
|
|
20
|
de Gruijl TD, van den Eertwegh AJ, Pinedo
HM and Scheper RJ: Whole-cell cancer vaccination: from autologous
to allogeneic tumor- and dendritic cell-based vaccines. Cancer
Immunol Immunother. 57:1569–1577. 2008.PubMed/NCBI
|
|
21
|
Chiang CL, Benencia F and Coukos G: Whole
tumor antigen vaccines. Semin Immunol. 22:132–143. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Copier J and Dalgleish A: Whole-cell
vaccines: a failure or a success waiting to happen? Curr Opin Mol
Ther. 12:14–20. 2010.PubMed/NCBI
|
|
23
|
Bodey B, Bodey B Jr, Siegel SE and Kaiser
HE: Failure of cancer vaccines: the significant limitations of this
approach to immunotherapy. Anticancer Res. 20:2665–2676.
2000.PubMed/NCBI
|
|
24
|
Cohen EP, Chopra A, O-Sullivan I and Kim
TS: Enhancing cellular cancer vaccines. Immunotherapy. 1:495–504.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fucikova J, Kralikova P, Fialova A, et al:
Human tumor cells killed by anthracyclines induce a tumor-specific
immune response. Cancer Res. 71:4821–4833. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu C, Sandford G, Fei G and Nicholas J: G
protein selectivity determinant specified by a viral chemokine
receptor-conserved region in the C tail of the human herpes virus 8
G protein-coupled receptor. J Virol. 78:2460–2471. 2004. View Article : Google Scholar
|
|
27
|
Cao CY, Han Y, Ren YS and Wang YL:
Apoptotic B16-F1 cells coated with recombinant calreticulin
mediated anti-tumor immune response in mice. Chin J Cancer Res.
22:253–259. 2010. View Article : Google Scholar
|