Introduction
One of the fundamental changes that occurs in cancer
cells is the shift in energy metabolism from the generation of ATP
from oxidative phosphorylation to glycolysis even in the presence
of sufficient oxygen (Warburg effect) (1,2).
Several agents that specifically inhibit glycolytic metabolism,
such as 2-deoxy-D-glucose (2-DG), have been used as effective
anticancer agents in cellular systems and in animal models
(3,4). Similar to glucose, 2-DG is taken up
through glucose transporters (GLUTs) and is phosphorylated by
hexokinase (HK) to form 2-DG-6-phosphate (2-DG-6-P). 2-DG-6-P
accumulates within the cell and is not metabolized further. Then,
2-DG-6-P induces cell growth arrest and cell death by inhibiting 2
glycolytic enzymes, HK and phosphoglucose isomerase (PGI) (5,6).
Although 2-DG has been undergoing clinical trials
for treatment of several types of cancers, its efficacy as a
monotherapy is limited by systemic toxicity at high doses (7–9).
However, 2-DG can sensitize tumors to other chemotherapeutic agents
or radiotherapy (10,11).
Here, we examined the metabolic changes induced by
2-DG in leukemia cells by metabolome analysis, and aimed to
identify the critical metabolic pathway which can be targeted in
conjunction with glycolysis inhibition.
Materials and methods
Cell lines
Two acute myelogenous leukemia (AML) cell lines were
used in this study. NB4, a t(15;17) APL cell line, was provided by
Dr M. Lanotte (Saint Louis Hospital, Paris, France). THP-1, a
monocytic AML monocytic cell line was obained from the Cell
Resource Center for Biomedical Research (Tohoku University, Japan).
Cell lines were grown in RPMI-1640 medium containing 10% fetal calf
serum (FCS, Thermo Electron, Melbourne, Australia) in a humidified
atmosphere of 5% CO2 and 95% air at 37°C.
Metabolome analysis
The cell lines were cultured in RPMI-1640 containing
3% FCS with or without the glycolysis inhibitor, 2-DG (0.5 mM), for
24 h. Then, 5×106 cells were centrifuged and washed in
5% mannitol. After centrifugation and removal of mannitol, cells
were suspended in methanol. The samples were analyzed by capillary
electrophoresis time-of-flight mass spectrometry (CE-TOFMS)
(12).
Glutathione measurement
After a 24-h culture with or without 2-DG, cells
were collected and assayed for glutathione (GSH) content using a
GSSG/GSH quantification kit (Dojindo, Japan).
Cell cultures
Cell lines were cultured in RPMI-1640 containing 3%
FCS with the glycolysis inhibitor, 2-DG (0.5 mM), the inhibitor of
G6P dehydrogenase [first step of the pentose phosphate pathway
(PPP)], dehydroepiandrosterone (DHEA) (20 μM), AMPK inhibitor
compound C (1 μM), or in combination for 48 h. MTS
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium]
(Promega, Madison, WI, USA) was then added to each culture. After 3
additional hours of incubation, absorbance at 490 nm was measured
by an ELISA plate reader. Cell lines were also cultured in
RPMI-1640 containing 3% FCS with or without DHEA (20 μM) and the
GSH synthesis inhibitor, buthylsulfoximine (BSO) (20 μM) for 48 h.
Then, MTS assay was also carried out.
Quantitative real-time PCR
Total RNA was isolated using the RNeasy Mini kit
(Qiagen, Germany). Random hexamer priming and PrimeScript reverse
transcriptase (Takara, Japan) were used to generate cDNA. Real-time
reverse transcriptase polymerase chain reaction (RT-PCR) was
carried out using a StepOne Plus Real-Time PCR system (Applied
Biosystems, USA). Primers for PCR were as follows: glutathione
synthetase forward, 5′-CCCTGCCCGAGTGGTCCAGT-3′; reverse,
5′-CACTCCCGCTGCCACACCAC-3′ and 18S rRNA (as a control gene)
forward, 5′-CGGCGACGACCCATTCGAAC-3′; reverse,
5′-GAATCGAACCCTGATTCCCCGTC-3′. The relative gene expression level
was determined by comparison with 18S rRNA.
Statistical analysis
Statistical analysis was carried out by the t-test
to examine the difference in growth of the cell lines, GSH content
or glutathione synthetase mRNA expression.
Results
Metabolome analysis of the leukemia cell
lines
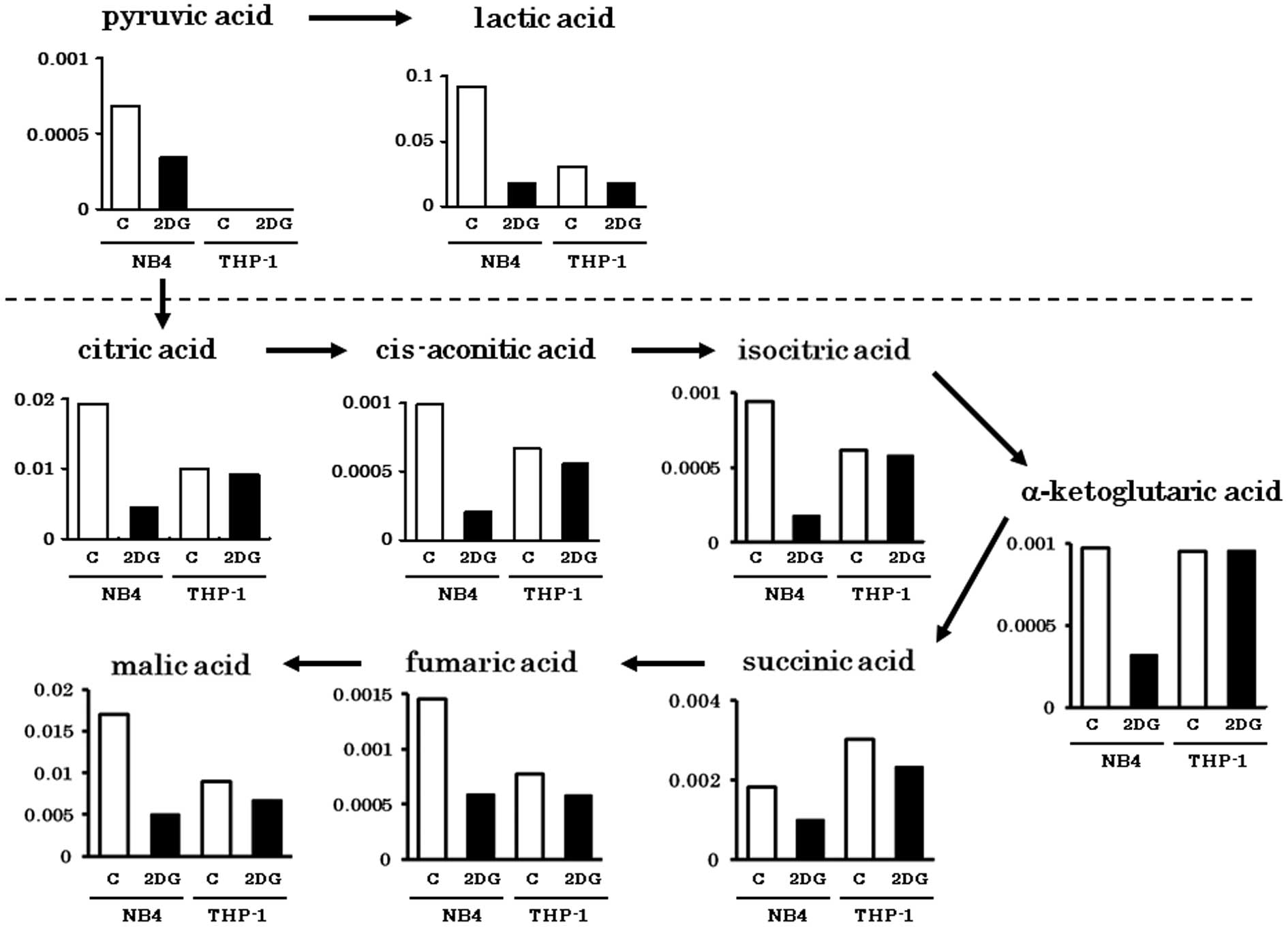
Metabolites in the glycolytic pathway (pyruvic acid
and lactic acid) were abundantly detected in the NB4 cells.
However, these were greatly decreased by 2-DG treatment (Fig. 1). Metabolites of the tricarboxylic
acid (TCA) cycle were comparably detected in both cell lines. The
amounts of TCA cycle metabolites in the THP-1 cells were not
obviously influenced by 2-DG as in the NB4 cells. This finding
indicates that TCA cycle metabolites in the THP-1 cells were
derived from non-glucose materials such as amino acids or fatty
acids. Metabolites of PPP (Ru5P, R5P, PRPP) were abundantly
detected in both NB4 and THP-1 cells, particularly when treated
with 2-DG (Fig. 2A). Carnitine and
its acetylated form, acetylcarnitine are important for
incorporation of fatty acids into mitochondria. Consistent with our
previous observation that THP-1 depends on fatty acid oxidation for
energy production (13), carnitine
and acetylcarnitine were abundantly detected in THP-1 cells even
with 2-DG treatment (Fig. 2B).
Synergistic effect of the inhibition of
glycolysis and PPP or AMPK
The addition of 2-DG (0.5 mM), DHEA (20 μM) or
compound C (1 μM) did not effectively inhibit the growth of both
cell lines. However, simultaneous addition of 2-DG and DHEA
synergistically inhibited the growth, particularly in the NB4 cells
(P=0.0011), while simultaneous addition of 2-DG and compound C
inhibited the growth of THP-1 cells more effectively than that of
NB4 cells (P=0.0319) (Fig. 3).
Activation of PPP by 2-DG treatment leads
to reduction in GSH
The first step of PPP is triggered by G6P
dehydrogenase, which produces NADPH. Since NADPH is utilized for
GSH reduction, the content of the reduced form of GSH in the cell
lines treated with or without 2-DG was determined. As shown in
Fig. 4, the reduced form of GSH was
upregulated following 2-DG treatment only in the NB4 cells
(P=0.0494).
BSO inhibits the synthesis of GSH
DHEA inhibits G6P dehydrogenase, resulting in
decreased NADPH and the reduced form of GSH. As shown in Fig. 5, the growth of NB4 cells was greatly
inhibited by the addition of BSO (P=0.033), DHEA (P=0.0254) or in
combination (P=0.0001) when compared with the effect in THP-1
cells.
Expression of glutathione synthetase is
upregulated by 2-DG in NB4 cells
Glutathione synthetase (GS) catalyzes the
condensation of γ-glutamylcysteine and glycine to form glutathione.
As shown in Fig. 6, real-time
quantitative RT-PCR study revealed that expression of GS was
upregulated following 2-DG treatment in NB4 cells (P=0.0007). This
may explain the finding that the reduced form of GSH was
upregulated by 2-DG in NB4 cells and that the growth of NB4 cells
was more effectively inhibited by BSO.
Discussion
Metabolome analysis revealed that NB4 cells mainly
utilized glucose as an energy source by glycolysis and oxidative
phosphorylation in mitochondria, as metabolites in the glycolytic
pathway and in the TCA cycle were significantly decreased by 2-DG,
a glycolysis inhibitor. In THP-1 cells, metabolites in the TCA
cycle were not decreased to the same extent by 2-DG as in the NB4
cells, which indicates that THP-1 cells utilized an energy source
other than glucose. TCA cycle metabolites in THP-1 cells may be
derived from acetyl-CoA by fatty acid β-oxidation, which was
supported by abundant detection of carnitine and acetylcarnitine in
the THP-1 cells (Fig. 2B). Our
previous observation (13) that
THP-1 depends on fatty acid oxidation for energy production also
corroborates of this finding. 2-DG treatment increased the PPP
metabolites in both cell lines. After entering the cell, 2-DG is
phosphorylated by hexokinase to form 2-DG-6-phosphate, which cannot
be further metabolized, and its accumulation leads to inhibition of
the glycolytic pathway and shunting through the PPP (5,14,15).
This PPP flux augments the generation of NADPH by
glucose-6-phosphate dehydrogenase (G-6-PDH). One of the uses of
NADPH is to prevent oxidative stress by reducing glutathione (from
GSSG to GSH). As shown in Fig. 4,
an increase in the reduced form of GSH following 2-DG treatment was
noted only in the NB4 cells. Upregulation of GS expression may
explain the increase in the reduced form of GSH by 2-DG in the NB4
cells. We demonstrated that the combination of 2-DG and inhibition
of PPP by DHEA effectively suppressed the growth of NB4 cells.
It has been reported that AMPK inhibits fatty acid
synthesis and activates fatty acid oxidation (16,17).
We previously demonstrated that 2-DG treatment activates AMPK in
THP-1 cells (13), which may
explain the replenishment of the TCA cycle by fatty acid oxidation
by carnitine palmitoyltransferase. Then, the combination of 2-DG
and inhibition of AMPK by compound C potently suppressed the growth
of THP-1 (Fig. 3).
Although 2-DG has been effective in preclinical and
clinical studies, this treatment has not been fully explored due to
concerns related to potential toxicities such as brain toxicity at
high doses (8,9). It is important to determine the
appropriate combination of metabolic inhibitors at low
concentrations which do not cause toxicities. Here, we demonstrated
that the combination of 2-DG and DHEA or compound C at a relatively
low concentration effectively inhibited the growth of NB4 and THP-1
cells, respectively (Fig. 7).
Further studies are warranted to ascertain the efficacy and safety
of these combinations.
Acknowledgements
We are grateful to Dr N. Kamada (Hiroshima
University, Japan) for Kasumi-1 and to Dr M. Lanotte (Saint Louis
Hospital, Paris, France) for the NB-4 cell line. We also thank Ms.
A. Usui and A. Nakamura for their technical and secretarial
assistance. This study was supported in part by the Ministry of
Education, Culture, Sports, Science and Technology, Japan
(MEXT)-Supported Program for the Strategic Research Foundation at
Private Universities, 2011–2015 (S1101027).
References
|
1
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gatenby RA and Gillies RJ: Why do cancers
have high aerobic glycolysis? Nat Rev Cancer. 4:891–899. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pelicano H, Martin DS, Xu RH and Huang P:
Glycolysis inhibition for anticancer treatment. Oncogene.
25:4633–4646. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
El Mjiyad N, Caro-Maldonado A,
Ramírez-Peinado S and Muñoz-Pinedo C: Sugar-free approaches to
cancer cell killing. Oncogene. 30:253–264. 2011.PubMed/NCBI
|
|
5
|
Sols A and Crane RK: Substrate specificity
of brain hexokinase. J Biol Chem. 210:581–595. 1954.PubMed/NCBI
|
|
6
|
Kurtoglu M, Gao N, Shang J, Maher JC,
Lehrman MA, Wangpaichitr M, Savaraj N, Lane AN and Lampidis TJ:
Under normoxia, 2-deoxy-D-glucose elicits cell death in select
tumor types not by inhibition of glycolysis but by interfering with
N-linked glycosylation. Mol Cancer Ther. 6:3049–3058. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tennant DA, Durán RV and Gottlieb E:
Targeting metabolic transformation for cancer therapy. Nat Rev
Cancer. 10:267–277. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Porporato PE, Dhup S, Dadhich RK, Copetti
T and Sonveaux P: Anticancer targets in the glycolytic metabolism
of tumors: a comprehensive review. Front Pharmacol. 2:492011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheong H, Lu C, Lindsten T and Thompson
CB: Therapeutic targets in cancer cell metabolism and autophagy.
Nat Biotechnol. 30:671–678. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maschek G, Savaraj N, Priebe W,
Braunschweiger P, Hamilton K, Tidmarsh GF, De Young LR and Lampidis
TJ: 2-Deoxy-D-glucose increases the efficacy of adriamycin and
paclitaxel in human osteosarcoma and non-small cell lung cancers in
vivo. Cancer Res. 64:31–34. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Singh D, Banerji AK, Dwarakanath BS,
Tripathi RP, Gupta JP, Mathew TL, Ravindranath T and Jain V:
Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose
escalation studies in patients with glioblastoma multiforme.
Strahlenther Onkol. 181:507–514. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ooga T, Sato H, Nagashima A, Sasaki K,
Tomita M, Soga T and Ohashi Y: Metabolomic anatomy of an animal
model revealing homeostatic imbalances in dyslipidaemia. Mol
Biosyst. 7:1217–1223. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Suganuma K, Miwa H, Imai N, Shikami M,
Gotou M, Goto M, Mizuno S, Takahashi M, Yamamoto H, Hiramatsu A,
Wakabayashi M, Watarai M, Hanamura I, Imamura A, Mihara H and Nitta
M: Energy metabolism of leukemia cells: glycolysis versus oxidative
phosphorylation. Leuk Lymphoma. 51:2112–2119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen W and Guéron M: The inhibition of
bovine heart hexokinase by 2-deoxy-D-glucose-6-phosphate:
characterization by 31P NMR and metabolic implications.
Biochimie. 74:867–873. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sandulache VC, Ow TJ, Pickering CR,
Frederick MJ, Zhou G, Fokt I, Davis-Malesevich M, Priebe W and
Myers JN: Glucose, not glutamine, is the dominant energy source
required for proliferation and survival of head and neck squamous
carcinoma cells. Cancer. 117:2926–2938. 2011.PubMed/NCBI
|
|
16
|
Hardie DG and Pan DA: Regulation of fatty
acid synthesis and oxidation by the AMP-activated protein kinase.
Biochem Soc Trans. 30:1064–1070. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jeon SM, Chandel NS and Hay N: AMPK
regulates NADPH homeostasis to promote tumour cell survival during
energy stress. Nature. 485:661–665. 2012. View Article : Google Scholar : PubMed/NCBI
|