Introduction
Breast cancer is a molecularly, biologically and
clinically heterogeneous disease. Previous microarray profiling
studies on breast cancer have identified subtypes that are
associated with different clinical outcomes (1,2). These
subtypes are classified as luminal A, luminal B, human epidermal
growth factor receptor 2 (HER2), basal-like and normal-like
subtypes. The identification of gene expression-based breast cancer
subtypes is considered a critical means of prognostication
(3) and the basal-like subtype is
an aggressive tumor that has a poor prognosis and no specific
targeted therapy. Since it is not always feasible to obtain gene
expression array information, a simple classification using a
combination of immunohistochemical markers has been proposed and
adopted in clinical practice (4,5).
However, there is still a need for well-defined biomarker panels
that allow breast cancer subtyping for clinical diagnostics and the
management of the disease.
Breast carcinogenesis is a multistep process
resulting from the accumulation of genetic alterations, as well as
epigenetic changes, such as promoter methylation and histone
modification (6,7). Promoter hypermethylation at specific
gene loci leading to gene silencing is a major mechanism of
epigenetic inactivation in cancer cells. A previous genome-wide
study on breast cancer has led to the identification of a number of
tumor suppressor genes that are inactivated by promoter
hypermethylation (8), and
epigenetic analyses have shown aberrant DNA methylation signatures
associated with the molecular subtypes of breast cancer (9,10).
However, limited information is available on the methylation status
of candidate genes associated with each molecular subtype.
The purpose of this study was to analyze the
methylation status of breast cancer-related genes according to the
molecular subtypes found in Korean women, and to investigate
whether the basal-like subtype displays distinct methylation
profiles compared with the other subtypes. We included 5 genes that
are involved in breast carcinogenesis and are commonly methylated
in breast cancer [cadherin 13 (H-cadherin; CDH13), secreted
frizzled-related protein 1 (SFRP1), fragile histidine triad
(FHIT), Syk and retinoblastoma protein-interacting
zinc-finger gene 1 (RIZ1)] and analyzed the methylation
status of these genes using a sensitive and quantitative
pyrosequencing assay.
Materials and methods
Patients and tumor characteristics
A total of 60 sporadic invasive ductal carcinoma
(IDC) tissue samples were obtained from the Daegu Catholic
University Hospital (Daegu, Korea). Each sample represented the 4
major molecular subtypes, encompassing the basal-like, HER2,
luminal A and luminal B subtypes. All specimens were reviewed by an
experienced pathologist. We subclassified the breast cancer samples
according to immunohistochemical findings for the estrogen receptor
(ER), progesterone receptor (PR), HER2 oncogene and Ki-67 labeling
index (11). The basal-like subtype
was defined as HER2-negative, ER- and PR-negative (triple-negative)
breast cancer. The HER2 subtype was defined as HER2-positive, ER-
and PR-negative. The luminal B subtype was defined as HER2-positive
and ER- and/or PR-positive breast cancer (HER2-positive). The
luminal A subtype was defined as ER- and/or PR-positive,
HER2-negative breast cancer with a low Ki-67 index. ER- and/or
PR-positive, HER2-negative breast cancer with a high Ki-67 status
was classified as the luminal B (HER2-negative) subtype. Ethics
approval for the study was obtained from the institutional review
board at Daegu Catholic University Hospital.
Construction of tissue microarrays
(TMAs)
Representative paraffin tumor blocks were selected
according to the primary evaluation of hematoxylin and eosin
(H&E)-stained slides before they were prepared for TMA
analysis. Two tumor tissue cores (1 mm in diameter) were taken from
each of the donor breast cancer tissue blocks using a manual punch
arrayer (Quick-Ray™; Uni-Tech Science, Seoul, Korea). The cores
were placed in a new recipient paraffin block that ultimately
contained 72–96 tissue cores. Each array block contained both tumor
and control tissue samples. Multiple sections (5-μm-thick) were cut
from the TMA blocks and then mounted onto microscope slides. The
TMA H&E-stained sections were reviewed under a light microscope
to confirm the presence of representative tumor areas.
Immuohistochemical staining and
interpretation
Immunohistochemical analysis was performed on
5-μm-thick TMA tissue sections using the Bond Polymer Intense
Detection system (Leica Microsystems, Mount Waverley, Victoria,
Australia) according to the manufacturer's instructions with minor
modifications. Briefly, the 5-μm-thick sections of formalin-fixed
and paraffin-embedded TMA tissues were deparaffinized with Bond
Dewax Solution (Leica Microsystems), and an antigen retrieval
procedure was performed using Bond ER Solution (Leica Microsystems)
for 30 min at 100°C. The endogenous peroxidase was quenched by a
5-min incubation with hydrogen peroxide. Sections were incubated
for 15 min at an ambient temperature with commercially available
primary monoclonal antibodies for ER (1:100, clone 6F11;
Novocastra), PR (1:100, clone 16; Novocastra), HER2 (1:250, A0485;
Dako), Ki-67 (1:200, MM1-L; Novocastra), Bcl-2 (1:4, clone 124;
Dako), p53 (1:200, BP53.12; Zymed) and epidermal growth factor
receptor (EGFR) (1:100, clone EGFR.25; Novocastra) using a
biotin-free polymeric horseradish peroxidase-linker antibody
conjugate system in a Bond-Max automatic slide stainer (Leica
Microsystems).
A cut-off value of 10% for the stained nuclei was
used to define ER and PR positivity. Cytoplasmic staining of any
intensity in >10% of the tumor cells was scored as positive for
Bcl-2. Membranous staining for HER-2 with strong complete staining
in 10% of the tumor cells was regarded as HER-2 overexpression. p53
staining was scored positive if >10% of the cells were stained
with a strong intensity. The Ki-67 labeling index was expressed as
a percentage and was graded as ‘high’ if the number of positive
cells was ≥14%.
DNA extraction and sodium bisulfate
treatment
For DNA extraction, 8 tissue sections
(5–10-μm-thick) were obtained from the paraffin-embedded primary
breast cancer tissues. Genomic DNA was isolated using the QIAamp
DNA FFPE Tissue kit (Qiagen, Hilden, Germany) following the
manufacturer's instructions. The purified DNA was quantified using
a ND-1000 spectrophotometer (NanoDrop Technologies, Inc.,
Wilmington, DE, USA). The quality of the DNA was verified by gel
electrophoresis. Sodium bisulfate modification of 200–500 ng
genomic DNA was performed using the EZ DNA Methylation-Gold kit
(Zymo Research, Orange, CA, USA) according to the manufacturer's
instructions.
Candidate selection
Over 100 individual candidate genes have been
reported to be commonly hypo- and hypermethylated in breast cancer.
We carried out literature searches on PubMed http://www.ncbi.nlm.nih.gov/pubmed for the keywords
(breast cancer, cancer and methylation) and searched serial
analysis of gene expression (SAGE) data (GeneCards, http://www.genecards.org/cgi-bin/cardsearch). We
selected 5 genes, cadherin 13 (H-cadherin; CDH13), secreted
frizzled-related protein 1 (SFRP1), FHIT, Syk
and RIZ1, and functional annotation of the candidate genes
was carried out using the functional annotation table function in
the DAVID database http://david.abcc.ncifcrf.gov (12).
Pyrosequencing
Methylation analysis was carried out using
pyrosequencing. Primers were designed using the PyroMark Assay
Design program version 2.0.1.15 (Qiagen) and the sequences are
presented in Table I. Polymerase
chain reaction (PCR) was carried out using bisulfate-treated DNA
under the following conditions: 95°C for 5 min; 45 cycles of 95°C
for 30 sec, 55°C for 30 sec and 72°C for 30 sec; and a final
extension of 5 min at 72°C. PCR was conducted using a PCR premix
(Enzynomics, Daejeon, Korea), and the quality and quantity of the
PCR product was confirmed by performing agarose gel (2%)
electrophoresis with loading 4 μl of 20 PCR products.
Pyrosequencing was performed using the Pyro Gold kit and PSQ 96MA
instrument (Qiagen) as instructed by the manufacturer. The
methylation index (MtI) of each gene in each sample was calculated
as the average value of mC/(mC
+ C) for all examined CpG sites in target regions. All
experiments included a negative control without a template.
 | Table IPrimer sequences used for PCR and
pyrosequencing. |
Table I
Primer sequences used for PCR and
pyrosequencing.
| Primer name | Primer sequence
(5′→3′) |
|---|
| CDH13_F |
TAAGGAAAATATGTTTAGTGTAGT |
| CDH13_R |
AAATTCTCCACTACATTTTATCC |
| CDH13_S |
GTGTAGTAGAGTGTATGAATGAAAA |
| SFRP1_F |
TTTTAGGAGGTTTTTGGAAGT |
| SFRP1_R |
ACTCTACCCCCTATTCTCC |
| SFRP1_S |
AGGTTTTTGGAAGTTTG |
| FHIT_F |
GGGAGGTAAGTTTAAGTGGAATATTG |
| FHIT_R |
CCACTAAACTCCCAAATAATAACCTAAC |
| FHIT_S |
GTAAGTTTAAGTGGAATATTGT |
| Syk_F |
TTAGTAGGGAGGGTTAGGG |
| Syk_R |
CTCATTTTAAACAACTTCCTTAAC |
| Syk_S |
ATATTGGGAGGAAGTG |
| RIZ1_F |
AGTAAGTTTTTTAAGGGTAGGATTAT |
| RIZ1_R |
CCCTAATACCCAAAAACAATAACCAA |
| RIZ1_S |
GTTTTTTAAGGGTAGGATTATTAT |
Statistical analysis
Statistical analyses were carried out using SPSS
version 15.0 software (SPSS Inc., Chicago, IL, USA). A one-sample
Kolmogorov-Smirnov test was used to evaluate the normal
distribution fit of continuous parameters. The clinicopathological
characteristics were compared across the 4 different breast cancer
subtypes using the χ2 test or Fisher's exact test for
categorical data, and ANOVA or the non-parametric Kruskal-Wallis
test for continuous data. A comparison of the mean methylation
frequencies across the subtypes was performed using ANOVA or the
Kruskal-Wallis test, and distributions of methylation levels across
the different subtypes were depicted for each gene using box plots.
Associations between methylation status and clinicopathological
characteristics were assessed using the Student's t-test or the
non-parametric Mann-Whitney U test for categorical variables, and
correlation between 2 continuous variables was assessed using
correlation analysis. All tests were two-sided and a P-value
<0.05 was considered to indicate a statistically significant
difference.
Clustering analyses were performed using GeneSpring
GX version 7.3 (Agilent Technologies Inc., Santa Clara, CA, USA) on
the basis of the mean methylation levels of genes and the CpG
sites. Hierarchical clustering was performed using Pearson's
correlation distance and average linkage.
Results
Clinicopathological characteristics
The patient characteristics are presented in
Table II. The average age of the
60 patients with invasive breast cancer was 51.77±13.22 years
(range, 26–90 years). A total of 15 cases were included for each
molecular subtype in the 60 breast cancer samples. TNM staging was
as follows: stage I, 29 patients (48.3%); stage II, 21 patients
(35.0%); stage III, 6 patients (10.0%); and stage IV, 4 patients
(6.7%).
| Table IIGeneral patient characteristics. |
Table II
General patient characteristics.
|
Characteristics | Value |
|---|
| Age (years), mean
(range) | 51.77±13.22
(26–90) |
| Menopausal status,
n (%) | |
|
Pre-menopausal | 27 (45.8) |
|
Post-menopausal | 32 (54.2) |
| Tumor size (cm),
mean (range) | 1.80±0.93
(0.10–4.50) |
| Histological grade,
n (%) | |
| I | 13 (21.7) |
| II | 11 (18.3) |
| III | 36 (60.0) |
| Nodal involvement,
n (%) | |
| Negative | 40 (69.0) |
| Positive | 18 (31.0) |
| Distant metastasis,
n (%) | |
| Negative | 58 (96.7) |
| Positive | 2 (3.3) |
| Molecular subtype,
n (%) | |
| Luminal A | 15 (25.0) |
| Luminal B | 15 (25.0) |
| HER2 | 15 (25.0) |
| Basal-like | 15 (25.0) |
| Methylation level
of candidate gene, mean % | |
| CDH13 | 15.66±13.84 |
| SFRP1 | 15.67±11.21 |
| FHIT | 3.43±0.97 |
| Syk | 8.73±5.32 |
| RIZ1 | 48.30±11.55 |
| Lymphovascular
invasion, n (%) | |
| Negative | 39 (66.1) |
| Positive | 20 (33.9) |
| ER, n (%) | |
| Negative | 31 (51.7) |
| Positive | 29 (48.3) |
| PR, n (%) | |
| Negative | 33 (55.0) |
| Positive | 27 (45.0) |
| HER2
overexpression, n (%) | |
| Negative | 30 (50.0) |
| Positive | 30 (50.0) |
| Ki-67, n (%) | |
| <14% | 25 (41.7) |
| ≥14% | 35 (58.3) |
Table III presents
the clinicopathological characteristics according to the 4 breast
cancer subtypes. The basal subtype was characterized by a high
histologicical grade (P<0.001), low extensive intraductal
component (EIC) (P<0.001), the presence of necrosis
(P<0.001), a high Ki-67 level (P<0.001) and a positive
expression of EGFR (P<0.001).
| Table IIIClinicopathological characteristics
according to breast cancer subtype. |
Table III
Clinicopathological characteristics
according to breast cancer subtype.
| Subtype |
|---|
|
|
|---|
|
Characteristics | Luminal A | Luminal B | HER2 | Basal-like | P-value |
|---|
| Age (years), mean ±
SD | 57.3±15.2 | 46.9±7.1 | 50.3±11.8 | 52.6±15.9 | 0.273 |
| Menopausal status,
n (%) |
|
Pre-menopausal | 7 (46.7) | 8 (57.1) | 7 (46.7) | 5 (33.3) | 0.643 |
|
Post-menopausal | 8 (53.3) | 6 (42.9) | 8 (53.3) | 10 (66.7) | |
| Tumor size (cm),
mean ± SD | 1.6±0.7 | 1.6±1.0 | 1.7±0.8 | 2.3±1.0 | 0.082 |
| Histological grade,
n (%) |
| I | 9 (60.0) | 2 (13.3) | 2 (15.4) | 0 (0.0) | <0.001 |
| II | 5 (33.3) | 4 (26.7) | 2 (13.3) | 0 (0.0) | |
| III | 1 (6.7) | 9 (60.0) | 11 (73.3) | 15 (100.0) | |
| Nodal involvement,
n (%) |
| Negative | 10 (66.7) | 10 (66.7) | 12 (85.7) | 8 (57.1) | 0.432 |
| Positive | 5 (33.3) | 5 (33.3) | 2 (14.3) | 6 (42.9) | |
| Distant metastasis,
n (%) |
| Negative | 15 (100.0) | 15 (100.0) | 14 (93.3) | 14 (93.3) | 1.000 |
| Positive | 0 (0.0) | 0 (0.0) | 1 (6.7) | 1 (6.7) | |
| Lymphovascular
invasion, n (%) |
| Negative | 10 (66.7) | 10 (66.7) | 11 (73.3) | 8 (57.1) | 0.844 |
| Positive | 5 (33.3) | 5 (33.3) | 4 (26.7) | 6 (42.9) | |
| EIC (%), mean ±
SD | 13.9±19.7 | 31.7±38.8 | 12.9±37.7 | 3.6±9.5 | <0.001 |
| Necrosis, n
(%) |
| Negative | 14 (100.0) | 11 (73.3) | 6 (40.0) | 1 (12.5) | <0.001 |
| Positive | 0 (0.0) | 4 (26.7) | 9 (60.0) | 7 (87.5) | |
| Ki-67, n (%) |
| <14% | 15 (100.0) | 6 (40.0) | 4 (26.7) | 0 (0.0) | <0.001 |
| ≥14% | 0 (0.0) | 9 (60.0) | 11 (73.3) | 15 (100.0) | |
| ER, n (%) |
| Negative | 0 (0.0) | 1 (6.7) | 15 (100.0) | 15 (100.0) | <0.001 |
| Positive | 15 (100.0) | 14 (93.3) | 0 (0.0) | 0 (0.0) | |
| PR, n (%) |
| Negative | 1 (6.7) | 2 (13.3) | 15 (100.0) | 15 (100.0) | <0.001 |
| Positive | 14 (93.3) | 13 (86.7) | 0 (0.0) | 0 (0.0) | |
| HER2, n (%) |
| Negative | 15 (100.0) | 0 (0.0) | 0 (0.0) | 15 (100.0) | <0.001 |
| Positive | 0 (0.0) | 15 (100.0) | 15 (100.0) | 0 (0.0) | |
| Bcl-2, n (%) |
| Negative | 13 (86.7) | 15 (27.3) | 13 (86.7) | 14 (93.3) | 0.740 |
| Positive | 2 (13.3) | 0 (0.0) | 2 (13.3) | 1 (6.7) | |
| p53, n (%) |
| Negative | 2 (13.3) | 1 (6.7) | 3 (20.0) | 5 (33.3) | 0.353 |
| Positive | 13 (86.7) | 14 (93.3) | 12 (80.0) | 10 (66.7) | |
| EGFR, n (%) |
| Negative | 14 (93.3) | 14 (93.3) | 7 (50.0) | 0 (90.0) | <0.001 |
| Positive | 1 (6.7) | 1 (6.7) | 7 (50.0) | 15 (100.0) | |
Methylation levels in different molecular
subtypes of breast cancer
A total of 59 cases showed aberrantly methylated
genes. Amplification of 1 sample failed during PCR analysis. The
mean methylation levels of CDH13, SFRP1, FHIT,
Syk and RIZ1 were 15.66±13.84, 15.67±11.21, 3.43±0.97,
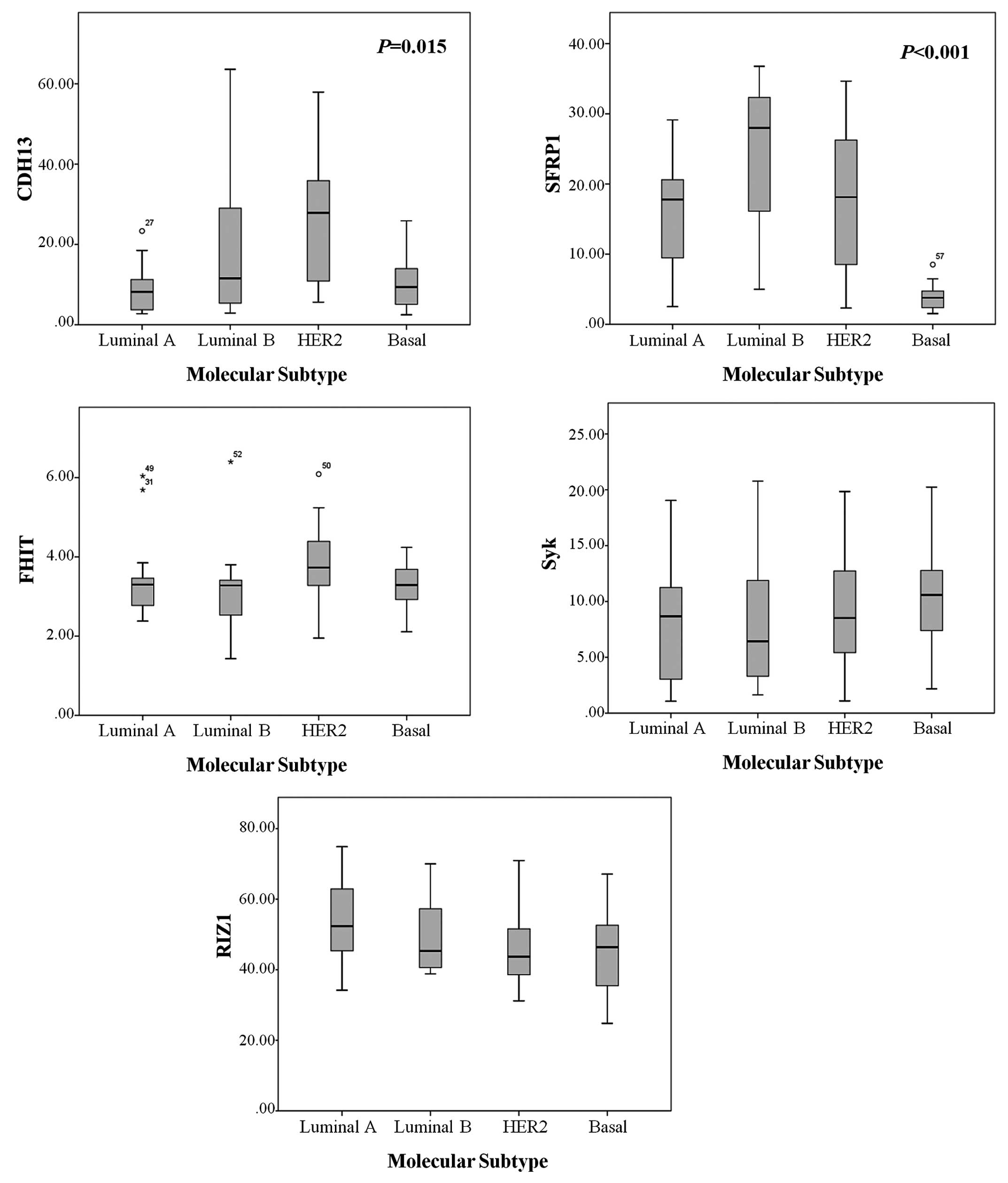
8.73±5.32 and 48.30%±11.55%, respectively. The methylation status
of CDH13 and SFRP1 was significantly different
according to breast cancer molecular subtypes (Table IV, Fig.
1). The CDH13 methylation level was significantly higher
in HER2 tumors compared to the luminal and basal-like subtypes
(P=0.006 and P=0.012, respectively) (Table V). The median methylation level and
average methylation ratio of the SFRP1 gene were
significantly lower in the basal-like subtype compared to the
luminal A, luminal B and HER2 subtypes (P=0.002, P<0.001 and
P=0.003, respectively). FHIT, Syk and RIZ1
methylation levels showed no significant differences among the
molecular subtypes.
| Table IVMethylation levels in breast cancer
subtypes. |
Table IV
Methylation levels in breast cancer
subtypes.
| Methylation level
(mean ± SD) |
|---|
|
|
|---|
| Gene | Luminal A | Luminal B | HER2 | Basal-like | P-value |
|---|
| CDH13 | 9.2±5.9 | 18.2±17.4 | 25.1±16.2 | 11.1±7.7.8 | 0.015 |
| SFRP1 | 15.5±7.8 | 24.3±10.7 | 18.0±10.7 | 4.0±2.0 | <0.001 |
| FHIT | 3.4±1.1 | 3.2±1.1 | 3.8±1.0 | 3.3±0.6 | 0.367 |
| Syk | 7.7±5.3 | 8.2±5.7 | 9.0±5.2 | 10.4±5.2 | 0.626 |
| RIZ1 | 53.0±11.5 | 48.7±10.9 | 46.2±11.4 | 44.5±12.1 | 0.257 |
| Table VComparison of methylation levels
between breast cancer subtypes. |
Table V
Comparison of methylation levels
between breast cancer subtypes.
| Basal-like vs.
Luminal | Basal-like vs.
HER2 | Luminal vs.
HER2 |
|---|
|
|
|
|
|---|
| Gene | Mean
difference | P-value | Mean
difference | P-value | Mean
difference | P-value |
|---|
| CDH13 | −2.611 | 0.533 | −14.022 | 0.006 | −11.411 | 0.012 |
| SFRP1 | −16.089 | <0.001 | −14.076 | 0.001 | 2.014 | 0.927 |
| FHIT | −0.047 | 0.879 | −0.541 | 0.137 | −0.494 | 0.121 |
| Syk | 2.376 | 0.198 | 1.392 | 0.510 | −0.984 | 0.571 |
| RIZ1 | −6.313 | 0.113 | −1.744 | 0.704 | 4.570 | 0.236 |
Gene-specific patterns of methylation for
classification of breast cancer
The varying methylation frequencies of the
CDH13 and SFRP1 genes in the different breast cancer
molecular subtypes provides evidence for subtype-specific
methylation profiling in breast cancer classification. In
particular, a low frequency of SFRP1 methylation was
significantly associated with the basal-like subtype (P<0.001).
We determined the area under the curve (AUC) of the receiver
operating characteristic (ROC) curve for the mean methylation level
of the SFRP1 gene. For the cut-off value of 7.50%, the AUC
was 0.941, with a sensitivity of 91.7% and a specificity of
84.2%.
Unsupervised hierarchical clustering based on the 5
genes and the variable methylated CpG loci of each gene resulted in
the formation of multiple small clusters that had no similar
biological patterns. As shown in Fig.
2, the results showed classes that were not well separated and
that did not appear to be associated with a molecular subtype.
Methylation status and
clinicopathological parameters
A comparison of the methylation status with the
clinicopathologic characteristics revealed that the negative
expression of ER, PR and HER-2, and the positive expression of
Bcl-2 and EGFR were associated with a low level of SFRP1
gene methylation (P=0.002, P=0.015, P<0.001, P=0.001 and
P<0.001, respectively) (Table
VI). The correlations between the methylation status of
RIZ1, Syk and FHIT and the clinicopathological
characteristics were dissimilar, RIZ1 was associated with
menopausal status and age (P=0.015 and P=0.042, respectively).
Syk was associated with histological grade (P=0.016).
| Table VIAssociation between
clinicopathological characteristics and DNA methylation levels
(P-value). |
Table VI
Association between
clinicopathological characteristics and DNA methylation levels
(P-value).
| CDH13 | SFRP1 | FHIT | Syk | RIZ1 |
|---|
| Age | 0.950 | 0.823 | 0.512 | 0.345 | 0.042 |
| Menopausal
status | 0.859 | 0.633 | 0.316 | 0.889 | 0.015 |
| Stage | 0.519 | 0.299 | 0.203 | 0.848 | 0.797 |
| Tumor size | 0.374 | 0.227 | 0.781 | 0.842 | 0.256 |
| LN(+) | 0.545 | 0.173 | 0.476 | 0.294 | 0.855 |
| Metastasis | 0.307 | 0.180 | 0.187 | 0.284 | 0.237 |
| Histological
grade | 0.158 | 0.168 | 0.758 | 0.016 | 0.807 |
| LVI | 0.608 | 0.355 | 0.788 | 0.896 | 0.983 |
| ER | 0.248 | 0.002 | 0.768 | 0.170 | 0.174 |
| PR | 0.138 | 0.015 | 0.428 | 0.176 | 0.380 |
| HER2 | 0.006 | <0.001 | 0.498 | 0.819 | 0.637 |
| Ki-67 | 0.194 | 0.162 | 0.759 | 0.027 | 0.125 |
| Bcl-2 | 0.308 | 0.001 | 0.545 | 0.550 | 0.933 |
| p53 | 0.271 | 0.347 | 0.802 | 0.761 | 0.945 |
| EGFR | 0.973 | <0.001 | 0.858 | 0.974 | 0.209 |
| Necrosis | 0.534 | 0.064 | 0.715 | 0.676 | 0.751 |
Discussion
Gene expression microarray analysis has made it
possible to identify distinct molecular subtypes that are
associated with different clinical outcomes (1–3).
Previous studies have documented the aberrant methylation of CpG
islands in gene promoters in breast carcinogenesis (7,8), and
have indicated that specific DNA methylation patterns may be
associated with some of the known breast cancer subtypes (9,10).
We observed a significantly lower methylation level
at a CpG site in the SFRP1 gene in the basal-like breast
cancer subtype when compared with other molecular subtypes,
consistent with the results of a previous study (13). SFRP1 is a member of the
SFRP family (14) and a
putative inhibitor of Wnt signaling (15,16)
that plays a key role in embryonic development, cell
differentiation and proliferation, as well as tumor development and
progression (17). It has been
described that SFRP1 expression is frequently lost or
downregulated in breast cancer (18,19),
and the loss of SFRP1 expression is associated with poor
prognosis, indicating a putative tumor suppressor gene function of
SFRP1(19). Furthermore, a
recent study indicated that SFRP1 has potential as a novel
biomarker for the triple-negative breast cancer phenotype (20). Thus, SFRP1 hypermethylation
may contribute to breast carcinogenesis, and may be useful as a
novel prognostic marker of breast cancer, particularly basal-like
tumors. In our study, 83.3% of the breast cancer cases had an
aberrant methylation of the SFRP1 gene. This result suggests
that promoter hypermethylation is the predominant mechanism of
SFRP1 gene silencing in human breast cancer, as shown in a
previous study (21), although
SFRP1 protein expression analysis was not included in this
study. Of the 60 cases that we analyzed, 15 were of the basal-like
subtype and tended to have decreased methylation of the
SFRP1 gene, suggesting that the DNA methylation of this gene
may contribute to the phenotype of breast cancer subtypes,
particularly the basal-like subtype.
The basal-like subtype of breast cancer has a
triple-negative phenotype with a poor prognosis and no specific
targeted therapy, despite an increased response to chemotherapy
compared to other breast cancer subtypes. Of note, a recent study
reported that, in triple-negative breast cancer, SFRP1
expression significantly correlates with an increased sensitivity
to neoadjuvant chemotherapy with taxane/anthracycline, and
preliminary experiments with siRNA-mediated knockdown of
SFRP1 showed a significantly decreased sensitivity to
paclitaxel, doxorubicin and cisplatin (20). In our study, the low frequency of
SFRP1 methylation in the basal-like subtype may account for
the higher expression of SFRP1 in basal-like tumors compared
to the other breast cancer subtypes and the associated increase in
response to chemotherapy in triple-negative breast cancer compared
to the other subtypes. These results suggest that SFRP1
signaling and methylation have potential as biomarkers tailored for
the basal-like subtype, and may be used to predict chemotherapy
response, as well as for treatment selection.
In addition, analysis of genes related with
subtype-specific methylation revealed that CDH13 was
specifically hypermethylated in HER2 tumors when compared with the
other molecular subtypes. This result is in accordance with a
previous study stating that the CDH13 gene is highly
methylated in HER2/neu-positive breast cancers (22). The CDH13 gene, coding for
H-cadherin, is a member of the cadherin family and a putative
mediator of cell-cell interaction and cancer cell invasion and
metastasis. It is considered as a tumor suppressor gene and may
contribute to the enhancement of tumor progression and invasion
(23). However, we did not observe
a correlation between CDH13 methylation and stage, tumor
grade, lymph node status and metastasis, which may reflect the
limited sample size used in this study.
Several genes have previously been shown to be
aberrantly methylated in breast cancer (8,24,25).
Furthermore, breast cancer subtype-specific epigenotypes have been
investigated through candidate gene approaches, as well as
genome-wide DNA methylation analysis. However, these observations
require further confirmation as the methylation frequency of a
candidate gene and subtype-specific methylation patterns vary
between independent studies. For example, Bediaga et
al(9) and Holm et
al(10) who used the
array-based DNA methylation-profiling approach, found that each
molecular subtype of breast cancer displayed specific methylation
profiles (9,10). However, these studies did not
identify and validate specific genes, such as SFRP1 and
CDH13, whose methylation status was used to discriminate
between the basal-like and HER2 subtypes in our study.
Nevertheless, our findings are consistent with those of previous
studies, stating that the HER2 subtype is associated with the
preferential hypermethylation of several genes, and that basal-like
tumors have several genes with low methylation levels (9,10,22,26).
Taken together, our results support the findings that methylation
may play a significant role in different breast tumor
phenotypes.
In the present study, we used quantitative DNA
methylation analysis to measure the methylation frequency in 5
genes known to be involved in breast carcinogenesis and are
commonly methylated in breast cancer. Pyrosequencing is uniquely
capable of quantifying methylation in explicit sequence context,
thereby enabling several consecutive CpG sites to be quantified
individually in a single assay. Although approaches for genome-wide
DNA methylation analysis hold promise for identifying novel
epigenetic targets, pyrosequencing is effective for identifying and
quantifying the aberrant methylation of breast cancer genes and
determining the criteria that may characterize and discriminate
specific molecular subtypes.
To detect possible common patterns of methylation
associated with the breast cancer molecular subtypes, hierarchical
clustering of the 5 genes was performed. However, the
discriminating ability of clustering analysis was poor compared
with using the methylation status of the SFRP1 gene alone.
This may be due to the relatively small sample size and the small
number of candidate genes, which were insufficient to produce
meaningful results (27).
Furthermore, 3 of the 5 candidate genes, FHIT, Syk
and RIZ1, displayed dissimilar methylation frequencies, and
the methylation status of these genes may not be useful for the
molecular classification of breast cancer.
Our study had several limitations. First, our study
included a relatively small number of samples and candidate genes.
These factors limit the application of our results to clinical
settings. Further studies with a larger number of breast cancer
cases are required to provide additional evidence. In addition, an
analysis of additional candidate genes may help define
subtype-specific methylation profiling for breast cancer
classification. Second, we did not include normal breast tissue in
the present study. Studies have shown that candidate genes are
hypermethylated in breast tumors, whereas matched normal breast
tissues have very low or no methylation levels (21,22,28–30).
However, direct comparisons of methylation levels in tumor tissue
with those of normal tissue would be required to prove that
epigenetic mechanisms may play a key role in the development of
breast cancer. Furthermore, comparisons of gene expression with
methylation levels in tumor and normal tissue would be helpful to
address this issue further.
In conclusion, our study revealed that the
basal-like subtype of breast cancer displays distinct methylation
profiles compared to other subtypes. We found that the basal-like
subtype is associated with low methylation levels of the
SFRP1 gene, and that the hypermethylation of CDH13
differs among the molecular subtypes of breast cancer. These
results suggest that the analysis of molecular markers, such as
gene hypermethylation are useful for the improved characterization
of molecular subtypes, and that the methylation levels of specific
genes in breast cancer may potentially serve as epigenetic
biomarkers and prognostic factors. Further studies with larger
sample sizes and more comprehensive DNA methylation profiling with
validation are required to clarify the predictive and prognostic
value of gene methylation patterns in breast cancer.
Acknowledgements
The present research has been supported by Korea
Breast Cancer Foundation.
References
|
1
|
Sørlie T, Perou CM, Tibshirani R, et al:
Gene expression patterns of breast carcinomas distinguish tumor
subclasses with clinical implications. Proc Natl Acad Sci USA.
98:10869–10874. 2001.PubMed/NCBI
|
|
2
|
Sørlie T, Tibshirani R, Parker J, et al:
Repeated observation of breast tumor subtypes in independent gene
expression data sets. Proc Natl Acad Sci USA. 100:8418–8423.
2003.PubMed/NCBI
|
|
3
|
Parker JS, Mullins M, Cheang MC, et al:
Supervised risk predictor of breast cancer based on intrinsic
subtypes. J Clin Oncol. 27:1160–1167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nielsen TO, Hsu FD, Jensen K, et al:
Immunohistochemical and clinical characterization of the basal-like
subtype of invasive breast carcinoma. Clin Cancer Res.
10:5367–5374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cheang MCU, Chia SK, Voduc D, et al: Ki67
index, HER2 status, and prognosis of patients with luminal B breast
cancer. J Natl Cancer Inst. 101:736–750. 2009. View Article : Google Scholar
|
|
6
|
Jaenisch R and Bird A: Epigenetic
regulation of gene expression: how the genome integrates intrinsic
and environmental signals. Nat Genet. 33:245–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dworkin AM, Huang TH and Toland AE:
Epigenetic alterations in breast: Implications for breast cancer
detection, prognosis and treatment. Semin Cancer Biol. 19:165–171.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Widschwendter M and Jones PA: DNA
methylation and breast carcinogenesis. Oncogene. 21:5462–5482.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bediaga N, Acha-Sagredo A, Guerra I, et
al: DNA methylation epigenotypes in breast cancer molecular
subtypes. Breast Cancer Res. 12:R772010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Holm K, Hegardt C, Staaf J, et al:
Molecular sybtypes of breast cancer are associated with
characteristic DNA methylation patterns. Breast Cancer Res.
12:R362010. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goldhirsch A, Wood WC, Coates AS, et al:
Strategies for subtypes - dealing with the diversity of breast
cancer: highlights of the St. Gallen International Expert Consensus
on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol.
22:1736–1747. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang DW, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
Bioinformatics Resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI
|
|
13
|
Wang S, Dorsey TH, Terunuma A, Kittles RA,
Ambs S and Kwabi-Addo B: Relationship between tumor DNA methylation
status and patient characteristics in African-American and
European-American women with breast cancer. PLoS One.
7:e379272012.PubMed/NCBI
|
|
14
|
Rattner A, Hsieh JC, Smallwood PM, et al:
A family of secreted proteins contains homology to the
cysteine-rich ligand-binding domain of frizzled receptors. Proc
Natl Acad Sci USA. 94:2859–2863. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Finch PW, He X, Kelley MJ, et al:
Purification and molecular cloning of a secreted, Frizzled-related
antagonist of Wnt action. Proc Natl Acad Sci USA. 94:6770–6775.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cadigan KM and Nusse R: Wnt signaling: a
common theme in animal development. Genes Dev. 11:3286–3305. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Polakis P: Wnt signaling and cancer. Genes
Dev. 14:1837–1851. 2000.
|
|
18
|
Ugolini F, Adélaïde J, Charafe-Jauffret E,
et al: Differential expression assay of chromosome arm 8p genes
identifies Frizzled-related (FRP1/FRZB) and Fibroblast Growth
Factor Receptor 1 (FGFR1) as candidate breast cancer genes.
Oncogene. 18:1903–1910. 1999. View Article : Google Scholar
|
|
19
|
Klopocki E, Kristiansen G, Wild PJ, et al:
Loss of SFRP1 is associated with breast cancer progression and poor
prognosis in early stage tumors. Int J Oncol. 25:641–649.
2004.PubMed/NCBI
|
|
20
|
Liedtke C, Ruckert C, Goette M, et al:
Secreted frizzled receptor protein 1 (sFRP-1) as both a potential
novel biomarker of triple negative breast cancer (TNBC), and its
sensitivity against taxane/anthracycline containing neoadjuvant
chemotherapy. Cancer Res. 69(24 Suppl): 40472009. View Article : Google Scholar
|
|
21
|
Veeck J, Niederacher D, An H, Klopocki E,
et al: Aberrant methylation of the Wnt antagonist SFRP1 in breast
cancer is associated with unfavourable prognosis. Oncogene.
25:3479–3488. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fiegl H, Millinger S, Goebel G, et al:
Breast cancer DNA methylation profiles in cancer cells and tumor
stroma: association with HER-2/neu status in primary breast cancer.
Cancer Res. 66:29–33. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Celebiler CA, Kilic Y, Saydam S, et al:
Predicting invasive phenotype with CDH1, CDH13, CD44, and TIMP3
gene expression in primary breast cancer. Cancer Sci.
100:2341–2345. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hinshelwood RA and Clark SJ: Breast cancer
epigenetics: normal human mammary epithelial cells as a model
system. J Mol Med. 86:1315–1328. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lo PK and Sukumar S: Epigenomics and
breast cancer. Pharmacogenomics. 9:1879–1902. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Feng W, Shen L, Wen S, et al: Correlation
between CpG methylation profiles and hormone receptor status in
breast cancers. Breast Cancer Res. 9:R572007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eisen MB, Spellman PT, Brown PO and
Botstein D: Cluster analysis and display of genome-wide expression
patterns. Proc Natl Acad Sci USA. 95:14863–14868. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yuan Y, Mendez R, Sahin A and Dai JL:
Hypermethylation leads to silencing of the SYK gene in human breast
cancer. Cancer Res. 61:5558–5561. 2001.PubMed/NCBI
|
|
29
|
Raish M, Dhillon VS, Ahmad A, et al:
Promoter hypermethylation in tumor suppressing genes p16 and FHIT
and their relationship with estrogen receptor and progesterone
receptor status in breast cancer patients from Northern India.
Transl Oncol. 2:264–270. 2009. View Article : Google Scholar
|
|
30
|
Du Y, Carling T, Fang W, Piao Z, Sheu JC
and Huang S: Hypermethylation in human cancers of the RIZ1 tumor
suppressor gene, a member of a histone/protein methyltransferase
superfamily. Cancer Res. 61:8094–8099. 2001.PubMed/NCBI
|