Introduction
Esophageal cancer is a malignant tumor of the
esophageal epithelial tissues, which occurs more frequently in
males than in females and is traditionally more prevalent in
subjects older than 40 years. However, in recent years there has
been an increased tendency for the disease to appear at a younger
age (1,2). The pathological classification
predominantly includes squamous cell carcinoma and adenocarcinoma,
which account for over 95% of all cases. Squamous cell carcinoma is
the more common of the two types. It has been estimated that ~80%
of all esophageal cancer cases occur in developing countries, with
the majority of the cases being squamous cell carcinoma (2). However, the incidence of esophageal
adenocarcinoma has been shown to be increasing in Western
industrialized countries (3).
The development of esophageal cancer is a complex
process involving many pathogenic factors, multiple stages, and
accumulation of multiple gene mutations and interactions. These
factors cause dysregulation of oncogenes, tumor-suppressor genes
and signaling protein molecules at the molecular level (4).
Raf kinase inhibitor protein (RKIP) is a family of
small, cytosolic phosphatidylethanolamine-binding proteins
originally purified from bovine brain. This protein family is
highly conserved and is rarely homologous to other types of
proteins. RKIP is widely distributed in many tissues in various
mammals such as mouse, monkey and human. It acts as a signal
regulator, which not only inhibits Raf-mediated MAPK and ERK
activities, but also inhibits the NF-κB signaling pathway and
regulates the activity of G protein-coupled receptors (5–7).
Reduced RKIP expression has been shown to affect
cell growth, angiogenesis, apoptosis and gene integrity (8). Our previous study found that RKIP may
prevent liver fibrosis, which is a precancerous lesion, through its
inhibition of hepatic stellate cell (HSC) proliferation (9). Increased RKIP expression was shown to
suppress invasion and reduce metastasis to basilar membranes in
mouse models of prostate cancer. It has also been shown to inhibit
growth and invasion of ovarian cancer. Reduced RKIP expression is
closely associated with progression and prognosis of hepatocellular
carcinoma, colorectal cancer, gastric cancer and gastrointestinal
stromal tumors (10–21). It has recently been proposed that
reduced RKIP expression is also related to progression and
pathological staging of esophageal cancer (22–24).
However, the underlying mechanisms of RKIP in esophageal cancer
cells remain unclear. The present study was, therefore, designed to
investigate the mechanisms involved in RKIP esophageal cancer
progression.
Materials and methods
Subjects
Surgical specimens from esophageal cancer patients
were classified into esophageal cancer tissues, tumor-adjacent
tissues (2 cm from the lesion) and normal esophageal tissues (5 cm
from the lesion). The specimens were collected from the Department
of Thoracic Surgery, The Second Hospital of Hebei Medical
University. Patient gender and age, post-surgical pathological
staging, lymph node status and distant metastasis were documented.
The tissues were either fixed in 4% paraformaldehyde solution for
hematoxylin and eosin (H&E) and immunohistochemical staining,
or were stored at −80°C for western blotting analysis.
The study was approved by the Ethics Committee of
The Second Hospital of Hebei Medical University. Informed consent
was obtained from all participants following a detailed description
of the purpose and potential benefits of the study.
Immunohistochemical staining
The tissues were cut into paraffin sections (4-μm),
dewaxed in xylene, dehydrated and high-pressure hot repaired in
citrate buffer. The sections were incubated in 3% hydrogen peroxide
and methanol at room temperature, and incubated in 1 μg/200 μl of
RKIP polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA) [phosphate-buffered saline (PBS) was used in the
negative controls] at 4°C overnight. The sections were then
incubated in 50 μl of polymer reinforcing agent at 37°C for 20 min,
incubated with 50 μl of rabbit-on-mouse HRP-polymer (Santa Cruz
Biotechnology, Inc.) at 37°C for 30 min and visualized using
3,3′-diaminobenzidine (DAB) for 3–5 min. Visualization was
terminated with tap water. The sections were stained with
hematoxylin, with a tan color indicating positive staining. Five
fields of view at high magnification (x400) were randomly selected
and observed, and the area of positive staining was measured.
Image-Pro Plus version 6.0 software was used for analysis. The RKIP
staining results were scored by a previously described method
(23). The IHC score (0–300) was
calculated as the product of the staining intensity (1, weak; 2,
moderate or 3, strong expression) and the staining rate (percentage
of positive cells in tissues, 0–100%). Tissues with final scores
exceeding the median score were determined to have high RKIP
expression; tissues equal or below the median were determined as
having downregulated RKIP expression. Correlations between RKIP
expression and lymph node or distant metastases were
investigated.
Cell culture and viral infection
The esophageal cancer cell line TE-1 was purchased
from the Cell Resource Center, Shanghai Institutes for Biological
Science of the Chinese Academy of Sciences (Shanghai, China). The
TE-1 cells were incubated in RPMI-1640 medium containing 10% fetal
bovine serum (FBS), 100 μg/ml streptomycin, 100 IU/ml penicillin, 1
mol/l HEPES and 4 mmol/l glutamine in 5% CO2 at 37°C.
When 90% single-layer saturation density was achieved, the cells
were digested with trypsin containing 0.5% EDTA and passaged at a
ratio of 1:2. The cell culture solution was replaced after 24, and
48 h later the cells were re-passaged.
Recombinant adenovirus was reconstructed by Shanghai
GeneChem Co. Ltd. (Shanghai, China). The TE-1 cells were exposed to
four different viruses: RKIP-RNAi-AD (which carried adenoviral RNAi
vector targeting RKIP); NC-RNAi-GFP-AD (a control viral vector for
RNAi without a specific target); RKIP-AD (a recombinant adenovirus
carrying the RKIP gene and expressing GFP); GFP-AD (a control
vector RKIP-AD expressing GFP). RPMI-1640 medium without serum and
antibiotics was used as a control. The TE-1 cells were seeded onto
plates at a 70% saturation density. They were synchronized with
RPMI-1640 medium without serum and antibiotics for 24 h prior to
treatment. The number of virus particles required was calculated as
the cell count × multiplicity of infection (MOI). The cell surface
was covered with RPMI-1640 medium without serum and antibiotics,
and incubated at 37°C for 2 h, with a further incubation for 46 h
after addition of complete medium. GFP expression was observed
under an inverted fluorescence microscope (Leica DMI3000 B
microscope), and determined by flow cytometry (FCM) to identify the
viral infection.
Determination of cell proliferation
TE-1 cells in an exponential growth phase were
seeded onto 96-well plates at a concentration of
5×104/ml/200 μl well. The cells were infected with
adenovirus as detailed above. Six replicate wells were used for
each group, with RPMI-1640 medium containing 2% FBS being used as a
negative control. After infection for 24, 48, 72 and 96 h, 20 μl of
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT;
Sigma Chemical Co., St. Louis, MO, USA) was added to each well and
incubated for 4 h, followed by the addition of 150 μl of dimethyl
sulfoxide (DMSO). The absorbance of each well at 492 nm
(A492 value) was determined using a microplate reader,
and the negative control well was adjusted to 0. The results were
expressed as the A492 value in the experimental
group/A492 value in the control group × 100%.
Determination of apoptosis
TE-1 cells in an exponential growth phase were
seeded onto 6-well plates at a concentration of
5×104/ml. The cells were infected with virus for 48 h,
digested with trypsin without EDTA, and re-suspended in PBS to
adjust the concentration to 5×105/ml. The cells were
then centrifuged at 2,000 × g for 5 min at 4°C, and the supernatant
was removed. The re-suspension and centrifugation procedure was
repeated twice. The cells were then re-suspended in 500 μl of
binding buffer, supplemented with 1 μl of Annexin V-PE, and
slightly mixed. Apoptosis was determined after incubation for 5 to
15 min at room temperature.
Detection of cell invasiveness
Matrigel (BD Biosciences, Franklin Lakes, NJ, USA)
and 24-well Transwells were stored at 4°C overnight. A total of 50
μl of dissolved Matrigel was evenly coated onto the bottom of the
upper chamber of each Transwell. The coated Transwells were
incubated at 37°C for at least 1 h in 5% CO2. After
interference for 48 h, the cells were digested with pancreatin,
centrifuged and re-suspended in RPMI-1640 medium containing 1%
bovine serum albumin (BSA), adjusting the cell concentration to
5×105/ml. The cell suspension (200 μl) was added to the
upper chamber of the Transwells, and 600 μl of RPMI-1640 medium
containing 10% FBS was added to the lower chamber. The Transwells
were then incubated at 37°C for 48 h in 5% CO2. The
non-invasive cells on the upper chamber were lightly removed with
cotton swabs, and the Transwell was inverted, and air-dried.
Anhydrous ethanol (600 μl) was added to the 24-well plates, and the
membranes were immersed in the well. After 30 min, the Transwells
were removed, air-dried, and rinsed three times in PBS for 10 min.
The cells were stained with crystal violet, counted and
photographed.
Western blot analysis
Frozen tissue samples (100 mg) were rinsed twice in
iced PBS, transferred to the ampulla of the homogenate tubes, and
cut into pieces. RIPA lysis buffer (1 ml) was added to the tubes,
and the tissues were prepared in the homogenate. The TE-1 cells
were cultured at a concentration of 1×107/ml, rinsed
with iced PBS and centrifuged at 3,000 × g for 10 min at 4°C. The
supernatant was removed, and 100 μl of modified RIPA lysis buffer
was added to the sediment. The tube was shaken for 15 sec, and the
cells were lysed on ice for 30 min. The supernatant was collected,
and the protein concentration was determined using the Bradford
method.
Sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) was performed with 80 μg of protein per
well. The gel was transferred to a PVDF membrane in an ice bath,
and blocked in 5% non-fat milk powder (for nonphosphorylated
antibody) or 5% BSA (for phosphorylated antibody) at room
temperature for 2 h.
The primary antibodies RKIP, phospho-RKIP, GRK2 and
GAPDH (Santa Cruz Biotechnology, Inc.), Raf-1 and phospho-Raf-1
(Bioworld Technology Inc., St. Louis Park, MN, USA), ERK1/2 and
phospho-ERK1/2 (Cell Signaling Technology, Inc., Beverly, MA, USA)
were diluted with blocking solution (Table I) and incubated at 4°C overnight.
The secondary antibody, HRP-conjugated goat anti-rabbit IgG, was
diluted with blocking solution, shaken and incubated at room
temperature for 2 h. Electrochemiluminescence (ECL) was added for
visualization, and X-ray images were generated. The visualized band
was scanned, and quantitative analysis was performed using NIH
ImageJ version 1.38 software. GAPDH was used as an internal
reference for RKIP, and RKIP served as an internal reference for
phospho-RKIP.
 | Table IConcentration of the primary
antibodies. |
Table I
Concentration of the primary
antibodies.
| Primary
antibodies | Dilution |
|---|
| RKIP | 1:300 |
| Phospho-RKIP (Ser
153) | 1:300 |
| GRK2 | 1:300 |
| Raf-1 | 1:500 |
| Phospho-Raf-1 (Ser
338/Tyr 341) | 1:600 |
| ERK1/2 | 1:2000 |
| Phospho-ERK1/2 (Thr
202/Tyr 204) | 1:2000 |
| GAPDH | 1:1000 |
Results were expressed as the percentage of the
optical density (OD) of the target band in relation to the OD of
the internal reference.
Quantitative RT-PCR assay
Approximately 1×107 of the wall-adherent
TE-1 cells were rinsed with sterile iced PBS, and suspended in 1 ml
of PBS after being scraped with cell scrapers. The cell suspension
was transferred to Eppendorf tubes, centrifuged at 3,000 × g for 10
min at 4°C, and the supernatant was removed. TRIzol (1 ml) was
immediately added to the cell sediment and shaken. RNA was
extracted following the manufacturer’s instructions. In brief, 2 μl
of RNA was added to 500 μl of diethylpyrocarbonate (DEPC)-treated
water, and the OD of the RNA samples was measured at 280 and 260 nm
on an ultraviolet spectrophotometer. RNA was reverse transcribed
into cDNA, and quantitative RT-PCR was performed on an ABI StepOne
Plus real-time PCR system device. GAPDH was used as an internal
reference gene. Primers were designed and synthesized according to
the gene sequence in GenBank (Table
II). The SYBR-Green reaction system was used for quantitative
RT-PCR assay, and the ΔCt value was calculated. Each reading was
carried out in triplicate. The difference in mRNA gene expression
was evaluated by relative quantification using the
2−ΔΔCt method. The mRNA expression of the gene in the
blank vector control group was defined as 1 (the blank vector
served as controls). The 2−ΔΔCt value indicated the
relative mRNA expression of the target gene in the RKIP-RNAi-AD or
RKIP-AD group.
| Table IIRNA oligonucleotides and qRT-PCR
primers. |
Table II
RNA oligonucleotides and qRT-PCR
primers.
| Target gene | Primer sequence
(5′-3′) |
|---|
| RKIP |
| Sense |
AGACCCACCAGCATTTCGTG |
| Antisense |
GCTGATGTCATTGCCCTTCA |
| MMP-14 |
| Sense |
GCTGAGATCAAGGCCAATGT |
| Antisense |
ATGTAGGCATAGGGCACCTC |
| LIN28 |
| Sense |
TCGGACTTCTCCGGGGCCAG |
| Antisense |
GCTGGTTGGACACGGAGCCC |
| GAPDH |
| Sense |
GAACGGGAAGCTCACTGGCATGGC |
| Antisense |
TGAGGTCCACCACCCTGTTGCTG |
Statistical analysis
Statistical analyses were performed using SPSS
version 13.0. Data are expressed as the means ± standard deviations
(SD). The Chi-square, Kruskal-Wallis, Spearman, one-way analysis of
variance (ANOVA), and Fisher’s least significant difference (LSD)
tests were used when appropriate. Values of P<0.05 were
considered statistically significant.
Results
Reduced RKIP expression in esophageal
cancer tissues in comparison with normal esophageal epithelium
tissues and tumor-adjacent tissues
H&E staining of the specimens from 40 patients
with esophageal cancer was able to distinguish the specimens of
esophageal squamous cell carcinoma, tumor-adjacent tissues and
normal squamous epithelium of the esophagus.
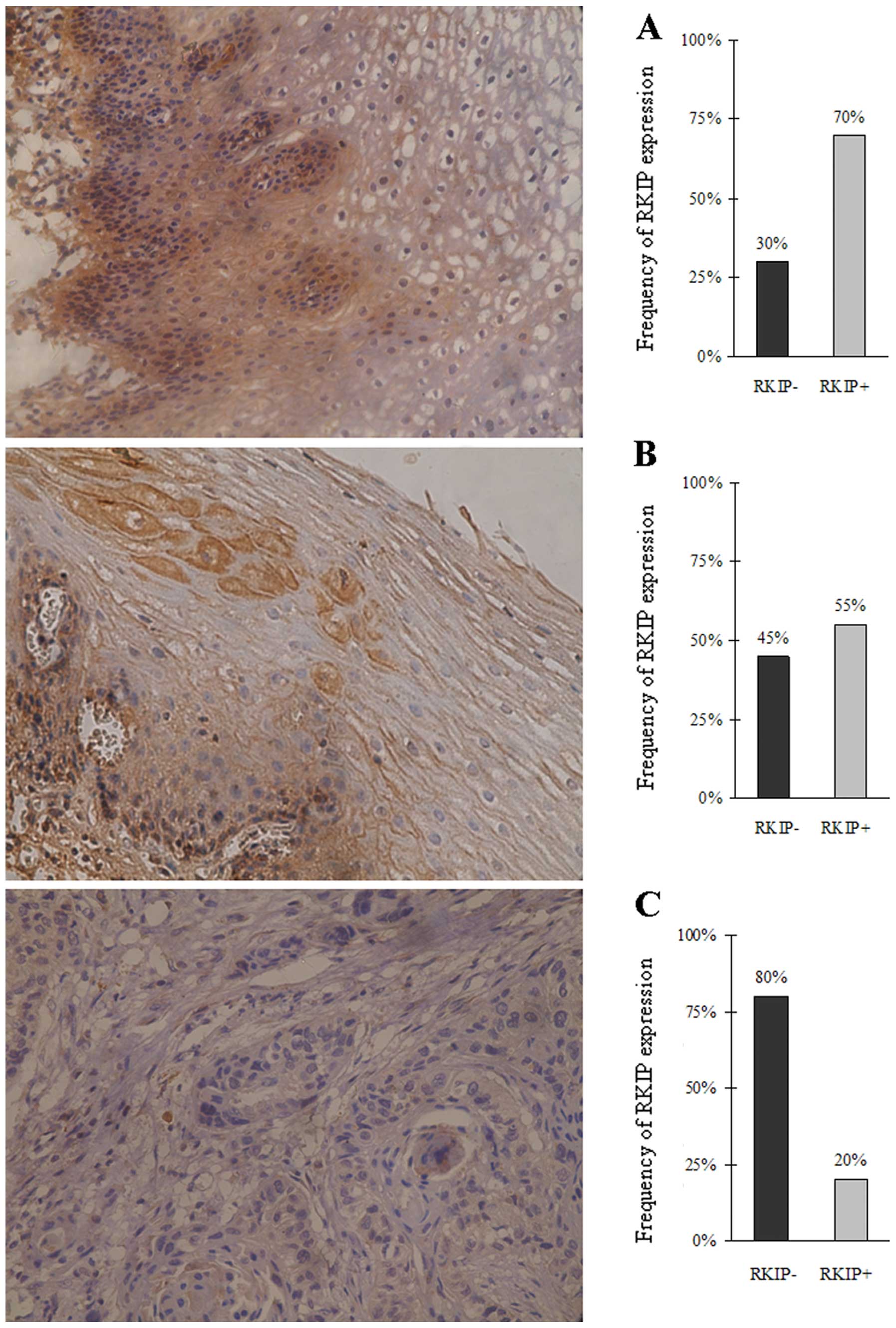
Immunohistochemical staining using the RKIP
polyclonal antibody showed high RKIP expression in normal
esophageal epithelia and tumor-adjacent tissues with abundant
yellowish-brown particle sediments noted in the cytoplasm. RKIP
expression was significantly lower in the esophageal squamous cell
carcinoma cells with no obvious yellowish-brown particle sediments
observed. The frequency of RKIP-positive expression was 70, 55 and
20% in the normal esophageal epithelia, tumor-adjacent tissues and
esophageal squamous cell carcinoma cases, respectively (Fig. 1). These results indicate that the
RKIP expression was significantly lower in the esophageal cancer
tissues than that in the normal esophageal epithelia (P<0.05,
Chi-square test).
Western blot analysis estimates of RKIP expression
in the esophageal squamous cancer tissues, tumor-adjacent tissues
and normal esophageal epithelia were 0.32±0.05, 0.56±0.08 and
0.70±0.11, respectively, also indicating significantly reduced RKIP
expression in the cancer tissues in comparison with the
tumor-adjacent tissues and the esophageal epithelia (P<0.05;
Kruskal-Wallis test) (Fig. 2).
RKIP expression is associated with lymph
node and distant metastasis of esophageal cancer
Immunohistochemical staining was performed to
measure the mean density of the RKIP-positive staining in the
cancer tissues. The mean rank of each parameter was calculated.
No correlation was observed between RKIP expression
and gender, age or pathological tumor staging (P>0.05). Eleven
of the 40 cases were well-differentiated carcinoma, 21 were
moderately differentiated carcinoma and 8 were poorly
differentiated carcinoma. Multiple independent sample nonparametric
testing resulted in mean ranks of 25.18, 19.69 and 16.19,
respectively (P>0.05), demonstrating that RKIP expression was
not associated with the degree of differentiation of esophageal
cancer tissues (Table III). These
results indicated low levels of RKIP expression in all 3 categories
of tumor differentiation.
| Table IIIAnalysis of the clinicopathological
parameters and RKIP expression. |
Table III
Analysis of the clinicopathological
parameters and RKIP expression.
| Clinicopathological
parameters | No. | Mean of rank | P-value |
|---|
| Gender | | | |
| Male | 27 | 20.15 | 0.404 |
| Female | 13 | 21.13 | |
| Age (years) | | | |
| >60 | 22 | 19.97 | 0.388 |
| ≤60 | 18 | 20.93 | |
| Pathological
stage | | | |
| Stage I–II | 16 | 24.41 | 0.084 |
| Above stage
II | 24 | 17.90 | |
| Tumor
differentiation | | | |
| Well
differentiated | 11 | 25.18 | 0.228 |
| Moderately
differentiated | 21 | 19.69 | |
| Poorly
differentiated | 8 | 16.19 | |
| Lymph node or
distant metastasis | | | |
| Metastasis | 15 | 11.40 | <0.01 |
| Without
metastasis | 25 | 15.96 | |
Fifteen of the 40 subjects had lymph node or distant
metastasis. Independent two sample Mann-Whitney tests provided mean
ranks of 11.4 and 15.96, respectively for patients with and without
lymph node involvement or metastases (P<0.01), indicating that
lower RKIP expression was associated with esophageal cancer
metastasis (Table III). These
findings further suggest that RKIP may act as a suppressor protein
for metastasis.
RKIP has no effects on proliferation and
apoptosis of the esophageal cancer cell line TE-1
The adenovirus infection efficiencies in the
different groups were determined using flow cytometry.
Adenovirus-mediated gene transfection at a MOI of 400 was employed
for the subsequent experiments. MTT assay demonstrated that
adenoviral infection for 24, 48, 72 and 96 h had no effect on the
viability of TE-1 cells (P>0.05) (Fig. 3A).
 | Figure 3Effects of Raf kinase inhibitor
protein (RKIP) on proliferation and apoptosis of the esophageal
cancer cell line TE-1 were investigated. (A) Cell viability was
assessed by MTT assay. Cell viability of TE-1 cells exposed to
recombinant adenoviruses at different time points was shown. TE-1
cells were transfected with recombinant adenovirus for 24, 48, 72
and 96 h, respectively. TE-1 cells were infected with RKIP-RNAi-AD,
NC-RNAi-GFP-AD, RKIP-AD and GFP-AD. Control cells were treated with
RPMI-1640 only. No difference in cell viability was noted in the
groups, P>0.05. (B) Apoptosis of TE-1 cells at 48 h after
recombinant adenoviral infection as detected by PE labeled flow
cytometry (FCM). Apoptosis of TE-1 cells transfected with
RKIP-RNAi-AD, NC-RNAi-GFP-AD, RKIP-AD and GFP-AD, respectively, was
detected by PE-labeled FCM. Control cells were treated with
RPMI-1640 only. No difference in apoptosis was noted among the
groups, P>0.05. |
Flow cytometry revealed that there was no
significant difference in the apoptotic rate of TE-1 cells between
the RKIP-AD (0.10±0.15%) and GFP-AD group (0.11±0.25%; P>0.05)
(Fig. 3B). Likewise, no significant
difference was observed in the apoptotic rate of TE-1 cells between
the RKIP-RNAi-AD (0.83±0.12%) and NC-RNAi-GFP-AD group (0.70±0.10%;
P>0.05) (Fig. 3B).
RKIP inhibits the invasive ability of the
esophageal cancer cell line TE-1
The effect of RKIP expression on the invasive
ability of esophageal cancer TE-1 cells was investigated (Fig. 4). The results showed that there were
no significant differences in the invasive ability of TE cells
among the GFP-AD (80.25±10.87), NC-RNAi-GFP-AD (67.75±13.30) and
control (75.50±17.48; P>0.05) groups. However, invasive
potential was significantly higher in the RKIP-RNAi-AD group
(127.25±16.62) than that in the NC-RNAi-GFP-AD group (67.75±13.30;
P<0.05), and the invasive ability was significantly lower in the
RKIP-AD group (9.50±7.14) than that in the GFP-AD group
(80.25±10.87; P<0.05) (Fig.
4).
RKIP has no effect on the MAPK signaling
pathway in esophageal TE-1 cells, but is involved in the G
protein-coupled signaling pathway
Western blot analysis was performed to determine the
expression of RKIP and its phosphorylation (p-RKIP) level in TE-1
cells following treatment with the adenoviruses. The results showed
that RKIP expression was significantly higher in the RKIP-AD group
(2.31±0.36) than that in the GFP-AD group (0.43±0.09; P<0.05),
and was significantly lower in the RKIP-RNAi-AD group (0.05±0.18)
than that in the NC-RNAi-GFP-AD group (0.38±0.08; P<0.05).
However, there was no significant difference in the p-RKIP/RKIP
level between the groups (P>0.05) (Fig. 5A and B).
 | Figure 5Western blot analysis was used to
detect Raf kinase inhibitor protein (RKIP), p-RKIP, Raf, p-Raf,
ERK, p-ERK, GRK2 and GAPDH protein expression in TE-1 cells at 48 h
after adenoviral infection. Lane 1, RKIP-RNAi-AD; lane 2,
NC-RNAi-GFP-AD; lane 3, RKIP-AD; lane 4, GFP-AD and lane 5, control
group. In the RKIP-AD group, a large amount of RKIP was expressed
as an exogenous protein (**P<0.05 compared with the
GFP-AD group). At the same time, RKIP expression was decreased in
the RKIP-RNAi-AD group (*P<0.05 compared with the
NC-RNAi-GFP-AD group). However, there were no differences in p-RKIP
expression among the groups (A and B). GRK2 expression was
increased in the RKIP-RNAi-AD group and decreased in the RKIP-AD
group (##P<0.05 compared with the NC-RNAi-GFP-AD
group; #P<0.05 compared with the GFP-AD group). No
differences in ERK, p-ERK, Raf and p-Raf were noted among the
groups (C and D). |
In contrast, GRK2 expression was significantly lower
in the RKIP-AD group (0.52±0.10) than that in the GFP-AD group
(1.01±0.11; P<0.05), and was significantly higher in the
RKIP-RNAi-AD group (1.37±0.15) than that in the NC-RNAi-GFP-AD
group (0.84±0.10; P<0.05). However, there was no significant
difference in Raf, p-Raf, ERK and p-ERK expression between the
groups (P>0.05) (Fig. 5C and
D).
RKIP inhibits the invasive and metastatic
ability of esophageal cancer cell line TE-1 by downregulating mRNA
expression of LIN28 and MMP-14
After increasing or inhibiting RKIP expression,
quantitative RT-PCR was performed, and the mRNA expression of
MMP-14, RKIP, LIN28 and GAPDH in the different groups was compared
using the relative quantitative method. RKIP mRNA expression was
significantly higher in the RKIP-AD group (166.46±0.09) than that
in the GFP-AD group (P<0.01), and was significantly lower in the
RKIP-RNAi-AD group (0.17±0.09) than that in the NC-RKIP-RNAi-AD
group (1.00±0.00; P<0.01). The mRNA expression of MMP-14 was
significantly lower in the RKIP-AD group (0.26±0.20) than that in
the GFP-AD group (1.00±0.00; P<0.01), and the mRNA expression of
MMP-14 was significantly higher in the RKIP-AD group (6.89±0.84)
than that in the GFP-AD group (1.00±0.00; P<0.01). In addition,
mRNA expression of LIN28 was significantly lower in the RKIP-AD
group (0.28±0.06) than that in the GFP-AD group (1.00±0.00;
P<0.01), and was increased in the RKIP-RNAi-AD group (1.86±0.12)
when compared with that in the NC-RKIP-RNAi-AD group (1.00±0.00;
P<0.01) (Fig. 6).
Discussion
Esophageal cancer is the eighth most common
malignant tumor in the world and the sixth most common cause of
tumor-related death. The incidence of esophageal cancer has
continued to increase during the last three decades (1,25). In
addition, there is regional variation in the incidence of
esophageal cancer with an incidence of ~4–10 cases/100,000 in
Western countries compared with an incidence of 50–100
cases/100,000 in China and South Africa (26,29).
RKIP is a regulator protein which is involved in
many signaling pathways where it mediates cell proliferation,
apoptosis and metastasis in order to regulate cell proliferation
balance. The present study demonstrated that RKIP expression was
reduced in esophageal cancer tissues in a manner that was
independent of gender, age, degree of tumor differentiation and
pathological tumor stage. However, we also demonstrated that
reduced RKIP expression was associated with an increased occurrence
of lymph node and distant metastases, suggesting that it may be
closely related with the invasive capacity of esophageal cancer
cells. Other studies have also shown that downregulation of RKIP
expression is related to regional lymph node metastasis and tumor
stage (22–24). The critical role of RKIP in tumor
metastasis was first identified in metastatic prostate cancer
tissues. It was shown that RKIP expression was lower in metastatic
prostate cancer than in primary tumor cells. Based on these
findings, blood levels of RKIP are used as a prognostic marker for
the malignant potential of prostate cancer. Other studies have
shown that RKIP expression is downregulated in metastatic lymph
nodes associated with human breast and metastatic colon cancer,
further suggesting that downregulation of RKIP expression is
involved in tumor metastasis. It has also been shown that
overexpression of RKIP is associated with reduced vascular invasion
in prostate cancer and melanoma. RKIP has also been reported to
inhibit tumor invasion, and significantly reduced RKIP expression
has been detected in insulinoma, colorectal carcinoma,
hepatocellular carcinoma and ovarian cancer (30–34).
In the present study, an adenovirus overexpressing
RKIP and a viral RNAi vector of RKIP was used to interfere with
RKIP expression in esophageal cancer cells in order to investigate
the role of RKIP in proliferation, apoptosis and invasion of
esophageal cancer cells. RKIP expression had no effect on the
proliferation and apoptosis of esophageal cancer cells. However,
in vitro overexpression of RKIP significantly inhibited the
invasive capacity of esophageal cancer cells.
Invasion is the most important phenotype of
malignant tumors, and it is the major factor that determines the
prognosis of malignant tumors. Specific microRNAs (miRNAs),
including Let-7, have been reported to play a role in the
maintenance of the invasiveness of cancer cells. It has been
demonstrated that Let-7 degrades target genes and inhibits their
transcription. In this way it functions at both the gene and
protein levels to promote tumor metastasis.
Increased RKIP expression has been shown to promote
Let-7 expression by inhibiting the MAPK signaling transduction
pathway, and thereby preventing cell invasion and metastasis
(31,35). When cells are stimulated by
extracellular growth factors, activated Ras activates Raf-1 which
in turn activates the MAPK signaling pathway. Subsequent activation
of ERK1/2 promotes Myc expression and further promotes LIN28
expression. LIN28 inhibits Let-7 expression resulting in elevated
expression of its target gene HMGA1, thereby activating genes such
as Snail, Twist and Slug which promote cell invasion and
metastasis. In the present study, we showed that RKIP
overexpression inhibited the invasiveness of esophageal cancer
cells and significantly reduced LIN28 transcription, suggesting
that RKIP inhibited esophageal cancer cell invasion by
downregulating LIN28 expression.
For tumors to develop, the normal cellular
homeostasis of the microenvironment is destroyed and is replaced by
one that facilitates tumor cell growth. The presence of
inflammatory cytokines in the tumor microenvironment also plays a
critical role in tumor invasion (36–39).
Thus, maintaining the tumor microenvironment is a prerequisite for
protecting or promoting the occurrence and development of
tumors.
It has been shown that RKIP prevents the invasion of
cancer cells by controlling the expression of matrix
metalloproteinases (MMPs), particularly MMP-1 and MMP-2 (40). Silencing RKIP expression results in
a highly invasive phenotype of the cancer cells with dramatically
elevated levels of MMP-1 and MMP-2 expression, whereas
overexpression of RKIP decreases cancer cell invasion in
vitro and reduces MMP-1 and MMP-2 expression. In accordance
with this finding, MMP-1 or MMP-2 in RKIP-knockout cells reverts
their invasiveness and metastastic potential to normal levels
(40). In the present study, we
demonstrated that overexpression of RKIP significantly inhibited
MMP-14 expression, indicating that it reduced the invasiveness of
esophageal TE-1 cancer cells by downregulating MMP-14 expression.
RKIP is a natural suppressor of Raf, which inhibits Raf
phosphorylation, thereby inhibiting the MAPK signaling transduction
pathway. It has been found that RKIP is highly expressed in Merkel
cell carcinoma cells, but the MAPK signaling transduction pathway
is not activated. These results indicate that RKIP does not affect
the ERK/MAPK signaling transduction pathway or proliferation or
apoptosis in Merkel cells.
In our studies with esophageal TE-1 cancer cells,
overexpression or low expression of RKIP for 24 h resulted in
stabilization of the MAPK signaling pathway, and either inhibition
or activation of the G protein-coupled signaling pathway. G protein
is a transmembrane protein, which acts as a second messenger,
converting extracellular information to intracellular information.
G-protein-coupled receptor kinase-2 (GRK-2) is therefore a negative
feedback inhibitory protein for G protein-coupled receptors
(GPCRs). GRK-2 has been shown to phosphorylate GPCRs, and separate
G protein from GPCRs.
Protein kinase C (PKC) has been shown to
phosphorylate RKIP at serine 153. Phosphorylated RKIP dissociates
from Raf-1, binds to GRK-2 and inhibits its activity (6). The binding conversion from Raf-1 to
GRK-2 suggests that RKIP serves as a signal regulator. In other
words, after PKC is activated, RKIP is phosphorylated, becomes
dissociated from Raf-1, and thereby prevents the inhibition of the
ERK signaling pathway and associated increases in cell
permeability. In this way it facilitates the binding of RKIP to
GRK-2, resulting in activation of the G protein-coupled signaling
pathway. It can, therefore, be hypothesized that RKIP acts as a key
that switches from one signaling pathway to another, depending on
its degree of phosphorylation.
In conclusion, our findings clearly demonstrate that
reduced RKIP expression is related to the development of lymph
nodes or distant metastasis associated with esophageal cancer. We
also provide evidence to suggest that RKIP inhibits esophageal
cancer cell invasion by downregulating the expression of GRK-2,
LIN28 and MMP-14.
Acknowledgements
This study was supported in part by grants to H.Q.J.
from the Natural Science Foundation of Hebei Province (China,
C2010000530), the Hebei Province Science and Technology Project
financed by the Department of Finance, and a grant to D.Q.Z. from
the Hebei Medical Research Key Project (China, 20110088), and a
grant to J.J.M. from the National Natural Science Foundation of
China (81200311).
References
|
1
|
Klein CA and Stoecklein NH: Lessons from
an aggressive cancer: evolutionary dynamics in esophageal
carcinoma. Cancer Res. 69:5285–5288. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mao WM, Zheng WH and Ling ZQ:
Epidemiologic risk factors for esophageal cancer development. Asian
Pac J Cancer Prev. 12:2461–2466. 2011.PubMed/NCBI
|
|
3
|
Lepage C, Rachet B, Jooste V, Faivre J and
Coleman MP: Continuing rapid increase in esophageal adenocarcinoma
in England and Wales. Am J Gastroenterol. 103:2694–2699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tew WP, Kelsen DP and Ilson DH: Targeted
therapies for esophageal cancer. Oncologist. 10:590–601. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yeung K, Seitz T, Li S, Janosch P,
McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H,
Sedivy JM and Kolch W: Suppression of Raf-1 kinase activity and MAP
kinase signalling by RKIP. Nature. 401:173–177. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yeung KC, Rose DW, Dhillon AS, Yaros D,
Gustafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W and Sedivy
JM: Raf kinase inhibitor protein interacts with NF-kappaB-inducing
kinase and TAK1 and inhibits NF-kappaB activation. Mol Cell Biol.
21:7207–7217. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Deiss K, Kisker C, Lohse MJ and Lorenz K:
Raf kinase inhibitor protein (RKIP) dimer formation controls its
target switch from Raf1 to G protein-coupled receptor kinase (GRK)
2. J Biol Chem. 287:23407–23417. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
al-Mulla F, Bitar MS, Taqi Z, Rath O and
Kolch W: RAF kinase inhibitory protein (RKIP) modulates cell cycle
kinetics and motility. Mol Biosyst. 7:928–941. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma J, Li F, Liu L, Cui D, Wu X, Jiang X
and Jiang H: Raf kinase inhibitor protein inhibits cell
proliferation but promotes cell migration in rat hepatic stellate
cells. Liver Int. 29:567–574. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Keller ET: Role of Raf kinase inhibitor
protein in pathophysiology of prostate cancer. For Immunopathol Dis
Therap. 2:89–94. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Khamis ZI, Iczkowski KA and Sang QX:
Metastasis suppressors in human benign prostate, intraepithelial
neoplasia, and invasive cancer: their prospects as therapeutic
agents. Med Res Rev. 32:1026–1077. 2012. View Article : Google Scholar
|
|
12
|
Bevilacqua E, Frankenberger CA and Rosner
MR: RKIP suppresses breast cancer metastasis to the bone by
regulating stroma-associated genes. Int J Breast Cancer.
2012:1247042012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Granovsky AE and Rosner MR: Raf kinase
inhibitory protein: a signal transduction modulator and metastasis
suppressor. Cell Res. 18:452–457. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Klysik J, Theroux SJ, Sedivy JM, Moffit JS
and Boekelheide K: Signaling crossroads: the function of Raf kinase
inhibitory protein in cancer, the central nervous system and
reproduction. Cell Signal. 20:1–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zeng L, Imamoto A and Rosner MR: Raf
kinase inhibitory protein (RKIP): a physiological regulator and
future therapeutic target. Expert Opin Ther Targets. 12:1275–1287.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Yang YH, Wang AQ, Yao B, Xie G,
Feng G, Zhang Y, Cheng ZS, Hui L, Dai TZ, Du XB and Wang D:
Immunohistochemical detection of the Raf kinase inhibitor protein
in nonneoplastic gastric tissue and gastric cancer tissue. Med
Oncol. 27:219–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chatterjee D, Sabo E, Tavares R and
Resnick MB: Inverse association between Raf kinase inhibitory
protein and signal transducers and activators of transcription 3
expression in gastric adenocarcinoma patients: implications for
clinical outcome. Clin Cancer Res. 14:2994–3001. 2008. View Article : Google Scholar
|
|
18
|
Zlobec I, Baker K, Minoo P, Jass JR,
Terracciano L and Lugli A: Node-negative colorectal cancer at high
risk of distant metastasis identified by combined analysis of lymph
node status, vascular invasion, and Raf-1 kinase inhibitor protein
expression. Clin Cancer Res. 14:143–148. 2008. View Article : Google Scholar
|
|
19
|
Minoo P, Zlobec I, Baker K, Tornillo L,
Terracciano L, Jass JR and Lugli A: Loss of raf-1 kinase inhibitor
protein expression is associated with tumor progression and
metastasis in colorectal cancer. Am J Clin Pathol. 127:820–827.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Al-Mulla F, Hagan S, Behbehani AI, Bitar
MS, George SS, Going JJ, García JJ, Scott L, Fyfe N, Murray GI and
Kolch W: Raf kinase inhibitor protein expression in a survival
analysis of colorectal cancer patients. J Clin Oncol. 24:5672–5679.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zlobec I, Baker K, Terracciano L, Peter S,
Degen L, Beglinger C and Lugli A: Two-marker protein profile
predicts poor prognosis in patients with early rectal cancer. Br J
Cancer. 99:1712–1717. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim HS, Won KY, Kim GY, Kim SC, Park YK
and Kim YW: Reduced expression of Raf-1 kinase inhibitory protein
predicts regional lymph node metastasis and shorter survival in
esophageal squamous cell carcinoma. Pathol Res Pract. 208:292–299.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Birner P, Jesch B, Schultheis A and
Schoppmann SF: RAF-kinase inhibitor protein (RKIP) downregulation
in esophageal cancer and its metastases. Clin Exp Metastasis.
29:551–559. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao C, Pang L, Ren C and Ma T: Prognostic
value of raf kinase inhibitor protein in esophageal squamous cell
carcinoma. Pathol Oncol Res. 18:471–477. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bray F, Jemal A, Grey N, Ferlay J and
Forman D: Global cancer transitions according to the Human
Development Index (2008–2030): a population-based study. Lancet
Oncol. 13:790–801. 2012.PubMed/NCBI
|
|
26
|
Lambert R, Saito H, Lucas E and
Sankaranarayanan R: Survival from digestive cancer in emerging
countries in Asia and Africa. Eur J Gastroenterol Hepatol.
24:605–612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bosetti C, Levi F, Ferlay J, Garavello W,
Lucchini F, Bertuccio P, Negri E and La Vecchia C: Trends in
oesophageal cancer incidence and mortality in Europe. Int J Cancer.
122:1118–1129. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brown LM, Devesa SS and Chow WH: Incidence
of adenocarcinoma of the esophagus among white Americans by sex,
stage, and age. J Natl Cancer Inst. 100:1184–1187. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shibata A, Matsuda T, Ajiki W and Sobue T:
Trend in incidence of adenocarcinoma of the esophagus in Japan,
1993–2001. Jpn J Clin Oncol. 38:464–468. 2008.PubMed/NCBI
|
|
30
|
Woods Ignatoski KM, Grewal NK, Markwart
SM, Vellaichamy A, Chinnaiyan AM, Yeung K, Ray ME and Keller ET:
Loss of Raf kinase inhibitory protein induces radioresistance in
prostate cancer. Int J Radiat Oncol Biol Phys. 72:153–160.
2008.PubMed/NCBI
|
|
31
|
Yun J, Frankenberger CA, Kuo WL, Boelens
MC, Eves EM, Cheng N, Liang H, Li WH, Ishwaran H, Minn AJ and
Rosner MR: Signalling pathway for RKIP and Let-7 regulates and
predicts metastatic breast cancer. EMBO J. 30:4500–4514. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zaravinos A, Bizakis J and Spandidos DA:
RKIP and BRAF aberrations in human nasal polyps and the adjacent
turbinate mucosae. Cancer Lett. 264:288–298. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li HZ, Gao Y, Zhao XL, Liu YX, Sun BC,
Yang J and Yao Z: Effects of raf kinase inhibitor protein
expression on metastasis and progression of human breast cancer.
Mol Cancer Res. 7:832–840. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li HZ, Wang Y, Gao Y, Shao J, Zhao XL,
Deng WM, Liu YX, Yang J and Yao Z: Effects of raf kinase inhibitor
protein expression on metastasis and progression of human
epithelial ovarian cancer. Mol Cancer Res. 6:917–928. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dangi-Garimella S, Yun J, Eves EM, Newman
M, Erkeland SJ, Hammond SM, Minn AJ and Rosner MR: Raf kinase
inhibitory protein suppresses a metastasis signalling cascade
involving LIN28 and let-7. EMBO J. 28:347–358. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Akutsu Y and Matsubara H: The significance
of lymph node status as a prognostic factor for esophageal cancer.
Surg Today. 41:1190–1195. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sgourakis G, Gockel I, Lyros O, Lanitis S,
Dedemadi G, Polotzek U, Karaliotas C and Lang H: The use of neural
networks in identifying risk factors for lymph node metastasis and
recommending management of t1b esophageal cancer. Am Surg.
78:195–206. 2012.PubMed/NCBI
|
|
38
|
Ben-Neriah Y and Karin M: Inflammation
meets cancer, with NF-κB as the matchmaker. Nat Immunol.
12:715–723. 2011.
|
|
39
|
Bromberg J and Wang TC: Inflammation and
cancer: IL-6 and STAT3 complete the link. Cancer Cell. 15:79–80.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Beshir AB, Ren G, Magpusao AN, Barone LM,
Yeung KC and Fenteany G: Raf kinase inhibitor protein suppresses
nuclear factor-κB-dependent cancer cell invasion through negative
regulation of matrix metalloproteinase expression. Cancer Lett.
299:137–149. 2010.
|