Introduction
Colon cancer is the second most common cancer
diagnosed in both men and women in the United States, resulting in
~55,000 deaths a year (1). It is
considered one of the most preventable types of cancers as several
modifiable environmental factors have been identified to play
important roles in the development of this disease. These include
specific dietary components such as selenium, an important
micronutrient that can protect colonic cells against a wide range
of external and internal stressors. Selenium has been reported to
inhibit the growth of malignant colonic cells as well as to induce
their demise.
Control of inflammatory conditions can also impact
colon carcinogenesis. Inflammation favors tumorigenesis by
stimulating angiogenesis (2),
damaging DNA (3,4), and chronically stimulating cell
proliferation (5,6). Proinflammatory genes have been shown
to be important for the maintenance and progression of colorectal
cancer (CRC) (7). Proinflammatory
cytokines bind to specific receptors and activate various signal
transduction pathways in the epithelial cells as well as in
interacting immune cells, leading to upregulation of prostaglandin
synthase 2 (PTGS2), interleukin (IL)-4, IL-6 and
IL-8 genes (8). These genes
have been shown to exhibit proinflammatory activity in the
intestine (9,10). These findings support the regular
use of non-steroidal anti-inflammatory drugs (NSAIDs) such as
aspirin or ibuprofen, which are associated with a 40–50% decrease
in the relative risk for colon cancer (11).
Nitric oxide (NO) has been implicated in the
carcinogenic process, particularly in colon or intestinal
epithelial cell lines (12,13) and in the known impairment of
regulatory processes in these cells (14). The calcium-independent, inducible
form of nitric oxide synthase (iNOS) can be induced in many
different cell types by cytokines and bacterial lipopolysaccharides
(LPS). Once expressed, iNOS can produce sustained and substantial
amounts of NO, which can be cytotoxic (15,16).
Recently, natural and synthetic drugs that inhibit iNOS and both
the NF-κB and STAT3 pathways (17)
have been shown to reduce intestinal inflammation in patients and
in rodents with inflammatory bowel disease (IBD). Chemically
induced colonic inflammation and formation of aberrant crypt foci
(ACF, early precursor lesions of colon cancer) also were diminished
in animal models administered the iNOS inhibitor
S,S′-1,4-phenylenebis(1,2-ethanediyl)bis-isothiourea
(PBIT) (18).
To create a more potent compound for inhibition of
colon cancer, a novel isosteric analog of PBIT,
S,S′-1,4-phenylenebis(1,2-ethanediyl)bis-isoselenourea
(PBIsSe) was synthesized, in which sulfur was replaced with
selenium. Incorporation of selenium into the structure of PBIT
provided the agent with additional novel properties at very low
concentrations. To investigate the efficacy of these iNOS
inhibitors for CRC, we first examined the effect of PBI-Se and PBIT
on the well-differentiated human intestinal epithelial Caco-2 cell
line stimulated with a defined cytokine mixture (consisting of
interferon γ (IFNγ), interleukin (IL)-1β and tumor necrosis factor
α (TNFα) or with lipopolysaccharide (LPS) and monitored inhibition
of growth, induction of apoptosis, and production of key
proinflammatory signaling molecules (IL-8 and IL-6). Most human
intestinal cell lines studied are poorly differentiated under
standard growth conditions. The Caco-2 cell line is considered to
resemble normal intestinal epithelial cells (19). LPS and the cytokine mixture have
been shown to increase IL-8, IL-6 and iNOS production (20,21).
We also examined the chemopreventive efficacy of PBIT and PBI-Se in
an azoxymethane (AOM)-induced rat colon carcinogenesis model using
ACF as an endpoint biomarker, and we tested the influence of PBI-Se
on immune cells.
Materials and methods
Cell culture and reagents
Caco-2 cells were obtained from the American Type
Culture Collection (ATCC, Manassas, VA, USA) and used between
passages 30 and 35. Cells were grown in Dulbecco’s modified Eagle’s
medium (DMEM) with 4 mM L-glutamine, 20% heat-inactivated foetal
bovine serum (FBS) and 1% non-essential amino acids. Cells were
cultured at 37°C in a water-saturated atmosphere of 95% air and 5%
CO2, re-fed every 2 days and passaged weekly. Caco-2
cells were used at 21 days after confluence as indicated, to permit
differentiation. PBIT and PBI-Se were provided by Dr Dhemant Desai
and were dissolved in phosphate-buffered saline (PBS). The primary
antibodies for cyclin D1 and proliferating cell nuclear antigen
(PCNA) were obtained from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). The anti-rabbit antibody and ECL detection system
were from Amersham (Arlington Heights, IL, USA). Ethidium bromide
and acridine orange were from Sigma (St. Louis, MO, USA).
Cell induction and treatment
Cells were grown as indicated above. At ~70%
confluence and 21 days, cultures were switched to serum-free medium
for 24 h. The serum-free medium was removed and the cell monolayer
was washed with PBS (pH 7.4) prior to addition of cytokines and LPS
with PBIT or PBI-Se in fresh medium without serum. Based on a
previous study (21), the cytokine
concentrations used for induction were IFNγ (200 μg/ml), IL-1β (5
ng/ml) and TNFα (100 ng/ml); the LPS concentration was 1 μg/ml.
Assay of the effect of PBI-Se and PBIT on
cell viability
After teatment with PBIT or PBI-Se, cells were
harvested and dissociated in a solution of 0.25% trypsin and 3 mM
ethylene diamine tetraacetic acid (EDTA) in PBS, pH 7.4 (without
calcium and magnesium). Trypan blue was added and cells were
counted with a hemocytometer. Only cells that excluded the dye were
counted as viable. Results are expressed as the number of viable
cells/ml.
Detection of apoptosis
Cells were exposed to PBIT (0, 30, 60 and 100 μM)
and PBI-Se (0, 2, 4 and 6 μM) for 24 h. Acridine orange/ethidium
bromide (1 μl of a stock prepared from one part each of 100 μg/ml
acridine orange and 100 μg/ml ethidium bromide in PBS) was added to
the cell suspension (25 μl; 5×106/ml) just before
microscopy. A 10-μl aliquot of the gently mixed suspension was
placed on microscope slides, covered with glass coverslips, and
examined under an Olympus AX71 microscope connected to a digital
imaging system with SPOT RT software version 3.0. Cells were scored
according to the following categories: C1, cells with large, green,
non-condensed nuclei defined as non-apoptotic, viable cells; C2,
cells with red/orange nuclei showing signs of nuclear bead
formation defined as apoptotic cells; and C3, cells with large red
nuclei not showing signs of nuclear condensation or bead formation
defined as necrotic cells. At least 200 cells per sample were
counted and scored. The apoptotic index (%) was calculated by
dividing the sum of apoptotic cells (C2) × 100 by the total number
of cells scored.
Reverse transcription-PCR for IL-6 and
IL-8
Caco2 cells were treated with cytokine mixture and
LPS plus subtoxic concentrations of PBIT and PBI-Se for 24 h. Total
RNA from the treated and untreated samples was extracted using
ToTally RNA™ kit (Ambion) as per the manufacturer’s instructions.
Equal quantities of DNA-free RNA were used for reverse
transcription reactions for cDNA conversion using the SuperScript™
Reverse Transcriptase (Invitrogen). PCR conditions for IL-8
amplifications were denaturation at 94°C for 2 min, followed by 50
cycles at 95°C for 15 sec, 55°C for 10 sec and 72°C for 20 sec; PCR
conditions for IL-6 consisted of denaturation at 95°C for 30 sec,
annealing at 62°C for 40 sec and extension at 72°C for 40 sec for
36 cycles. PCR was performed using the Taq Polymerase Master
Mix (Qiagen, Inc.). The PCR products were visualized and
photographed under UV illumination. The primer sequences used were
as follows: IL-6 (628 bp), 5′-ATG AAC TCC TTC TCC ACA AGC GC-3′
(sense) and 5′-G AAG AGC CCT CAG GCT GGA CTG-3′ (antisense); IL-8
(295 bp), 5′-ACT TCC AAG CTG GCC GTG GCT CTC TTG GCA-3′ (sense) and
5′ TGA ATT CTC AGC CCT CTT CAA AAA CTT CTC-3′ (antisense).
Detection of PCNA and cyclin D1 by
western blotting
Expression of PCNA and cyclin D1 proteins was
analyzed in CaCo2 cells. After treatment with PBIT and PBI-Se for
24 h, cells were harvested and lysed in lysis buffer [50 mM
Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1% NP-40, 1 mM
phenylmethylsulfonyl fluoride (PMSF)] on ice. After centrifugation,
supernatants were collected and the protein content was measured
using a Bio-Rad protein assay kit with bovine serum albumin (BSA)
as a standard. Equal amounts of protein from each extract were
separated via polyacrylamide gel electrophoresis (PAGE) in 8%
sodium dodecyl sulfate (SDS) and transferred onto nitrocellulose
membranes (Toyo Roshi Kaisha, Ltd., Tokyo, Japan) using the Bio-Rad
electrotransfer system. Blots were blocked by incubating in 5% milk
with Tris-HCl (pH 7.5) and 0.1% Tween-20 for 1 h at room
temperature and probed overnight at 4°C with rabbit anti-cyclin D1
polyclonal antibody (Santa Cruz Biotechnology) for cyclin D1
protein and mouse anti-PCNA monoclonal antibody (Santa Cruz
Biotechnology) for PCNA. Antibodies were diluted 1:1,000 with 5%
milk in Tris-HCl (pH 7.5) and 0.1% Tween-20. The immunoblots were
then probed with horseradish peroxidase-conjugated anti-rabbit IgG
for cyclin D1 and horseradish peroxidase-conjugated anti-mouse IgG
for PCNA [1:2,000 diluted with 5% milk in Tris-HCl (pH 7.5)]. After
the final wash, the signal was detected with an enhanced
chemiluminescence kit (Pierce Biotechnology, Inc., Rockford, IL,
USA).
In vivo experiments
Animals, diet and care
All animal experiments were carried out in
accordance with the NIH guidelines and the University of Oklahoma
Health Sciences Center Institutional Animal Care and Use Committee
approved protocol. Male F344 rats were obtained from Harlan
Laboratories, housed under standardized conditions (21°C, 60%
relative humidity, 12 h light/12 h dark cycle, 20 air changes/h),
and fed a standard laboratory rodent chow and drinking water
through reverse osmosis until initiation of the experiment.
Prepared diets were based on the modified AIN-76A containing 5%
corn oil by weight (American Institute of Nutrition). The
experimental diets contained 0.001% (10 ppm) or 0.002% (20 ppm)
PBI-Se. PBI-Se was premixed with a small quantity of casein and
then blended into the bulk diet using a Hobart Mixer. Both control
and experimental diets were prepared weekly and stored in a cold
room. Rats were allowed ad libitum access to the respective
diets and tap water.
Experimental design for the efficacy
of PBI-Se
The experiment was designed to evaluate the efficacy
of 0, 10 and 20 ppm of PBI-Se administered continuously starting 3
days after carcinogen treatment. The dose selection was based on
our maximum tolerated dose (MTD) study. At 7 weeks of age, groups
of rats [n=12 rats per treatment group, 12 AOM-treated plus 6
vehicle (saline)-treated] were fed the control diet. At 8 weeks of
age, rats intended for carcinogen treatment were injected s.c. with
azoxymethane (AOM; Midwest Research Institute, Kansas City, MO,
USA) at a dose rate of 15 mg/kg body weight once weekly for 2
weeks, and those intended for vehicle treatment received an equal
volume of normal saline. The experimental diets were initiated 3
days later and continued until termination of the experiment 8
weeks later (Fig. 1). Rats were
sacrificed by CO2 euthanasia, and all organs were
examined grossly. Colons were evaluated for aberrant crypt foci
(ACF). For this evaluation, they were slit open lengthwise from the
anus to the ceacum and then fixed flat with the mucosa on the upper
side between filter papers in 10% buffered formalin.
Quantification of ACF
Topographical analysis of the colonic mucosa was
carried out according to Bird (22)
and our previous report (23).
After a minimum of 24 h, fixed colons were stained with 0.2%
methylene blue solution for 5–10 min, placed mucosal side up on a
microscopic slide, and viewed under a light microscope. The total
number of ACF in the entire colon was determined in every 2-cm
section of the colon, starting from the distal (taken as 0 cm) to
the proximal end of the colon. Aberrant crypts were distinguished
from the surrounding normal crypts by their increased size,
increased distance of cells from lamina to basal surfaces, and
easily discernible pericryptal zone. The variables used to assess
the aberrant crypts were incidence and multiplicity. Aberrant crypt
multiplicity was determined as the number of crypts in each focus
and categorized as containing up to 4 or more aberrant crypts per
focus.
Giemsa staining of the spleens
Spleens from untreated and treated animals were
fixed in formalin, sectioned (4 μm), and stained with Giemsa to
enumerate the number of polymorphic nuclear cells (PMNs). The
percentages of PMNs were calculated relative to control untreated
spleens.
Statistical analysis
All the results are reported as the means ± SE.
Statistical differences between control and treated groups were
evaluated using the unpaired t-test with Welch’s correction.
Differences between groups were considered significant at
P<0.05.
Results
Effect of PBIT and PBI-Se on the
proliferation of Caco2 cells
Caco2 cells were exposed for 24 h in the presence or
absence of a cytokine mixture (IL-1β, TNFα + IFNγ), or to LPS and
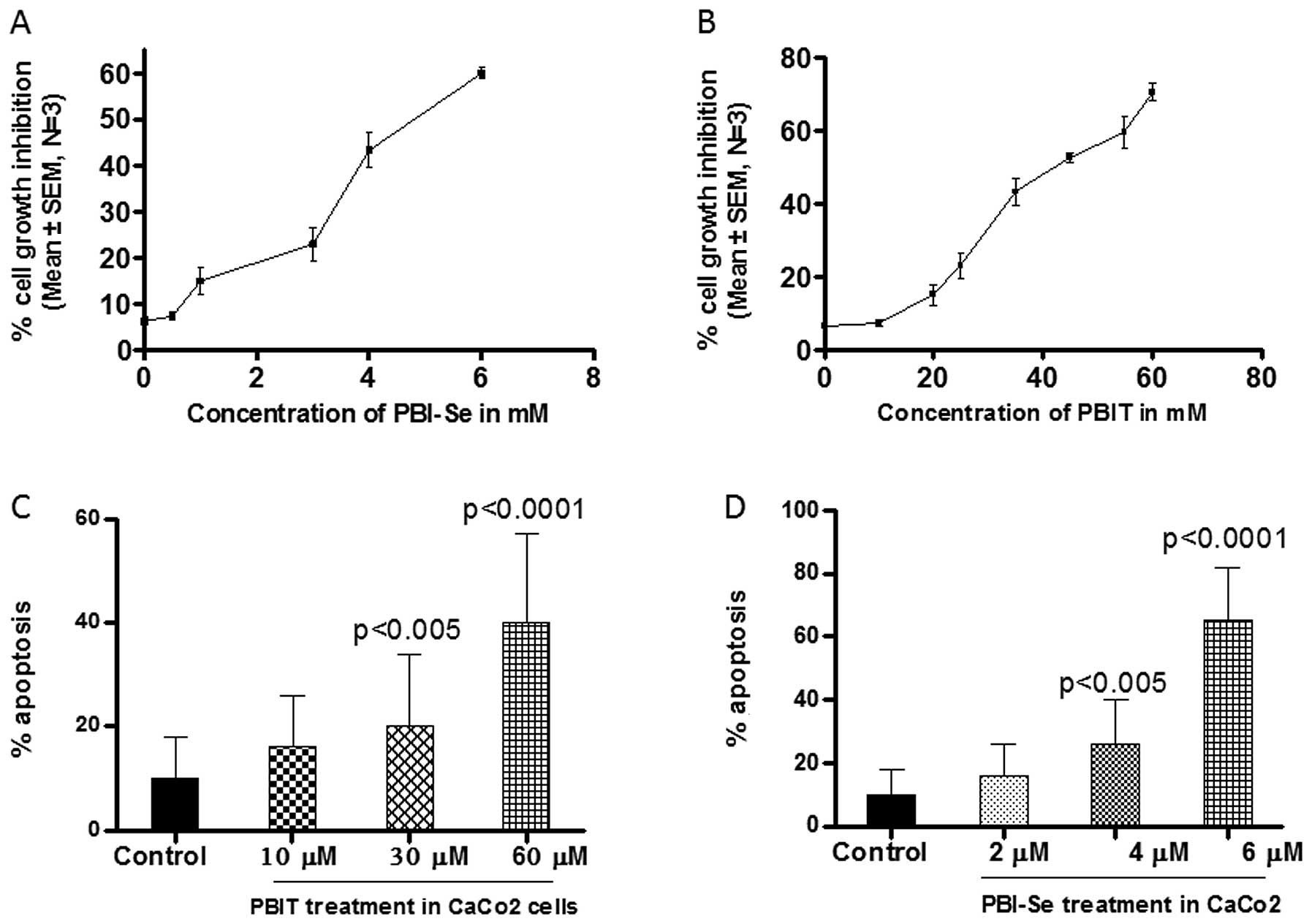
to PBIT (0–100 μM) or PBI-Se (1–12 μM). As shown in Fig. 2A and B, both iNOS inhibitors caused
dose-dependent growth inhibition as determined using a Trypan blue
dye exclusion assay. After 24 h of exposure, 50% inhibition of cell
growth was achieved at 40 μM PBIT or at 4 μM PBI-Se. PBI-Se induced
significant toxicity in Caco2 cells at or above 6 μM, whereas PBIT
induced significant toxicity at or above 60 μM. These results
demonstrate that PBI-Se is more potent than PBIT.
Effect of PBI-Se and PBIT on apoptosis in
Caco2 cells
We observed apoptotic bodies by staining the cells
exposed to PBIT or PBI-Se with ethidium bromide and acridine
orange. The population of apoptotic cells was enhanced
significantly (P<0.0002) in the PBI-Se-treated cells. As shown
in Fig. 2D PBI-Se induced apoptosis
in Caco2 cells in a dose-dependent manner (5.6% at 2 μM, 24% at 4
μM and 60% at 6 μM vs. the untreated cells).
Effect of PBI-Se and PBIT on IL-6 and
IL-8 in Caco2 cells induced with a cytokine mixture
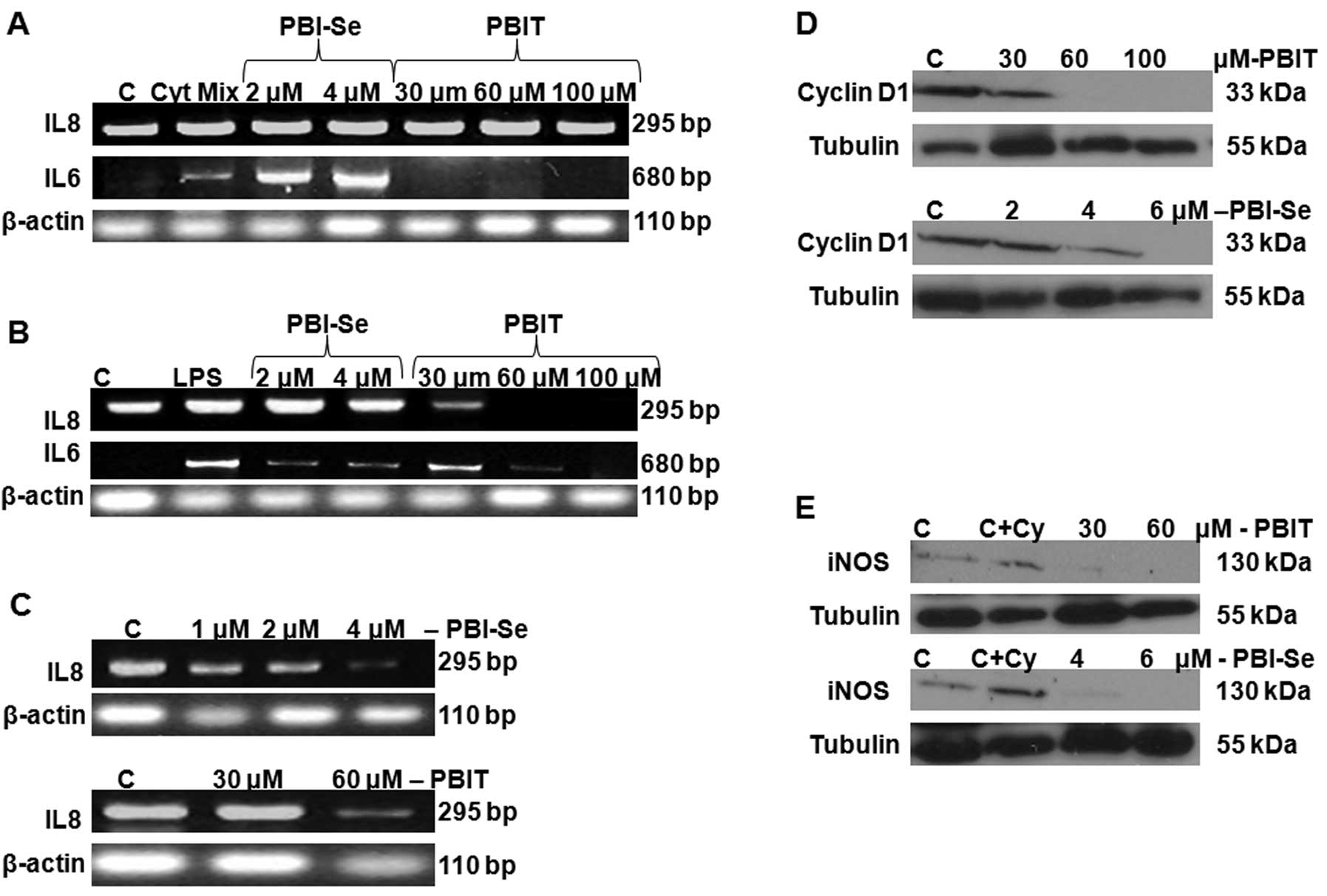
The inflammatory cytokine response (IL-6 and IL-8)
was measured in 3-week cultured Caco-2 cells induced with the
cytokine mixture (TNFα plus IL-1β + IFNγ). We identified basal IL-8
production in CaCo2 cells, whereas no IL-6 production was noted in
the control cells (in the absence of stimulation with the cytokine
mixture). As shown in Fig. 3A,
stimulation with the cytokine mixture for 24 h caused a significant
induction in IL-6 and ~50-fold increase in IL-8. Treatment with
PBIT (30 and 60 μM) significantly decreased IL-6 at both doses
tested, whereas treatment with PBI-Se caused a significant
dose-dependent increase in cytokine-induced IL-6. There was no
blockage of IL-8 by either drug. Taken together, the results
indicate that inflammatory responses are increased by treatment of
Caco-2 cells with the cytokine mixture and that PBIT, but not
PBI-Se, can block the IL-6 component of the response.
Effect of PBI-Se and PBIT on expression
of IL-6 and IL-8 in CaCo2 cells induced with LPS
We also incubated CaCo2 cells with LPS, in the
presence or absence of NOS inhibitors PBIT or PBI-Se for 24 h. PBIT
significantly attenuated the LPS-induced IL-6 and IL-8 production
in a dose-dependent manner. PBI-Se also dose-dependently attenuated
LPS-stimulated IL-6 production but not LPS-stimulated IL-8
production (Fig. 3B), whereas, both
the drugs decreased endogenous production of IL-8 in CaCo2 cells
(Fig. 3C).
Effect of PBI-Se and PBIT on iNOS and
cyclin D1 in CaCo2 cells
A significant dose-dependent decrease in cyclin D1
protein expression was observed with both iNOS inhibitors,
suggesting inhibition of cell cycle progression (Fig. 3D). As expected, treatment of Caco2
cells with PBI-Se and PBIT caused a significant inhibition of
cytokine-induced iNOS protein expression (Fig. 3E).
Effect of PBI-Se on ACF formation in
rats
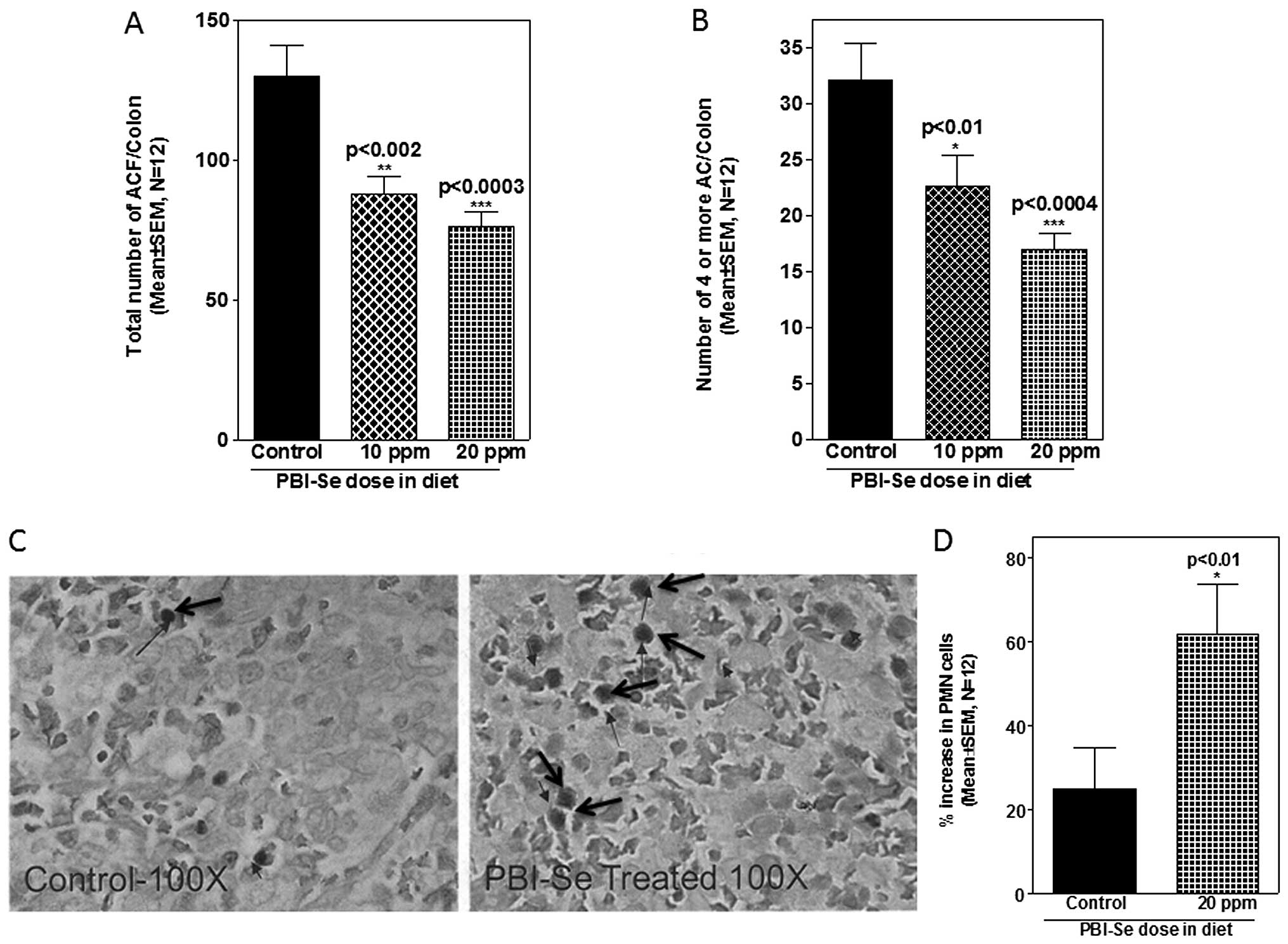
An initial examination of the effect of PBI-Se on
AOM-induced colon carcinogenesis was carried out in rats. Efficacy
endpoints used were inhibition of total ACF number as well as
reduction in the number of multicrypt clusters (4 or more) of
aberrant crypts. Rats that were treated with saline and fed the
control or experimental diets showed no evidence of ACF formation
in the colon (data not shown). In rats fed the control diet, AOM
treatment induced, on average, ~130 ACF/colon, out of which 32 foci
contained multiple (4 or more) aberrant crypts/focus (Fig. 4A). ACF were observed predominantly
in the distal colons. PBI-Se was found to be an effective inhibitor
of total ACF/colon (32–41%, P<0.001) and of multicrypt clusters
containing 4 or more crypts/focus (29–47%, P<0.001) (Fig. 4B).
Effect of PBI-Se on PMNs in spleen
Selenium is essential for cell mediated immunity
which includes destruction of neoplastic cells (24). Hence, the effect of selenium in the
diet on the number of PMNs was analyzed in the spleens of rats fed
with the PBI-Se diet. Supplementation of rat diets with PBI-Se
increased the percentage of PMNs in the spleens (P<0.002;
Fig. 4C and D).
Discussion
In a quest to identify and characterize better
compounds for chemoprevention, an isosteric selenium analog of the
known iNOS inhibitor PBIT, was synthesized and tested in
vitro and in vivo along with PBIT for comparison. PBI-Se
and PBIT both caused a decrease in proliferation of Caco2
intestinal epithelial cells attributable to inhibition of DNA
synthesis (Fig. 2). The
IC50 for inhibition by PBI-Se was 3–4 μM, which is
consistent with previously published data showing growth inhibition
in melanoma cell lines (25). Cell
cycle arrest can be achieved by a decrease in the levels of cyclins
and/or associated cyclin-dependent kinases (CDKs) (26) and this is evident in the Caco2 cell
line by a decrease in cyclin D1 protein expression with both PBI-Se
and PBIT (Fig. 3D).
Intestinal epithelial cells have the potential to
express and secrete a wide array of inflammatory cytokines
(including chemokines such as IL-8), which may function as signals
to neighboring immune and inflammatory cells, initiating and
amplifying an ongoing acute immune response (27–29).
Numerous studies have shown that IL-8 and iNOS may be expressed in
a variety of cells in the setting of inflammation. It has been
reported that selenium supplementation in individuals results in
induction of TNFα, IL-1β, IL-8, superoxide dismutase (SOD)2,
chemokine (C-X-C motif) ligand 2 (CXCL2) and several other
immunological and oxidative stress-related genes (30). IL-8 was reported to inhibit
significantly the growth of intraperitoneal and subcutaneous tumors
induced by transplantation of K562 cells and to induce cell death
by apoptosis in these cells in vivo(31,32).
IL-8 production can be increased after stimulation with a mixture
of IL-1β, IFNγ and TNFα in a variety of cells (33,34).
Previous studies also demonstrated that Caco-2 cells
grown in a non-co-culture system increased IL-8 protein and mRNA in
response to a wide range of LPS concentrations (20). We observed high endogenous
expression of IL-8 in unstimulated Caco2 cells and enhanced
expression when the cells were stimulated with a mixture of IL-1β,
IFNγ and TNFα or with LPS (Fig. 3A and
B). Both PBIT and PBI-Se inhibited control endogenous IL-8
production (Fig. 3C). PBIT
inhibited LPS-stimulated IL-8 production but not the
cytokine-stimulated IL-8 production, whereas comparable growth
inhibitory concentrations of PBI-Se did not decrease IL-8 in either
stimulation condition (Fig. 3A and
B). These results suggest that LPS and proinflammatory
cytokines use different cell signaling pathways in colon cancer
epithelial cells and that PBI-Se, unlike PBIT, does not interfere
with LPS-enhanced IL-8 production. The induction of IL-8 has
important biologic consequences for control of tumor growth. Lee
et al(35) showed that the
introduction of IL-8, a chemo-attractant for neutrophils, basophils
and a subset of T-cells, into human ovarian tumors resulted in
massive granulocytic and monocytic infiltration and subsequent
reduction or elimination of tumor growth. Very high levels of IL-8
can be associated with increased risk of death (36) (in which cases, PBIT but not PBI-Se
might be efficacious in decreasing the toxicity). IL-8 is an
important mediator of the innate immune system response and
maintenance of this chemokine by PBI-Se but not PBIT in the
presence of LPS supports previous findings of the beneficial
effects of selenium on the immune system. The results also are
consistent with the observation that people with acute and severe
illness who develop inflammation and widespread infection often
have decreased levels of selenium in their blood (37).
A more dramatic difference between PBI-Se and PBIT
was observed in their effects on IL-6 production. PBIT decreased
both LPS- and cytokine-stimulated IL-6 production. At comparable
growth inhibitory concentrations, PBI-Se had a significant effect
on the large LPS-induced IL-6 production and actually increased the
lesser cytokine-induced IL-6 production. IL-6 is one of the major
mediators of the inflammatory response and is implicated in
inflammation, tumor growth and angiogenesis. In patients with
active ulcerative colitis, there are increased concentrations of
IL-6 and IL-8 in the mucosa (38).
These results suggest that PBI-Se is able to inhibit only the
LPS-induced activation pathway. These divergent effects of PBIT and
PBI-Se on IL-6 production require further investigation but suggest
that PBI-Se and PBIT have different mechanisms of action and may
have different chemopreventive/therapeutic niches. Blunting
production of proinflammatory cytokines and/or enhancing
anti-inflammatory factors to obtain a balance may become very
important. Our results confirm previous findings that show that
PBIT suppressed the inducible isoforms of NOS and cyclooxygenase
(COX) and also that PBI-Se was effective in inhibiting both iNOS
and cyclin D1. These two agents demonstrate that they reduce
inflammatory responses in intestinal epithelial cells.
There is a great deal of evidence indicating that
selenium supplementation at high levels reduces the incidence of
cancer in animals. More than two-thirds of over 100 published
studies in 20 different animal models of spontaneous, viral, and
chemically induced cancers found that selenium supplementation
significantly reduces tumor incidence (39). Observational studies indicate that
death from cancer, including lung, colorectal and prostate cancers,
is lower among people with higher blood levels or intake of
selenium (40–46). The incidence of prostate, colorectal
and lung cancer was notably lower in the group given selenium
supplements (47). In the present
study, PBI-Se was more potent than PBIT at inhibiting Caco2 growth
in vitro and administration of PBI-Se provided up to 41%
inhibition of AOM-induced total ACF formation, and suppression in
growth of foci with 4 or more crypts by ≥47%. These results clearly
support the potential colon tumor inhibitory properties of PBI-Se
(Fig. 4A and B). However, a
previous study showed that dietary administration of Se-free PBIT
at 50 ppm could suppress the total number of ACF/colon by ~58% and
aberrant crypt multiplicities by 78%, compared with a control diet
(18). Although differential
inhibition of IL-6 and IL-8 by the two inhibitors may contribute to
the apparent reversal in order of efficacy of the two iNOS
inhibitors in vivo, the difference may be due to different
experimental designs. In the present study, animals were fed PBI-Se
after the AOM injections, whereas in the previous study, PBIT was
administered before AOM treatments. Also, the PBI-Se dose used here
post-initiation was 50% less than the dose of PBIT used previously
in the chemoprevention protocol. Our pre-clinical results are
consistent with previous clinical observations with other forms of
selenium (selenomethionine) showing lowered risk of developing
prostate, lung and colorectal cancer in a large scale cancer
prevention trial (48). Although
the exact mechanism by which PBI-Se acts cannot be determined, the
present findings highlight the need for further research on the
potential interactions between epithelial cells and immune cells
under different inflammatory conditions with activation of varied
signaling pathways.
Certain breakdown products of selenium have been
suggested to prevent tumor growth by enhancing immune cell activity
and suppressing development of blood vessels to the tumor (49). There is accumulating evidence that
selenium deficiency may contribute to the development of a form of
heart disease, hypothyroidism, and a weakened immune system
(50,51). At the doses used, the selenium in
PBI-Se may have enhanced immune system function and affected
carcinogen metabolism. Selenium is an essential component of
thyroid metabolism and antioxidant defense, as well as immune
function. It appears to improve activation and proliferation of
B-lymphocytes and to enhance T-cell function. It is also involved
in several key metabolic activities through selenoprotein enzymes
that protect against oxidative damage (52). In one randomized study of
free-living, healthy humans (57–84 years of age), those given 400
mcg/day of selenium for 6 months had a 65% increase in T cells,
particularly CD4 cells, and a 58% increase in NK cell cytotoxicity
(53). Studies on the effects of
selenium deficiency on HIV connected selenium levels to T cell
function and apoptosis suggesting that selenium may enhance
resistance to infection by modulating IL production and T helper
cell responses (54). The increased
number of PMNs in the PBI-Se-treated rat spleens suggests an effect
on immune cells which may have an important role in inducing
apoptotic effects in the colon cancer cells.
Recently, a large randomized, placebo-controlled
intervention study, the SELECT study, found that 200 mcg/day of
selenium did not alter the risk of prostate cancer (55). However, a phase III clinical trial
at NIH using selenium for chemoprevention therapy to try to prevent
the development of neoplasia in the prostate is underway. Animal
studies suggest that mammary tumors are significantly reduced by
selenium, and a study in women that should yield more definitive
information on this relationship is presently underway (56). The present study shows that PBI-Se
is a more potent growth inhibitory agent in vitro than its
isosteric sulfur analog PBIT and that it does not decrease
cytokine- or LPS-stimulated IL-8 production. Both in vivo
and in vitro data support the development of PBI-Se for
prevention and treatment of colon cancer.
Acknowledgements
The authors thank Dr Julie Sando for her valuable
suggestions and for editing the article. The authors also thank the
OUHSC Rodent Barrier Facility staff for their support for bioassay
studies.
References
|
1
|
American Cancer Society. Colorectal cancer
key statistics. Available at: http://www.cancer.org/Cancer/ColonandRectumCancer/DetailedGuidecolorectal-cancer-key-statistics.
|
|
2
|
Jackson JR, Seed MP, Kircher CH,
Willoughby DA and Winkler JD: The codependence of angiogenesis and
chronic inflammation. FASEB J. 11:457–465. 1997.PubMed/NCBI
|
|
3
|
Phoa N and Epe B: Influence of nitric
oxide on the generation and repair of oxidative DNA damage in
mammalian cells. Carcinogenesis. 23:469–475. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jaiswal M, LaRusso NF, Burgart LJ and
Gores GJ: Inflammatory cytokines induce DNA damage and inhibit DNA
repair in cholangiocarcinoma cells by a nitric oxide-dependent
mechanism. Cancer Res. 60:184–190. 2000.PubMed/NCBI
|
|
5
|
Moore MA: Cytokine and chemokine networks
influencing stem cell proliferation, differentiation, and marrow
homing. J Cell Biochem. 38:29–38. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nakajima N, Kuwayama H, Ito Y, Iwasaki A
and Arakawa Y: Helicobacter pylori, neutrophils,
interleukins, and gastric epithelial proliferation. J Clin
Gastroenterol. 25:S198–S202. 1997. View Article : Google Scholar
|
|
7
|
Eberhart CE, Coffey RJ, Radhika A,
Giardiello FM, Ferrenbach S and DuBois RN: Up-regulation of
cyclooxygenase 2 gene expression in human colorectal adenomas and
adenocarcinomas. Gastroenterology. 107:1183–1188. 1994.PubMed/NCBI
|
|
8
|
Rhodes JM and Campbell BJ: Inflammation
and colorectal cancer: IBD-associated and sporadic cancer compared.
Trends Mol Med. 8:10–16. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Keshavarzian A, Fusunyan RD, Jacyno M,
Winship D, MacDermott RP and Sanderson IR: Increased interleukin-8
(IL-8) in rectal dialysate from patients with ulcerative colitis:
evidence for a biological role for IL-8 in inflammation of the
colon. Am J Gastroenterol. 94:704–712. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nusrat A, Sitaraman SV and Neish A:
Interaction of bacteria and bacterial toxins with intestinal
epithelial cells. Curr Gastroenterol Rep. 3:392–398. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weiss HA and Forman D: Aspirin,
non-steroidal anti-inflammatory drugs and protection from
colorectal cancer: a review of the epidemiological evidence. Scand
J Gastroenterol. 220:137–141. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jenkins DC, Charles IG, Thomsen LL, et al:
Roles of nitric oxide in tumour growth. Proc Natl Acad Sci USA.
92:4392–4396. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ambs S, Merriam WG, Bennett WP, et al:
Frequent nitric oxide synthase-2 expression in human colon
adenomas: implication for tumour angiogenesis and colon cancer
progression. Cancer Res. 58:334–341. 1998.PubMed/NCBI
|
|
14
|
Jobin C, Haskill S, Mayer L, Panja A and
Sartor RB: Evidence for altered regulation of I kappa B alpha
degradation in human colonic cells. J Immunol. 158:226–234.
1997.PubMed/NCBI
|
|
15
|
Knowles RG and Moncada S: Nitric oxide
synthases in mammals. Biochem J. 298:249–258. 1994.PubMed/NCBI
|
|
16
|
Forstermann U, Gath I, Schwarz P, Closs EI
and Kleinert H: Isoforms of nitric oxide synthase. Properties,
cellular distribution and expressional control. Biochem Pharmacol.
50:1321–1332. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pana MH and Ho CT: Chemopreventive effects
of natural dietary compounds on cancer development. Chem Soc Rev.
37:2558–2574. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rao CV, Kawamori T, Hamid R and Reddy BS:
Chemoprevention of colonic aberrant crypt foci by an inducible
nitric oxide synthase-selective inhibitor. Carcinogenesis.
2:641–644. 1999.PubMed/NCBI
|
|
19
|
Briske-Anderson MJ, Finley JW and Newman
SM: The influence of culture time and passage number on the
morphological and physiological development of CaCo2 cells. Proc
Soc Exp Biol Med. 214:248–257. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang Y, Li N, Liboni K and Neu J:
Glutamine decreases lipopolysaccharide-induced IL-8 production in
Caco-2 cells through a non-NF-kappaB p50 mechanism. Cytokine.
22:77–83. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chittezhath M, Deep G, Singh RP, Agarwal C
and Agarwal R: Silibinin inhibits cytokine-induced STATs, MAPKs,
NF-κB and AP-1 activation, and down-regulates HIF-1α and iNOS in
human lung carcinoma A549 cells. Mol Cancer Ther. 7:1817–1826.
2008.
|
|
22
|
Bird RP: Observation and quantification of
aberrant crypts in the murine colon treated with a colon
carcinogen: preliminary findings. Cancer Lett. 37:147–151. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Janakiram NB, Mohammed A, Zhang Y, Choi
CI, Woodward C, Collin P, Steele VE and Rao CV: Chemopreventive
effects of Frondanol A5, a Cucumaria frondosa extract,
against rat colon carcinogenesis and inhibition of human colon
cancer cell growth. Cancer Prev Res. 3:82–91. 2010.PubMed/NCBI
|
|
24
|
Arthur JR, McKenzie RC and Beckett GJ:
Selenium in the immune system. J Nutr. 133:S1457–S1459. 2003.
|
|
25
|
Chung CY, Madhunapantula SV, Desai D, Amin
S and Robertson GP: Melanoma prevention using topical PBI-Se.
Cancer Prev Res. 4:935–948. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: a review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fusunyan RD, Quinn JJ, Fujimoto M,
MacDermott RP and Sanderson IR: Butyrate switches the pattern of
chemokine secretion by intestinal epithelial cells through histone
acetylation. Mol Med. 5:631–640. 1999.PubMed/NCBI
|
|
28
|
Ogle CK, Guo XL, Hasselgren PO, Ogle JD
and Alexander JW: The gut as a source of inflammatory cytokines
after stimulation with endotoxin. Eur J Surg. 163:45–51.
1997.PubMed/NCBI
|
|
29
|
Haller D, Bode C, Hammes WP, Pfeifer AMA,
Schiffrin EJ and Blum S: Non-pathogenic bacteria elicit a
differential cytokine response by intestinal epithelial
cell/leucocyte co-cultures. Gut. 47:79–87. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kibriya MG, Jasmine F, Argos M, Verret WJ,
Rakibuz-Zaman M, Ahmed A, Parvez F and Ahsan H: Changes in gene
expression profiles in response to selenium supplementation among
individuals with arsenic-induced pre-malignant skin lesions.
Toxicol Lett. 169:162–176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Terui Y, Ikeda M, Tomizuka H, et al:
Identification of a novel apoptosis-inducing factor derived from
leukemic cells: endothelial interleukin-8, but not
monocyte-derived, induces apoptosis in leukemic cells. Biochem
Biophys Res Commun. 243:407–411. 1998. View Article : Google Scholar
|
|
32
|
Terui Y, Ikeda M, Tomizuka H, et al:
Activated endothelial cells induce apoptosis in leukemic cells by
endothelial interleukin-8. Blood. 92:2672–2680. 1998.PubMed/NCBI
|
|
33
|
Hoffmann E, Dittrich-Breiholz O, Holtmann
H and Kracht M: Multiple control of interleukin-8 gene expression.
J Leukoc Biol. 72:847–855. 2002.PubMed/NCBI
|
|
34
|
Garat C and Arend WP: Intracellular IL-1Ra
type 1 inhibits IL-1-induced IL-6 and IL-8 production in Caco-2
intestinal epithelial cells through inhibition of p38
mitogen-activated protein kinase and NF-kappaB pathways. Cytokine.
23:31–40. 2003. View Article : Google Scholar
|
|
35
|
Lee Li-F, Hellendall RP, Wang Y, Stephen
Haskill J, Mukaida N, Matsushima K and Ting JP: IL-8 reduced
tumorigenicity of human ovarian cancer in vivo due to neutrophil
infiltration. J Immunol. 164:2769–2775. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Molica S, Vitelli G, Levato D, Levato L,
Datillo A and Gandolfo GM: Clinico-biological implications of
increased serum levels of interleukin −8 in B cell chronic
lymphocytic leukemia. Haematologica. 84:208–211. 1999.PubMed/NCBI
|
|
37
|
Gartner R, Albrich W and Angstwurm MW: The
effect of a selenium supplementation on the outcome of patients
with severe systemic inflammation, burn, and trauma. Biofactors.
14:199–204. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Murata Y, Ishiguro Y, Itoh J, Munakata A
and Yoshida Y: The role of pro-inflammatory and immunoregulatory
cytokines in the pathogenesis of ulcerative colitis. Gastroenterol
Clin North Am J. 30:56–60. 1995.
|
|
39
|
Combs GF Jr and Gray WP: Chemopreventive
agents: selenium. Pharmacol Ther. 79:179–192. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Russo MW, Murray SC, Wurzelmann JI,
Woosley JT and Sandler RS: Plasma selenium levels and the risk of
colorectal adenomas. Nutr Cancer. 28:125–129. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Patterson BH and Levander OA: Naturally
occurring selenium compounds in cancer chemoprevention trials: a
workshop summary. Cancer Epidemiol Biomarkers Prev. 6:63–69.
1997.PubMed/NCBI
|
|
42
|
Knekt P, Marniemi J, Teppo L, Heliovaara M
and Aromaa A: Is low selenium status a risk factor for lung cancer?
Am J Epidemiol. 148:975–982. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fleet JC: Dietary selenium repletion may
reduce cancer incidence in people at high risk who live in areas
with low soil selenium. Nutr Rev. 55:277–279. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shamberger RJ: The genotoxicity of
selenium. Mutat Res. 154:28–48. 1985.
|
|
45
|
Young KL and Lee PN: Intervention studies
on cancer. Eur J Cancer Prev. 8:91–103. 1999. View Article : Google Scholar
|
|
46
|
Burguera JL, Burguera M, Gallignani M,
Alarcon OM and Burgueera JA: Blood serum selenium in the province
of Merida, Venezuela, related to sex, cancer incidence and soil
selenium content. J Trace Elem Electrolytes Health Dis. 4:73–77.
1990.PubMed/NCBI
|
|
47
|
Combs GF Jr, Clark LC and Turnbull BW:
Reduction of cancer risk with an oral supplement of selenium.
Biomed Environ Sci. 10:227–234. 1997.PubMed/NCBI
|
|
48
|
Clark LC, Combs GF Jr, Turnbull BW, et al:
Effects of selenium supplementation for cancer prevention in
patients with carcinoma of the skin. A randomized controlled trial.
JAMA. 276:1957–1963. 1996. View Article : Google Scholar
|
|
49
|
Combs GF Jr, Clark LC and Turnbull BW: An
analysis of cancer prevention by selenium. Biofactors. 14:153–159.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Combs GF Jr: Food system-based approaches
to improving micronutrient nutrition: the case for selenium.
Biofactors. 12:39–43. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zimmerman MB and Kohrle J: The impact of
iron and selenium deficiencies on iodine and thyroid metabolism:
biochemistry and relevance to public health. Thyroid. 12:867–878.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ryan-Harshman M and Aldoori W: The
relevance of selenium to immunity, cancer and
infectious/inflammatory diseases. Can J Diet Pract Res. 66:98–102.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wood SM, Beckham C, Yosioka A, Darban H
and Watson RR: Beta-carotene and selenium supplementation enhances
immune response in aged humans. Integr Med. 2:85–92. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Baum MK, Miguez-Burbano MJ, Campa A and
Shor-Posner G: Selenium and interleukins in persons infected with
human immunodeficiency virus type 1. J Infect Dis. 182:S69–S73.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lippman SM, Klein EA, Goodman PJ, et al:
Effect of selenium and vitamin E on risk of prostate cancer and
other cancers: the Selenium and Vitamin E Cancer Prevention Trial
(SELECT). JAMA. 301:39–51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Whanger PD: Selenium and its relationship
to cancer: an update. Br J Nutr. 91:11–28. 2004. View Article : Google Scholar : PubMed/NCBI
|