Introduction
Ascorbic acid (AsA) (also known as vitamin C)
therapy has been considered a therapeutic option for cancer, and
has few side-effects when administered intravenously in
pharmacologic concentrations (1).
Chen et al reported that AsA is selectively toxic for some
cancer cells but it is not toxic to normal cells (2). AsA exhibits cytotoxic effects in tumor
cells, which have a low concentration of intracellular catalase
that degrades hydrogen peroxide (H2O2)
(3). Studies have shown that tumor
cells are easily damaged by H2O2, and
production of the adenosine triphosphate (ATP) is decreased due to
mitochondrial damage, thus leading to tumor cell death (3–7).
H2O2 is produced during radiation therapy and
some antineoplastic drugs kill the tumor cell by its cytotoxic
activity (8). The generation of
reactive oxygen species (ROS) derived from
H2O2 is thought to be involved in the
cytotoxicity. Vitamin C therapy can be used alone or in combination
with chemotherapy (9–11). AsA in combination with radiation
therapy is also expected to be effective in cancer therapy since it
is considered to have few side-effects (12,13).
Koyama et al reported that AsA does not inhibit the fatal
effects of radiation, but inhibits carcinogenesis and mutation
(30). Therefore, AsA may reduce
the risk of a second cancer in normal cells during combined AsA and
radiation therapy.
AsA is also known as a radical scavenger (14,15),
and it scavenges O2•−, • OH,
1O2, and NO under in vitro conditions;
thus, it can scavenge ROS generated by antineoplastic drugs or
X-irradiation (16). Therefore,
conflicting data exist regarding AsA inhibiting the cytotoxic
effects generated by the action of antineoplastic drugs or
X-irradiation (17–21).
The majority of cell deaths induced by X-irradiation
depend on the production of intracellular ROS, which is generated
during irradiation. Within several hours after irradiation,
secondary ROS production occurs intracellularly, and it induces
apoptosis (22,23). Mitochondria are well known as a
major source of intracellular ROS and produce ROS during
intracellular ATP synthesis. Therefore, the source of secondary ROS
as a result of irradiation is thought to be the mitochondria
(22–24). The generation of ROS from
mitochondria and the loss of the mitochondrial membrane potential
play an important role in inducing cell death (23,25,26).
In this study, we examined the mechanism underlying
cell death caused by a combination treatment of AsA and
X-irradiation from the viewpoint of ROS generation by using HL-60
human promyelocytic leukemia cells and we clarified that AsA does
not inhibit the cytotoxic effects of X-irradiation.
Materials and methods
Cell culture, X-irradiation, and drug
treatment
The HL-60 human promyelocytic leukemia cell line
(RIKEN BioResource Center, Tsukuba, Japan) was used in these
experiments. Cells were cultured in RPMI-1640 (Gibco, Grand Island,
NY, USA) supplemented with 10% fetal bovine serum and 1%
penicillin-streptomycin, and were maintained at 37°C with 95% air
and 5% CO2. The passage duration was 3–5 days, and the
density was not allowed to exceed 1×106 cells/ml.
X-irradiation was delivered using an X-ray machine MBR-1520R-3
(Hitachi, Tokyo, Japan) at 150 kV and 20 mA through a 0.5-mm Al and
0.3-mm Cu filter at a dose rate of 1.0 Gy/min. L(+)-ascorbic acid
was purchased from Wako (Tokyo, Japan). The AsA was dissolved in
RPMI-1640 medium and deacidified with sodium hydrate before
treatment. Catalase (Sigma Aldrich, St. Louis, MO, USA) was added
to the culture to achieve a final concentration of 1,300 U/ml.
Cell viability assay
HL-60 cells (4.0×105 cells/ml) were
cultured for 6 h. A final concentration of 0.01–10.0 mM AsA was
then added to the culture and the number of viable cells was
counted using trypan blue dye exclusion method after 24 h. For the
next experiment, 4×105 cells/ml HL-60 cells were
cultured with or without catalase for 6 h, and 1.0 or 2.5 mM AsA
was added to the cells in combination with 2 Gy X-irradiation. The
viable cells were counted by trypan blue dye exclusion method after
24 h.
Measurement of intracellular ROS
Subsequently, HL-60 cells (1.5×105
cells/ml) were cultured with or without catalase for 6 h, and 2.5
mM AsA was added to the cells in combination with 2 Gy
X-irradiation. The intracellular ROS production was measured using
a flow cytometer (Cytomics FC500, Beckman Coulter, Fullerton, CA)
using the ROS-sensitive probe 2′,7-dichlorofluorescin diacetate
(H2DCFDA; Molecular Probes, Invitrogen Corp., CA, USA)
at the indicated times after exposure to X-irradiation. The cells
were washed with phosphate buffered saline without Ca2+
and Mg2+ [PBS(−)], incubated at 37°C with 5 μM
H2DCFDA in PBS(−) for 15 min, washed in PBS(−), and then
resuspended in PBS(−) containing 5 mg/l propidium iodide (PI; Sigma
Aldrich) to exclude dead cells. Sample data were analyzed using
FlowJo software (Treestar, Inc., San Carlos, CA, USA). The median
H2DCFDA fluorescence intensity of each sample was
normalized to that of control sample to calculate the relative
H2DCFDA intensity.
For the precise evaluation of ROS production
immediately following X-irradiation, we labeled the cells with
H2DCFDA prior to AsA and X-irradiation treatments
(27,28). In brief, cells were incubated at
37°C with 5 μM H2DCFDA in PBS(−) for 15 min. After
labeling, the cells were treated with AsA and/or X-irradiation in
the presence of H2DCFDA, washed in PBS(−), and then
resuspended in PBS(−) containing 5 mg/l PI. Samples were analyzed
using a flow cytometer immediately after the treatment.
Measurement of mitochondrial superoxide
and mitochondrial membrane potential
The cells were treated with AsA and/or X-irradiation
as described above without the addition of catalase. The
mitochondrial superoxide levels were measured using the flow
cytometer with mitochondrial superoxide indicator MitoSOX Red
(Molecular Probes, Invitrogen Corp.) at the indicated times after
exposure to X-irradiation. The cells were washed with PBS(−),
incubated at 37°C with 5 μM MitoSOX Red in Hanks’ Balanced Salt
Solutions (HBSS; with Ca2+ and Mg2+) for the
15 min, and then washed and resuspended in PBS(−). Sample data were
analyzed using the FlowJo software. The median MitoSOX Red
fluorescence intensity of each sample was normalized to that of
control sample to calculate the relative MitoSOX Red intensity.
The changes in the mitochondrial membrane potential
were measured using the flow cytometer with
3,3′-dihexyloxacarbocyanine iodide [DiOC6(3)]; Molecular Probes, Invitrogen Corp.) at
indicated times after exposure to X-irradiation. The cells were
washed with PBS(−), incubated at 37°C with 40 nM H2DCFDA
in HBSS for 15 min, washed in HBSS, and then resuspended in
serum-free RPMI-1640 medium containing 5 mg/l PI to exclude dead
cells. Sample data were analyzed using FlowJo software. The median
DiOC6(3) fluorescence intensity of
each sample was normalized to that of control sample to calculate
the relative DiOC6(3)
intensity.
Statistical analysis
Statistical comparisons were performed using the
Tukey-Kramer test. All results are presented as the mean ± SD from
the results of at least three independent experiments. p-values
>0.01 or 0.05 were considered to indicate statistically
significant differences. Statistical analysis was performed using
the Excel 2007 software program (Microsoft, USA) with Statcel 2
add-in software (29).
Results
Cell death by AsA treatment and
X-irradiation
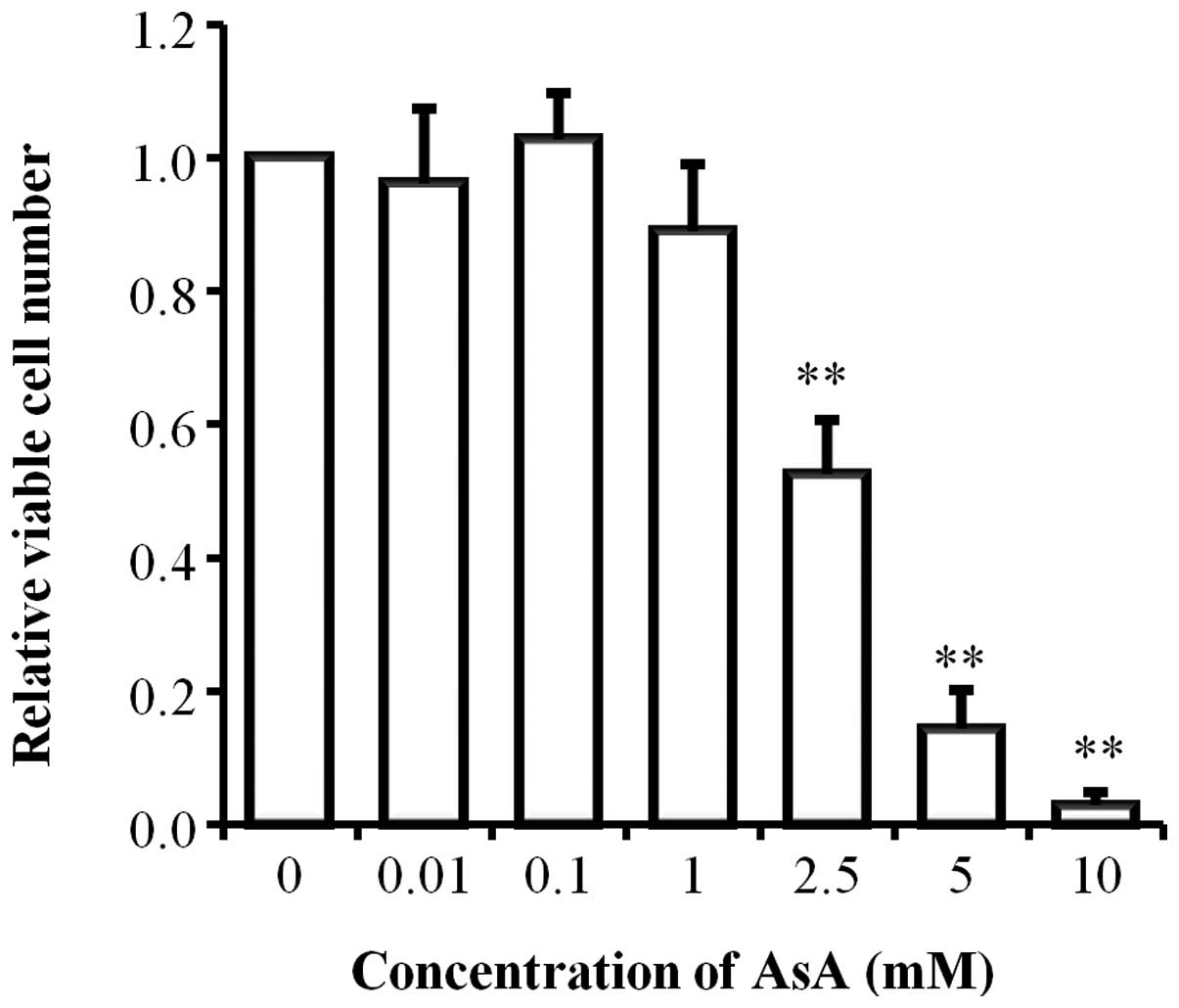
In the initial experiment, we examined the AsA
sensitivity of HL-60 cells. After AsA treatment for 24 h, we
counted the viable HL-60 cells by trypan blue dye exclusion method.
AsA showed cytotoxic effects on the growth of HL-60 cells in a
dose-dependent manner from ~1 mM concentration (Fig. 1). Enhanced cell growth was not
observed in AsA treatment at low concentrations (0.01 or 0.1
mM).
Additive cytotoxic effects were observed when the
cells were exposed to 2 Gy X-irradiation after 2.5 mM AsA treatment
(Fig. 2). When 2.5 mM AsA and 2 Gy
X-irradiation were used in combination, a significant decrease in
the relative viable cell number was observed when compared to
application of 2 Gy X-irradiation alone (p<0.05). Fig. 2 also shows the cytotoxic effect of
AsA alone and in combination with X-irradiation in the presence of
catalase. When AsA was added to the culture in the presence of
catalase, the cytotoxic effects of AsA disappeared. Moreover, the
additive cytotoxic effects of 2.5 mM AsA and 2 Gy X-irradiation in
combination decreased to the same level as those obtained by 2 Gy
X-irradiation alone.
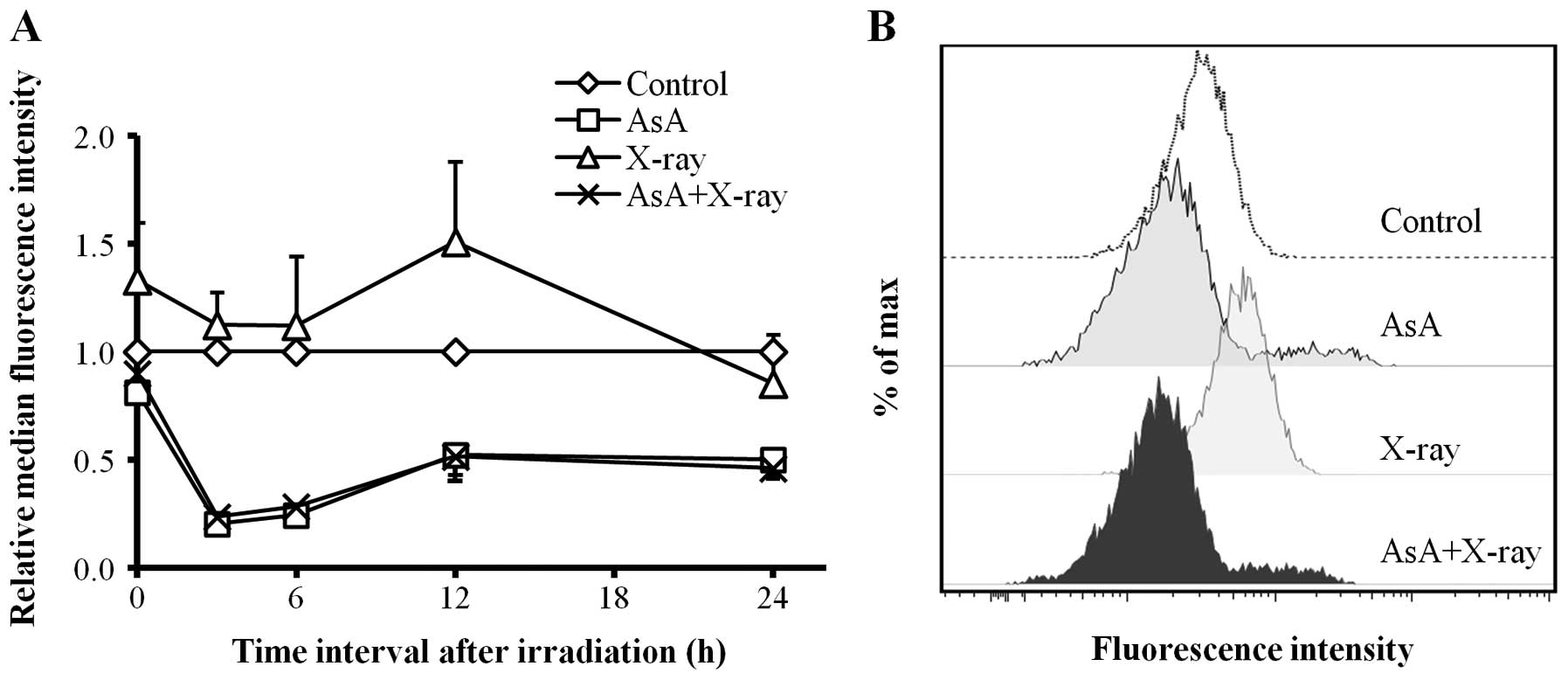
Kinetics of intracellular ROS
Fig. 3A shows the
changes in intracellular ROS levels as analyzed using a flow
cytometer. In cells treated with X-irradiation alone (2 Gy), the
intracellular ROS levels increased and reached a peak at 12 h
(p<0.01) compared to control, and decreased slowly thereafter.
By contrast, treatment with AsA alone (2.5 mM) and in combination
with X-irradiation (2 Gy) significantly decreased intracellular ROS
levels at 3–24 h after X-irradiation. A representative histogram of
DCF fluorescence intensity at 12 h is shown in Fig. 3B.
Fig. 3C shows the
changes in the intracellular ROS levels in the presence of
catalase. The intracellular ROS levels of X-irradiated cells
increased slightly and reached a peak after 6 h, but there was no
significant difference when compared to control cells. The ROS
levels of cells treated with AsA alone and in combination with
X-irradiation were also not significantly altered compared to those
of the control cells. A representative histogram of DCF
fluorescence intensity at 6 h is shown in Fig. 3D.
In the absence of catalase, the ROS levels of cells
treated with a combination of AsA and X-irradiation were
significantly higher than those of the control cells at 0 h after
X-irradiation, and this difference was statistically significant
(p<0.05). For the precise evaluation of ROS production
immediately following X-irradiation, we labeled the cells with
H2DCFDA prior to AsA and X-irradiation treatment. The
ROS levels of cells treated with AsA alone and in combination with
X-irradiation significantly increased immediately following
X-irradiation, compared to control cells (Fig. 4A and B).
Kinetics of mitochondrial superoxide and
mitochondrial membrane potential
The changes in mitochondrial superoxide levels were
analyzed by flow cytometry (Fig.
5A). When X-irradiation was used alone, mitochondrial
superoxide levels increased slightly and reached a peak at 12 h
(p<0.01) compared to control (Fig.
5B). By contrast, AsA alone and in combination with
X-irradiation significantly decreased the mitochondrial superoxide
levels at 3–24 h after X-irradiation. These changes were similar to
the changes in the intracellular ROS levels.
In order to investigate whether AsA scavenged
intracellular ROS or whether generation of ROS from mitochondria
was decreased, we used DiOC6(3) (a
carbocyanine dye that accumulates in active mitochondria) to
measure the mitochondrial membrane potential by flow cytometry.
When X-irradiation was used alone, the mitochondrial membrane
potential increased slightly and reached a peak at 12 h (p<0.01)
compared to control (Fig. 6A).
Contrary to this, in the presence of only AsA or in combination
with X-irradiation, the mitochondrial membrane potential gradually
decreased and became ~40% of control value at 12 h after
X-irradiation (Fig. 6B).
Discussion
In the present study, we described the potential of
AsA and X-irradiation combination treatment, particularly against
intracellular ROS in HL-60 cells. We demonstrated that AsA, a
radical scavenger, did not inhibit the cytotoxic effect when used
in combination with X-irradiation although it resulted in
intracellular ROS reduction.
In this study, additive cytotoxic effects were
observed when the cells were exposed to 2 Gy X-irradiation after
2.5 mM AsA treatment. When a combination of 1 mM AsA and 2 Gy
X-irradiation was applied, the protective effect of AsA against 2
Gy X-irradiation was not observed and no significant cytotoxic
effects were found. These results are consistent with the studies
that AsA does not inhibit the fatal effects of radiation (12,30).
When AsA was added to the culture in the presence of catalase, the
cytotoxic effects of AsA disappeared. Moreover, the additive
cytotoxic effects decreased to the same level obtained by
X-irradiation alone. These results suggest that the action pathway
of hydroxyl radicals derived from H2O2 is
different in AsA and X-irradiation treatments. Catalase, being a
large protein, does not penetrate cell membranes and, therefore, it
is not taken up by cells. Catalase neutralizes the
H2O2 derived from AsA in the extracellular
fluids. Therefore, it is considered that the cytotoxic effect of
extracellular H2O2 that AsA generates is much
more effective than the cytotoxic effect of intracellular AsA
(3,31).
Our present study showed that AsA significantly
decreased intercellular ROS production. Hence, such a result might
indicate that combined AsA and X-irradiation treatment may not be
effective as cell death due to signal transduction by ROS is
inhibited (20,21). In AsA treatment, a significant
change in the intercellular ROS level was not observed in the
presence of catalase as compared with the significant reduction of
intracellular ROS in the absence of catalase. For this reason, our
results suggest that the slight change in intracellular ROS is due
to the neutralizing effect of extracellular
H2O2 by catalase in preference to scavenging
intercellular ROS by AsA. It is thought that extracellular
H2O2 generated by AsA treatment might mainly
decrease intercellular ROS or both cytotoxic effects and radical
scavenger are necessary for significant reduction of ROS. Some
studies have reported that antineoplastic drugs exhibit cytotoxic
effects with reduction of ROS; hence, it showed the ability of
scavenging ROS such as AsA (32,33).
It was also reported that intercellular ROS slightly decreased when
cells were treated with H2O2(34).
When AsA alone and in combination with X-irradiation
was used, large quantities of intracellular ROS were observed
immediately following AsA and X-irradiation treatment, which was
observed by labeling the cells with H2DCFDA prior to
treatment. It is reported that H2O2
generation was dependent on the presence of trace amounts of serum
in media (2). When the cells were
treated with AsA and/or X-irradiation in the presence of
H2DCFDA in 10% FBS growth medium, but not in PBS,
~12-fold ROS production in the control was observed for AsA alone
and in combination with X-irradiation (data not shown). Since
H2O2 can easily permeate cell membranes
(35), large quantities of
H2O2 derived from AsA might damage HL-60
cells immediately following AsA treatment, after which the
intercellular ROS production is decreased. Frömberg et al
showed that AsA or dehydroascorbate (DHA) is important for
cytotoxic efficiency in the redox state of vitamin C, and they
report higher therapeutic efficacy of AsA over DHA in various cell
lines (31). Furthermore, it was
reported that tumor cells take up DHA, but not AsA, in large
quantities. However, a moderate change in the intracellular ROS
levels was observed in the order of mM DHA (18), although DHA turn into AsA in cells
(16), contrary to our results of
AsA treatment. Therefore, the redox state of vitamin C agents may
lead to contradictory results in vitamin C treatment.
Mitochondria release cytochrome c, which
activates caspase for apoptosis, leading to changes in
mitochondrial respiratory chain, and the mitochondrial membrane
potential is depolarized. Therefore, mitochondrial membrane
potential is used as an indicator for evaluating cell life and
death (36). In our study,
mitochondrial membrane potential gradually disappeared in cells
treated with AsA. However, it is reported that an increase in
intracellular ROS is observed when the mitochondrial membrane
potential is decreased by several antineoplastic agents (37). Some studies reported the existence
of ROS-independent mitochondrial pathway, since reduction of the
mitochondrial membrane potential is observed without an increase in
intracellular ROS (32,33,38,39).
AsA might induce apoptosis in HL-60 cells through a ROS-independent
mitochondrial pathway as intracellular ROS was significantly
decreased as compared with the control, and a reduction in the
mitochondrial membrane potential was observed in AsA treatment.
Some studies reported that the antineoplastic agent
that has the ability of scavenging ROS induces apoptosis through a
ROS-independent mitochondrial pathway with reduction in ROS
(32,33). Furthermore, an increase in
intracellular ROS and superoxide levels derived from mitochondria
was reported and mitochondrial membrane potential hyperpolarization
was observed after X-ray irradiation. It is thought that
X-irradiation arrests cell cycle, inhibits cell division and
increases in the mitochondrial content, leading to the activation
of mitochondrial respiratory chain, resulting in the increase of
mitochondrial ROS (24,40). Our data are consistent with these
studies, but changes in the X-ray irradiated cells were contrary to
the changes in cells treated with AsA. When a combination of AsA
and X-irradiation was used, ROS levels decreased to the same level
obtained by AsA alone, but AsA did not inhibit the cytotoxic
effects of X-irradiation. While considering ROS generation, these
indicate that additive cytotoxic effects were observed since AsA
and X-irradiation follow different signaling pathways. Shinozaki
et al reported that the involvement of Bax and caspase 8
were different following X-irradiation or AsA treatment alone as
compared with those following combined X- irradiation and AsA
treatment against the apoptosis mechanism (12).
In the present study, we examined the mechanism
underlying cell death caused by a combination of AsA and
X-irradiation from the viewpoint of ROS generation by HL-60 cells
to reveal the clinical possibility of a combination therapy. The
combination decreased intracellular ROS generation, but additive
cytotoxic effects and reduction of mitochondrial membrane potential
were observed in cells. These results suggest that AsA, which is a
radical scavenger, did not exert protective effects against ROS
production by X-irradiation and the signaling pathway in
mitochondria was different for AsA and X-irradiation. Our results
suggest that combination therapy of AsA and X-irradiation does not
have an effect on cancer cell death while considering ROS
generation.
References
|
1
|
Padayatty SJ, Sun AY, Chen Q, Espey MG,
Drisko J and Levine M: Vitamin C: intravenous use by complementary
and alternative medicine practitioners and adverse effects. PLoS
One. 5:e114142010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen Q, Espey MG, Krishna MC, et al:
Pharmacologic ascorbic acid concentrations selectively kill cancer
cells: action as a pro-drug to deliver hydrogen peroxide to
tissues. Proc Natl Acad Sci USA. 102:13604–13609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen Q, Espey MG, Sun AY, et al: Ascorbate
in pharmacologic concentrations selectively generates ascorbate
radical and hydrogen peroxide in extracellular fluid in vivo. Proc
Natl Acad Sci USA. 104:8749–8787. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takemura Y, Satoh M, Satoh K, Hamada H,
Sekido Y and Kubota S: High dose of ascorbic acid induces cell
death in mesothelioma cells. Biochem Biophys Res Commun.
394:249–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hsieh BS, Huang LW, Su SJ, et al: Combined
arginine and ascorbic acid treatment induces apoptosis in the
hepatoma cell line HA22T/VGH and changes in redox status involving
the pentose phosphate pathway and reactive oxygen and nitrogen
species. J Nutr Biochem. 22:234–241. 2011. View Article : Google Scholar
|
|
6
|
Hampton MB and Orrenius S: Dual regulation
of caspase activity by hydrogen peroxide: implications for
apoptosis. FEBS Lett. 414:552–556. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hyslop PA, Hinshaw DB, Halsey WA Jr, et
al: Mechanisms of oxidant-mediated cell injury. The glycolytic and
mitochondrial pathways of ADP phosphorylation are major
intracellular targets inactivated by hydrogen peroxide. J Biol
Chem. 263:1665–1675. 1998.PubMed/NCBI
|
|
8
|
Lamson DW and Brignall MS: Antioxidants in
cancer therapy; their actions and interactions with oncologic
therapies. Altern Med Rev. 4:304–329. 1999.PubMed/NCBI
|
|
9
|
Padayatty SJ, Riordan HD, Hewitt SM, Katz
A, Hoffer LJ and Levine M: Intravenously administered vitamin C as
cancer therapy: three cases. CMAJ. 174:937–942. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Riordan HD, Casciari JJ, González MJ, et
al: A pilot clinical study of continuous intravenous ascorbate in
terminal cancer patients. PR Health Sci J. 24:269–276.
2005.PubMed/NCBI
|
|
11
|
Monti DA, Mitchell E, Bazzan AJ, et al:
Phase I evaluation of intravenous ascorbic acid in combination with
gemcitabine and erlotinib in patients with metastatic pancreatic
cancer. PLoS One. 7:e297942012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shinozaki K, Hosokawa Y, Hazawa M, et al:
Ascorbic acid enhances radiation-induced apoptosis in an HL60 human
leukemia cell line. J Radiat Res. 52:229–237. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Herst PM, Broadley KW, Harper JL and
McConnell MJ: Pharmacological concentrations of ascorbate
radiosensitize glioblastoma multiforme primary cells by increasing
oxidative DNA damage and inhibiting G2/M arrest. Free Radic Biol
Med. 52:1486–1493. 2012. View Article : Google Scholar
|
|
14
|
Rose RC and Bode AM: Biology of free
radical scavengers: an evaluation of ascorbate. FASEB J.
7:1135–1142. 1993.PubMed/NCBI
|
|
15
|
Li Y and Schellhorn HE: New developments
and novel therapeutic perspectives for vitamin C. J Nutr.
137:2171–2184. 2007.PubMed/NCBI
|
|
16
|
Reth M: Antioxidant defenses - endogenous
and diet derived. Free Radicals in Biology and Medicine. Halliwell
B and Gutteridge JMC: 4th edition. Oxford University Press; New
York: pp. 160–166. 2007
|
|
17
|
Lawenda BD, Kelly KM, Ladas EJ, Sagar SM,
Vickers A and Blumberg JB: Should supplemental antioxidant
administration be avoided during chemotherapy and radiation
therapy? J Natl Cancer Inst. 100:773–783. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Heaney ML, Gardner JR, Karasavvas N, et
al: Vitamin C antagonizes the cytotoxic effects of antineoplastic
drugs. Cancer Res. 68:8031–8038. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yamamoto T, Kinoshita M, Shinomiya N, et
al: Pretreatment with ascorbic acid prevents lethal
gastrointestinal syndrome in mice receiving a massive amount of
radiation. J Radiat Res. 51:145–156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Witenberg B, Kalir HH, Raviv Z, Kletter Y,
Kravtsov V and Fabian I: Inhibition by ascorbic acid of apoptosis
induced by oxidative stress in HL-60 myeloid leukemia cells.
Biochem Pharmacol. 57:823–832. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Witenberg B, Kletter Y, Kalir HH, et al:
Ascorbic acid inhibits apoptosis induced by X irradiation in HL60
myeloid leukemia cells. Radiat Res. 152:468–478. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee YJ, Lee DH, Cho CK, et al: HSP25
inhibits radiation-induced apoptosis through reduction of
PKCδ-mediated ROS production. Oncogene. 24:3715–3725.
2005.PubMed/NCBI
|
|
23
|
Ni Y, Gong XG, Lu M, Chen HM and Wang Y:
Mitochondrial ROS burst as an early sign in sarsasapogenin-induced
apoptosis in HepG2 cells. Cell Biol Int. 32:337–343. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ogura A, Oowada S, Kon Y, et al: Redox
regulation in radiation-induced cytochrome c release from
mitochondria of human lung carcinoma A549 cells. Cancer Lett.
277:64–71. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grad JM, Cepero E and Boise LH:
Mitochondria as targets for established and novel anti-cancer
agents. Drug Resist Updat. 4:85–91. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yoshino H, Kiminarita T, Matsushita Y and
Kashiwakura I: Response of the Nrf2 protection system in human
monocytic cells after ionising irradiation. Radiat Prot Dosimetry.
152:104–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hachiya M and Akashi M: Catalase regulates
cell growth in HL60 human promyelocytic cells: evidence for growth
regulation by H2O2. Radiat Res. 163:271–282.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yanai H: Statcel-The Useful Add-in
Software Forms on Excel. 2nd edition. OMS; Tokyo: 2006
|
|
30
|
Koyama S, Kodama S, Suzuki K, Matsumoto T,
Miyazaki T and Watanabe M: Radiation-induced long-lived radicals
which cause mutation and transformation. Mutat Res. 421:45–54.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Frömberg A, Gutsch D, Schulze D, et al:
Ascorbate exerts anti-proliferative effects through cell cycle
inhibition and sensitizes tumor cells towards cytostatic drugs.
Cancer Chemother Pharmacol. 67:1157–1166. 2011.PubMed/NCBI
|
|
32
|
Wang XH, Jia DZ, Liang YJ, et al: Lgf-YL-9
induces apoptosis in human epidermoid carcinoma KB cells and
multidrug resistant KBv200 cells via reactive oxygen
species-independent mitochondrial pathway. Cancer Lett.
249:256–270. 2007. View Article : Google Scholar
|
|
33
|
Zhang JY, Wu HY, Xia XK, et al:
Anthracenedione derivative 1403P-3 induces apoptosis in KB and
KBv200 cells via reactive oxygen species-independent mitochondrial
pathway and death receptor pathway. Cancer Biol Ther. 6:1413–1421.
2007. View Article : Google Scholar
|
|
34
|
Lin KT, Xue JY, Sun FF and Wong PY:
Reactive oxygen species participate in peroxynitrite-induced
apoptosis in HL-60 cells. Biochem Biophys Res Commun. 230:115–119.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Antunes F and Cadenas E: Estimation of
H2O2 gradients across biomembranes. FEBS
Lett. 475:121–126. 2000.
|
|
36
|
Griffiths EJ: Mitochondria-potential role
in cell life and death. Cardiovasc Res. 46:24–27. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hara K, Kasahara E, Takahashi N, et al:
Mitochondria determine the efficacy of anticancer agents that
interact with DNA but not the cytoskeleton. J Pharmacol Exp Ther.
337:838–845. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hou DX, Uto T, Tong X, et al: Involvement
of reactive oxygen species-independent mitochondrial pathway in
gossypol-induced apoptosis. Arch Biochem Biophys. 428:179–187.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ko CH, Shen SC, Hsu CS and Chen YC:
Mitochondrial-dependent, reactive oxygen species-independent
apoptosis by myricetin: roles of protein kinase C, cytochrome c,
and caspase cascade. Biochem Pharmacol. 69:913–927. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yamamori T, Yasui H, Yamazumi M, et al:
Ionizing radiation induces mitochondrial reactive oxygen species
production accompanied by upregulation of mitochondrial electron
transport chain function and mitochondrial content under control of
the cell cycle checkpoint. Free Radic Biol Med. 53:260–270. 2012.
View Article : Google Scholar
|