Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common cancer worldwide (1) and its
frequency is increasing in Southeast Asia, Africa and Western
countries. In particular, the mortality rate of HCC in Taiwan has
not decreased due to limited treatment options (2,3). HCC
occurs more often in men than in women; in addition, males have a
poorer prognosis in comparison with females (4). However, little is known about the
underlying molecular mechanisms of HCC.
The role of nuclear receptors in HCC development has
drawn considerable attention (5).
One such example is peroxisome proliferator-activated receptor γ
(PPARγ), a ligand-activated transcription factor that is involved
in tumor promotion, cellular differentiation and apoptosis
(6,7). Several other studies also focused on
PPARγ as their target gene to treat various types of cancer, such
as colon, thyroid, lung, breast, prostate and liver cancer
(8). For example, troglitazone
inhibited the growth of human liver cancer cells by inducing
apoptosis through caspase-3 activation (9). In breast cancer cells, estrogen
receptor (ER)α binds to peroxisome proliferator-activated receptor
response element and negatively interferes with PPARγ signaling
(10). Similarly, in preadipocytes
cells, ERβ overexpression inhibits ligand-mediated PPARγ activity,
which further results in a blockade of PPARγ-induced adipocytic
gene expression (11).
Expression ratio of ERα and ERβ apparently changes
during hepatocarcinogenesis (12).
A large body of evidence has shown decreased ERα in HCC patients
(13,14); similarly, loss of ERβ expression has
been indicated as a common step in the development of colorectal
cancer (15). Activation of these
ERs controls several biological processes, including cell growth,
differentiation and apoptosis. However, the effect of ERα or ERβ on
PPARγ expression in HCC is not well studied. In the present study,
either ERα or ERβ is overexpressed by transient transfection and
then receptor is activated by 17β-estradiol. At the same time, we
conducted the assay with 17β-estradiol alone to elucidate whether
ligand alone can induce ERα or ERβ expression in ER-negative Hep3B
cells. These results showed that ERα or ERβ may act as a tumor
suppressor in downregulating PPARγ expression in Hep3B cells and
were further accelerated by ligand addition.
Materials and methods
Specimen collection and
immunohistochemistry
Written consent was obtained from all patients.
Surgical specimens of human liver cancer tissues were obtained by
mastectomy from the operating rooms of the Changhua Christian
Hospital in Changhua and the China Medical University Hospital in
Taichung, Taiwan. Following resection, these specimens were stored
at −70°C before being used for the analysis. The tissue biopsy was
dried at 58°C overnight, dewaxed in xylene for 40 min and
rehydrated in ethanol. Blocking with 3% H202
in 50% methanol/50% phosphate-buffered saline (PBS) and incubated
with 5% cosmic calf serum to reduce non-specific staining of the
secondary antibody. Tissue sections were incubated overnight at 4°C
with PPARγ (1:100). The sections were washed with PBS and incubated
for 1 h at room temperature with the peroxidase-conjugated
secondary antibody. Immunoreactivity was visualized with
3,3′-diaminobenzidine (DAB) substrate (Roche Diagnostics, Mannheim,
Germany). After coloring and rinsing with distilled water, the
sections were counterstained slightly with Mayer’s hematoxylin,
dehydrated in graded alcohols, cleared in xylene and detected using
microscopy (Olympus, Tokyo, Japan).
Cell culture
The Chang liver cell line, HepG2, Hep3B, Huh-7 and
HA22T cells were purchased from ATCC. Chang liver cells were grown
in DMEM, HepG2 and Hep3B were grown in MEM (Gibco, Grand Island,
NY, USA) and Huh-7 and HA22T cells were grown in DMEM. All media
were supplemented with 10% fetal bovine serum (FBS) (Biochrom AG,
Berlin, Germany) and 1% penicillin streptomycin (Gibco).
Establishment of the double-stable
Tet-On/ERα and ERβ Hep3B cell line
The double-stable Tet-On/ERα or ERβ Hep3B cell line,
which grows well in the presence of both G418 and hygromycin, was
established by plasmid transfection using the Lipofectamine method.
Briefly, the primary Tet-On Hep3B cell line was generated by
transfecting Hep3B cells with 10 μg Tet-On (Clontech Laboratories,
Worcester, MA, USA), a regulator plasmid encoding the G418
resistance gene. The primary Tet-On Hep3B cells were then
transfected with 10 μg of pTRE2/ERα or ERβ plasmid encoding the
hygromycin resistance gene. Double-stable cells were selected with
700 μg/ml G418 and 100 μg/ml hygromycin and further screened for
ERα mRNA using DNA sequencing.
Transfection
Hep3B cells were transfected with a plasmid carrying
the ERα and ERβ gene using 10 and 100 μM of Lipofectamine
(Invitrogen, Auckland, New Zealand) according to the manufacturer’s
guidelines. After 6 h of transfection, MEM supplemented with 10%
charcoal/dextran (CD)-FBS (Sigma, St. Louis, MO, USA) was added for
12 h, and MEM containing 1% FBS and antibiotics were added for 6 h.
Prior to treatment, the cells were starved in MEM (no phenol red)
with 1% antibiotics for 6 h and then replaced with phenol red-free
MEM containing 1% FBS and vehicle or 17β-estradiol (E2) (Sigma),
doxycycline or fenofibrate (Clontech, Mountain View, CA, USA) for
different times. These transfection experiments were repeated three
times with consistent results.
Reverse transcription (RT)
Total RNA was extracted using an Ultraspec™ kit
(Biotecx, Houston, TX, USA) according to the manufacturer’s
instructions. A total of 4 μg of RNA was used for the RT reaction.
RT was performed at 37°C for 60 min using 55.5 μl DEPC
H2O, 4 μg total RNA, 0.5 μl of RNase inhibitor (40 U/μl)
(Promega, Madison, WI, USA), 20 μl of 5X RT buffer, 8 μl of dNTP
(2.5 mM), 10 μl of oligo(dT) (5 μM/ml) (Mission Biotech, Taipei,
Taiwan) and 2 μl of MMLV reverse transcriptase (200 U/μl)
(Promega). The resulting cDNA was added to the PCR mixture
containing 9.5 μl of DEPC water, 2.5 μl of 10X PCR buffer (MD Bio,
Taipei, Taiwan), 2.5 μl of dNTP (10 mM) (Promega), 2.5 μl of each
primer (5 μM), 0.5 μl of Taq (2 U/μl) (MD Bio) and 4 μl of 2.5
mMdNTP mixture.
Western blotting
Cells were lysed at each time-point with lysis
buffer [50 mM Tris base (pH 7.4), 0.5 M NaCl, 1 M
ethylenediamine-mercaptoethanol (BME), 1% NP-40%, 10% glycerol,
Igepal CA-630] (Sigma) and protease inhibitor cocktail tablets
(Roche). Proteins were analyzed and separated by 10% SDS-PAGE,
transferred to nitrocellulose membranes and probed with antibodies
against the following proteins: PPARγ, ERα, ERβ and α-tubulin
(Santa Cruz Biotechnology, Santa Cruz, CA, USA). The blots were
incubated with peroxidase-conjugated secondary antibody for 1 h.
Bands were monitored using western blot chemiluminescence reagent
(Santa Cruz Biotechnology).
Statistical analysis
All data are expressed as percentages of the control
and mean ± SD. The results are based on three independent
experiments. Student’s unpaired t-test was used to compare the
differences between groups. Experimental group vs. control group:
P-value <0.05 was considered to indicate a statistically
significant difference; *P<0.05 and
**P<0.01.
Results
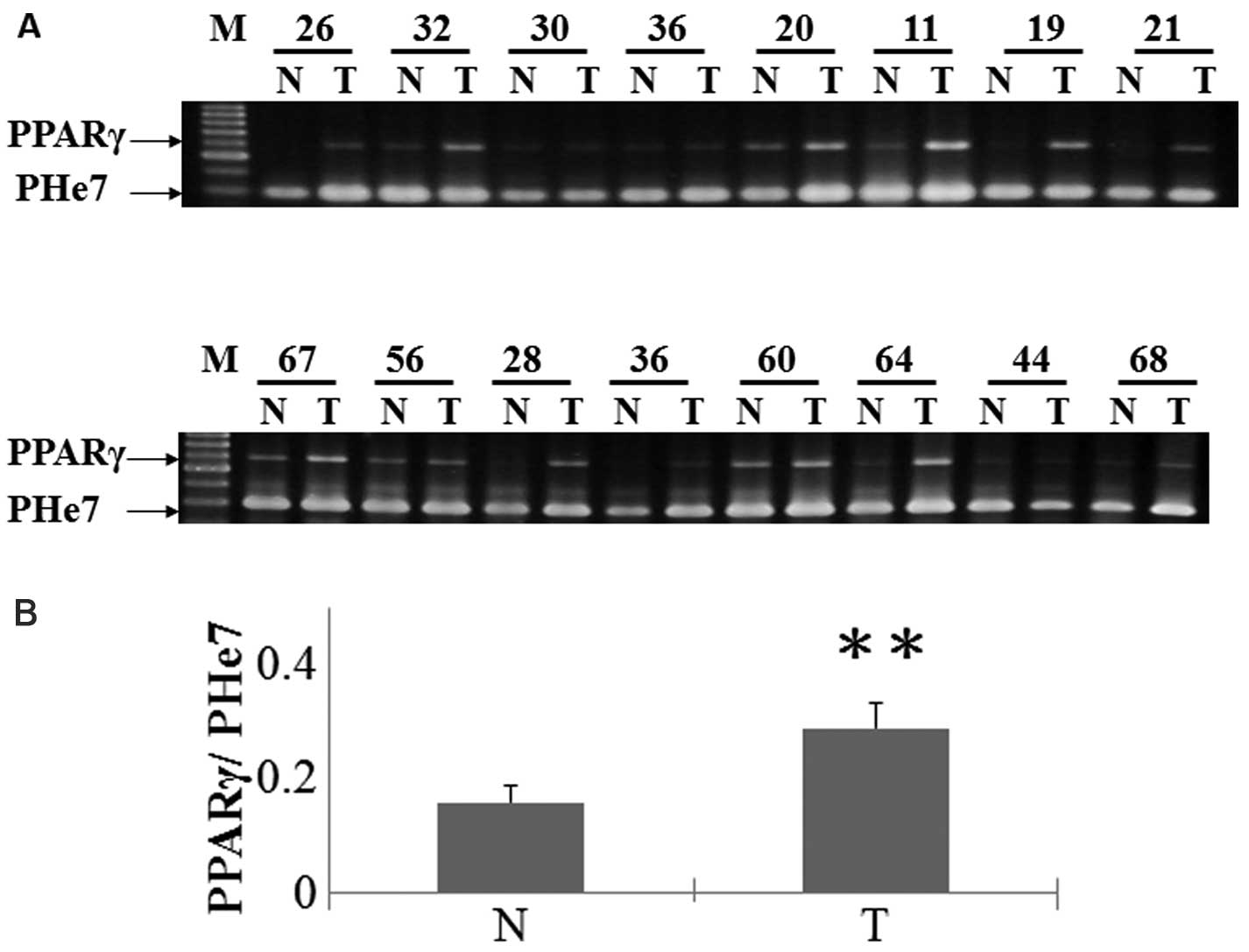
PPARγ expression in HCC is significantly increased
in tumor tissues compared with surrounding non-tumorous liver,
particularly in poorly differentiated tumor compared to
well-differentiated tumor (16). To
assess the role that PPARγ in liver cancer in vivo, we
analyzed tumor and non-tumor patient tissues for PPARγ expression.
The majority of normal liver tissues do not express PPARγ, whereas
tumor tissues from the liver cancer patients showed a significant
increase in PPARγ expression (Fig.
1).
Next, we analyzed the expression of PPARγ in HCC
cells in vitro using HepG2, Hep3B, HuH-7 and HA22T cell
lines. As shown in Fig. 2A, RT-PCR
analysis readily detected the expression of PPARγ mRNA in all cell
lines. Western blot analysis did not display exactly same
expression pattern when compared with the mRNA expression. Compared
with other cell lines used, only Hep3B cells expressed PPARγ
protein (Fig. 2B). These results
were consistent with the hypothesis that mRNA levels do not
necessarily correlate with the protein expression data (17).
Estrogen exerts its biological function by binding
to one of two specific ERs, ERα and ERβ. Thus, the level of
exogenous ERα and endogenous PPARγ in Hep3B cells transfected with
empty vector or ERα expression vectors was examined in the presence
of E2. As shown in Fig. 3A, ERα
containing Hep3B cells induced ERα mRNA expression and further
decreased PPARγ expression. On the other hand, E2 treatment altered
the expression level of ERα and PPARγ in ERα overexpressing Hep3B
cells. However, in empty vector transfected Hep3B cells, E2
treatment reduced PPARγ mRNA levels without increasing ERα
expression. This was further confirmed by western blot analysis
(Fig. 3B). We then verified the
possibility of ERα in inhibiting PPARγ expression using a stable
cell line that expresses ERα. Tet-On/ERα Hep3B cells were treated
with a range (0–1.5 μg/ml) of Dox for 24 h and then analyzed for
PPARγ expression. As shown in Fig.
3C, a dose-dependent decrease in PPARγ was observed in response
to Dox. This was more noticeable at the 0.2 μg/ml
concentration.
In order to elucidate whether exogenous expression
of ERβ inhibits PPARγ expression, vector or ERβ transfected Hep3B
cells were exposed to E2 treatment. No activation of ERβ and PPARγ
was observed in cells transfected with empty vector and in E2
exposed cells. Fig. 4 shows that
ERβ overexpression plus E2 treatment effectively inhibited PPARγ
mRNA and protein expression.
Discussion
The nuclear receptor superfamily (estrogen, thyroid,
glucocorticoid receptors and peroxisome proliferator-activated
receptors) plays an important role in controlling cellular
homeostasis, and administration of its ligand has been effectively
used in cancer treatment (18–21).
The role of PPARγ in tumor development is controversial as fewer
studies showed ligand activated PPARγ promotes growth inhibition
and apoptosis in human esophageal (22) breast (23) ovarian and liver cancer (24) and other reports showed PPARγ ligand
could inhibit growth and metastasis of PPARγ positive cancer cells
(25). In the present study, we
found a significant increase in PPARγ mRNA expression in HCC
tissues compared with non-cancerous tissues. Similarly, in human
lung cancer tissues, increased PPARγ expression was observed
compared with non-tumor tissues (25). These findings suggest that PPARγ was
involved in hepatocarcinogenesis. However, decreased PPARγ
expression in tumor tissue has been observed in human esophageal,
breast and ovarian cancer (22,26,27).
Therefore, a better understanding of the PPARγ mechanism in
different cancer tissues is required.
PPARγ expression has been demonstrated in
vitro in several cell lines; particularly in liver cancer cell
lines, PPARγ mRNA was expressed at various levels (9). Similarly, we analyzed the effects of
PPARγ activation in four human HCC cell lines, compared with the
HepG2, Huh-7 and HA22T cells, Hep3B cells constitutively express
PPARγ expression at the RNA and protein levels. Having observed
significant upregulation of PPARγ expression in Hep3B cells, we
next conducted experiments to test the potential role of ERs in
inhibiting PPARγ expression in Hep3B cells.
Previous studies showed overexpression of ERα
inhibits growth of ECV304 and the Ishikawa cell line by decreasing
endothelin-1 and VEGF expression (28). Our results, consistent with a
previous report (29), showed that
ERα binds with PPARγ and functionally interferes with PPARγ
signaling in a ligand-dependent manner. Compared with ERα
expression, decreased ERβ was found in patients with chronic
hepatitis or cirrhosis and in those with HCC. In normal breast
cells, ERβ was found to negatively regulate cellular proliferation.
Our data are in agreement with these results, showing ERβ
overexpression decreased PPARγ expression in an E2-dependent
manner. Collectively, the present study provided a basic
understanding of ERα and ERβ in PPARγ expression; further studies
using these ERs are currently being conducted to elucidate how
these ERs control Hep3B cell molecular mechanisms.
Acknowledgements
This research was funded by the China Medical
University (grant no. CMU 101-AWARD-04 and CMU 101-S-18).
References
|
1
|
Greten TF, Papendorf F, Bleck JS, et al:
Survival rate in patients with hepatocellular carcinoma: a
retrospective analysis of 389 patients. Br J Cancer. 92:1862–1868.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB: Hepatocellular carcinoma:
recent trends in the United States. Gastroenterology. 127:S27–S34.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weng CJ, Chau CF, Hsieh YS, Yang SF and
Yen GC: Lucidenic acid inhibits PMA-induced invasion of human
hepatoma cells through inactivating MAPK/ERK signal transduction
pathway and reducing binding activities of NF-κB and AP-1.
Carcinogenesis. 29:147–156. 2008.PubMed/NCBI
|
|
4
|
El-Serag HB, Mason AC and Key C: Trends in
survival of patients with hepatocellular carcinoma between 1977 and
1996 in the United States. Hepatology. 33:62–65. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vacca M, Degirolamo C, Massafra V, et al:
Nuclear receptors in regenerating liver and hepatocellular
carcinoma. Mol Cell Endocrinol. 368:108–119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Koeffler HP: Peroxisome
proliferator-activated receptor γ and cancers. Clin Cancer Res.
9:1–9. 2003.
|
|
7
|
Vanden Heuvel JP: Peroxisome
proliferator-activated receptors (PPARS) and carcinogenesis.
Toxicol Sci. 47:1–8. 1999.PubMed/NCBI
|
|
8
|
Panigrahy D, Singer S, Shen LQ, et al:
PPARγ ligands inhibit primary tumor growth and metastasis by
inhibiting angiogenesis. J Clin Invest. 110:923–932. 2002.
|
|
9
|
Toyoda M, Takagi H, Horiguchi N, et al: A
ligand for peroxisome proliferator activated receptor γ inhibits
cell growth and induces apoptosis in human liver cancer cells. Gut.
50:563–567. 2002.
|
|
10
|
Wang X and Kilgore MW: Signal cross-talk
between estrogen receptor alpha and beta and the peroxisome
proliferator-activated receptor gamma1 in MDA-MB-231 and MCF-7
breast cancer cells. Mol Cell Endocrinol. 194:123–133. 2002.
View Article : Google Scholar
|
|
11
|
Foryst-Ludwig A, Clemenz M, Hohmann S, et
al: Metabolic actions of estrogen receptor beta (ERβ) are mediated
by a negative cross-talk with PPARγ. PLoS Genet.
4:e10001082008.
|
|
12
|
Wu X, Subramaniam M, Grygo SB, et al:
Estrogen receptor-beta sensitizes breast cancer cells to the
anti-estrogenic actions of endoxifen. Breast Cancer Res.
13:R272011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ng IO, Ng M and Fan ST: Better survival in
women with resected hepatocellular carcinoma is not related to
tumor proliferation or expression of hormone receptors. Am J
Gastroenterol. 92:1355–1358. 1997.PubMed/NCBI
|
|
14
|
Jonas S, Bechstein WO, Heinze T, et al:
Female sex hormone receptor status in advanced hepatocellular
carcinoma and outcome after surgical resection. Surgery.
121:456–461. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kennelly R, Kavanagh DO, Hogan AM and
Winter DC: Oestrogen and the colon: potential mechanisms for cancer
prevention. Lancet Oncol. 9:385–391. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu J, Shen B, Chu ES, et al: Inhibitory
role of peroxisome proliferator-activated receptor gamma in
hepatocarcinogenesis in mice and in vitro. Hepatology.
51:2008–2019. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Borbath I and Horsmans Y: The role of
PPARγ in hepatocellular carcinoma. PPAR Res. 2008:2095202008.
|
|
18
|
Li MY, Deng H, Zhao JM, Dai D and Tan XY:
Peroxisome proliferator-activated receptor gamma ligands inhibit
cell growth and induce apoptosis in human liver cancer BEL-7402
cells. World J Gastroenterol. 9:1683–1688. 2003.PubMed/NCBI
|
|
19
|
Lovat PE, Oliverio S, Ranalli M, et al:
GADD153 and 12-lipoxygenase mediate fenretinide-induced apoptosis
of neuroblastoma. Cancer Res. 62:5158–5167. 2002.PubMed/NCBI
|
|
20
|
Galbiati E, Caruso PL, Amari G, et al:
Pharmacological actions of a novel, potent, tissue-selective
benzopyran estrogen. J Pharmacol Exp Ther. 303:196–203. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen GG, Lee JF, Wang SH, Chan UP, Ip PC
and Lau WY: Apoptosis induced by activation of
peroxisome-proliferator activated receptor-gamma is associated with
Bcl-2 and NF-kappaB in human colon cancer. Life Sci. 70:2631–2646.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Terashita Y, Sasaki H, Haruki N, et al:
Decreased peroxisome proliferator-activated receptor gamma gene
expression is correlated with poor prognosis in patients with
esophageal cancer. Jpn J Clin Oncol. 32:238–243. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cui Y, Miyoshi K, Claudio E, et al: Loss
of the peroxisome proliferation-activated receptor gamma (PPARγ)
does not affect mammary development and propensity for tumor
formation but leads to reduced fertility. J Biol Chem.
277:17830–17835. 2002.
|
|
24
|
Yu J, Qiao L, Zimmermann L, et al:
Troglitazone inhibits tumor growth in hepatocellular carcinoma in
vitro and in vivo. Hepatology. 43:134–143. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li MY, Hsin MK, Yip J, Mok TS, Underwood
MJ and Chen GG: PPARgamma activation extinguishes smoking
carcinogen by inhibiting NNK-mediated proliferation. Am J Respir
Cell Mol Biol. 42:113–122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Badawi AF and Badr MZ: Expression of
cyclooxygenase-2 and peroxisome proliferator-activated receptor-γ
and levels of prostaglandin E2 and
15-deoxy-Δ12,14-prostaglandin J2 in human
breast cancer and metastasis. Int J Cancer. 103:84–90. 2003.
|
|
27
|
Sakamoto A, Yokoyama Y, Umemoto M, et al:
Clinical implication of expression of cyclooxygenase-2 and
peroxisome proliferator activated-receptor gamma in epithelial
ovarian tumours. Br J Cancer. 91:633–638. 2004.PubMed/NCBI
|
|
28
|
Upla P, Marjomaki V, Kankaanpaa P, et al:
Clustering induces a lateral redistribution of α2β1 integrin from
membrane rafts to caveolae and subsequent protein kinase
C-dependent internalization. Mol Biol Cell. 15:625–636. 2004.
|
|
29
|
Bonofiglio D, Gabriele S, Aquila S, et al:
Estrogen receptor α binds to peroxisome proliferator-activated
receptor response element and negatively interferes with peroxisome
proliferator-activated receptor γ signaling in breast cancer cells.
Clin Cancer Res. 11:6139–6147. 2005.
|