Introduction
microRNAs (miRNAs) are an endogenous conserved class
of non-coding 20–22 nt small RNAs that regulate gene expression at
the post-transcriptional level by mostly binding to 3′-UTR of
target mRNAs, leading to mRNA degradation or translation
inhibition. Recent reports demonstrate a role for miRNA expression
in disease progression and outcome. To date, several miRNA
expression profiles in esophageal squamous cell carcinoma (ESCC)
have been reported (1,2), and several specific miRNAs have been
proven to be involved in ESCC tumorigenesis, including miR-21
(3) and miR-129–2 (4).
The role of miR-27a in tumorigenesis differs in
various cells and tissues. It is regarded as an oncogene in several
types of tumors. Liu et al(5) reported that suppression of miR-27a
inhibits gastric cancer cell growth by targeting prohibitin; Zhang
et al(6) confirmed that the
overexpression of miR-27 promotes the metastasis of the human
gastric cancer cell line AGS, whereas its depletion decreases cell
metastasis. However, miR-27a was found to be downregulated in
several other tumor types (reviewed in ref. 7), including acute promyelocytic leukemia
(8), colorectal cancer (9,10),
malignant melanoma (11), oral
squamous cell carcinoma (12) and
prostate cancer (13), indicating
it may be a possible tumor suppressor. Most recently, miR-27a was
found to target epidermal growth factor receptor (EGFR) (14,15),
which is activated and promotes tumorigenesis in numerous types of
tumors.
To date, there are no report on the role of miR-27a
in the tumorigenesis of ESCC. Although Zhang et al(16) showed that the downregulation of
miR-27a reversed the multidrug resistance of ESCCs, they did not
investigate the expression level of miR-27a in ESCC and its
possible role in tumorigenesis.
In the present study, we evaluated the expression of
miR-27a in ESCC specimens and cell lines and studied the role of
miR-27a in cell growth and migration of esophageal carcinoma cell
line TE-1. We also investigated the mechanisms of miR-27a
modulation during TE-1 cell growth.
Materials and methods
Cell lines
Primary cultures of normal esophageal epithelial
cells (NEECs) were established in our laboratory. NEECs were
established from fresh biopsies of adjacent non-cancerous
esophageal tissue, according to a previous report (17). The NEECs and ESCCs were grown at
37°C in 5% CO2 with keratinocyte serum-free medium, with
40 μg/ml bovine pituitary extract, 1.0 ng/ml epidermal growth
factor, 100 U/ml penicillin and 100 μg/ml streptomycin.
The esophageal cancer cell lines, including NECC,
TE1, TE13, Eca109, EC9706, KYSE140 and KYSE30, were obtained from
the Cell Bank of the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China). All cell lines were grown in
RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with
10% fetal bovine serum, 100 μg/μl streptomycin and 100 μg/μl
penicillin in a humidified incubator containing 5% CO2
at 37°C.
Patient information and tissue
specimens
The present study was conducted on a total of 25
samples from ESCC patients with nodal involvement, which were
histopathologically and clinically diagnosed at the First
Affiliated Hospital of Zhengzhou University in 2012. For the use of
these clinical materials for research purposes, prior patient
consent and approval from the Institutional Research Ethics
Committee were obtained.
Quantitative RT-PCR analysis
(qRT-PCR)
Total RNAs were extracted from cells with TRIzol
reagent (Invitrogen). For the detection of KRAS mRNA, cDNA was
synthesized from 1 mg of total RNA by means of a reverse reaction
kit according to the manufacturer’s instructions (Promega, Madison,
WI, USA). Human GAPDH was amplified in parallel as an internal
control. For miR-27a, reverse transcription and qRT-PCR reactions
were performed by means of a qSYBR-Green-containing PCR kit
(Shanghai Genepharma Co., Ltd., Shanghai, China), and U6 snRNA was
used as an endogenous control for miRNA detection. Expression of
each gene was quantified by measuring cycle threshold (Ct) values
and normalized using the ΔCt method [2Ct(reference) −
Ct(target)] relative to U6 snRNA or GAPDH.
Constructs
luc-UTR vectors were constructed by cloning the
predicted miR-27a target region or its mutant control into the
NheI and SalI sites of the pmirGLO luciferase vector
(Promega) using the PCR generated fragments. The oligonucleotide
pairs contained the Kpn1 internal site for clone
confirmation: sense-wt: 5′-CTAGCTAGGTACCTTGAACT
AGCAATGCCTGTGAAAG-3′ and antisense-wt: 5′-TCG
ACTTTCACAGGCATTGCTAGTTCAAGGTACCTAG-3′; sense-mut:
5′-CTAGCTAGGTACCTTATTATAGCAATG
CACACAGAAG-3′ and antisense-mut:
5′-TCGACTTCTGT GTGCATTGCTATAATAAGGTACCTAG-3′. Bold
indicates NheI and SalI sites; underlining indicates
the Kpn1 site; italics indicates the mutated sites.
Synthesized RNA duplexes of scramble miRNA, miR-27a,
and their inhibitors anti-scramble and anti-miR-27a were obtained
from Baoxin Bio-Technology Co., Ltd. (Zhengzhou, China). To
construct a vector expressing miR-27a, the precursor sequence of
miR-27a (MI0000085) was synthesized, annealed and then inserted
into the BamHI-HindIII fragment of the pGCsi/U6
vector (GeneChem, Shanghai, China). A construct including the
non-specific miRNA was used as a negative control. The miR-27a
knockdown lentivirus was purchased from Baoxin Bio-Technology.
The KRAS-expressing vector was constructed by
cloning full-length KRAS cDNA into the eukaryotic expression vector
pcDNA3.1(+) (Invitrogen). The empty pcDNA3.1(+) vector was used as
a negative control.
Cell transfection and infection
TE-1 cells were infected with the miR-27a lentivirus
or the control lentivirus expressing a scrambled miRNA. All cells
were selected with 500 mg/l G418 to generate two stable monoclonal
cell lines (a stable cell line expressing miR-27a, TE-1-miR-27a and
a control stable cell line, TE-1-scramble).
To establish stable miR-27a knockdown cell lines,
TE-1 cells were transduced with the miR-27a knockdown lentivirus or
the control lentivirus and selected with 5 mg/l puromycin.
For miRNA and pcDNA3.1-KRAS combination experiments,
TE-1-miR-27a and TE-1-scramble cells were transfected with
pcDNA3.1-KRAS or empty vector using Lipofectamine 2000
(Invitrogen).
Luciferase assay
TE-1-miR-27a cells were transfected with
pmirGLO-KRAS-wt, pmirGLO-KRAS-mut or pmirGLO-ctrl using
Lipofectamine 2000 (Invitrogen). Luciferase activity was measured
24 h after transfection using the Dual-Glo luciferase assay system
(Promega). The Renilla luciferase activity served as
internal control.
MTT assay
Cells were seeded onto 96-well plates at a density
of 5×104 cells/well in 100 μl medium. All cells were
maintained in a humidified 37°C incubator with 5% CO2.
20 μl 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) solution (5 g/l in phosphate-buffered saline) was added to
each well of the microplate, and the absorbance at 570 nm was
measured by a microplate reader. After a 4-h incubation, the number
of viable cells was measured a point, was set 5 re-wells.
In vitro scratch assay
TE-1 cells (5×106/well) were seeded to
90% confluence in a 6-well plate for overnight culture. The
following day a scratch was made through the center of each well
using a 200-μl pipette tip, creating an open ‘scratch’ or ‘wound’
that was clear of cells. The dislodged cells were removed by three
washes with complete culture media, and cells were incubated under
standard conditions. Migration into the open area was documented at
72 h post-scratching.
Western blot analysis
Western blot analyses were performed as previously
described (18). Antibodies against
KRAS (Sigma-Aldrich), ERK (Santa Cruz Biotechnology),
phosphorylated ERK (Santa Cruz Biotechnology), and c-Fos (Santa
Cruz Biotechnology) were obtained from Cell Signaling Technology.
The anti-β-actin antibody was from Santa Cruz Biotechnology.
In vivo tumorigenesis
Five-week-old male nude athymic BALB/c nu/nu mice
were used for examining tumorigenicity. To evaluate the role of
miR-27a in tumor formation, TE-1-miR-27a cells, TE-1-scramble
control, TE-1-anti-miR-27a cells, or TE-1-anti-scramble control
were propagated and inoculated subcutaneously into the dorsal
flanks of nude mice (2×106 cells in a 0.2-ml volume).
Tumor size was measured every 5 days. After 30 days, the mice were
sacrificed, necropsies were performed and the tumors were weighed.
Tumor volumes were determined according to the following formula: A
× B2/2, where A is the largest diameter and B is the
diameter perpendicular to A. The experiments were performed using
five mice per group, and all animal procedures were performed in
accordance with institutional guidelines.
Statistical analysis
Statistical evaluation of data was performed using
SPSS 13 analysis software (SPSS, Chicago, IL, USA).
Independent-samples t-test, or paired-samples t-test were used to
evaluate statistical significance. Spearman’s correlation tests
were used to evaluate the pair-wise expression correlation between
miR-27a and KRAS. Data are shown as means ± SEM. The significant
level was set at P<0.05.
Results
miR-27a is downregulated in ESCC cell
lines and clinical specimens
Published datasets archived in the publicly
available Gene Expression Omnibus (GEO) repository were used for
re-analysis using GEO2R (NCBI online gene expression tool)
(19). Datasets published in GEO
reference series GSE13937 (20)
were used for microRNA expression analysis of hsa-miR-27a in ESCC
compared with adjacent normal esophageal tissues. These datasets
used OSU-CCC Human and Mouse MicroRNA Microarray version 3.0 array
platform. Normalized signal intensity value was analyzed using the
paired-samples t-test analysis. The expression of hsa-miR-27a was
decreased in tumors when compared with that in adjacent normal
tissues from ESCC patients with nodal involvement (n=20; P=0.041).
However, in ESCC patients without nodal involvement, this
difference was not significant (n=24; P=0.785) (Fig. 1A).
We used quantitative real-time PCR (qRT-PCR) to
measure mature miR-27a expression levels in ESCC tissues of
patients with nodal involvement and cell lines. miR-27a was
significantly downregulated in ESCC tissues when compared with the
paired adjacent normal tissues (paired-samples t-test, n=25;
P=0.025) (Fig. 1B). In six
esophageal carcinoma cell lines, the expression of miR-27a was
decreased, particularly in TE-1 cells (independent-samples t-test;
P=0.003) (Fig. 1C).
miR-27a directly targets and inhibits
KRAS
To determine the role of miR-27a in ESCCs, we
performed a bioinformatic search (TargetScan and PicTar) to
identify putative mRNA 3′-UTR targets for the mature miR-27a. We
found that miR-27a had a seed region that matched the 3′-UTR of
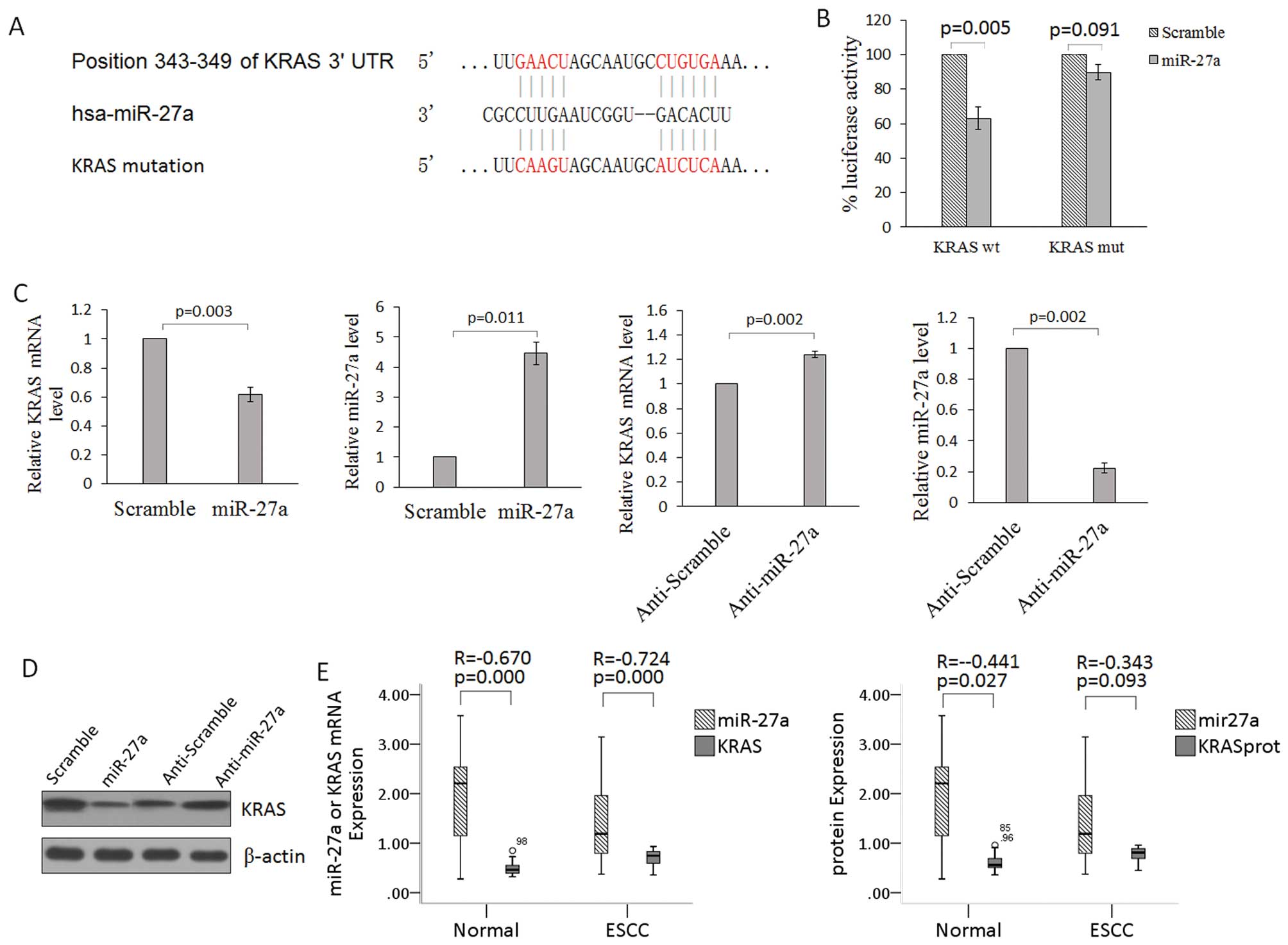
human KRAS (nucleotides 343–349; NM_033360) (Fig. 2A). To verify that KRAS is a direct
target of miR-27a, the KRAS 3′-UTR containing the miR-27a binding
site was cloned into the pmirGLO control vector downstream of the
luciferase ORF. This reporter construct was used to transfect 293
cells. Co-transfection of miR-27a with the wt KRAS 3′-UTR construct
in 293 cells resulted in a significant inhibition of luciferase
activity compared with the negative control (Fig. 2B). Mutagenesis of the miR-27a
binding site within the KRAS 3′-UTR abolished the ability of
miR-27a to regulate the luciferase expression (Fig. 3B). In addition, overexpression of
miR-27a in TE-1 cells strongly reduced the endogenous protein and
mRNA levels of KRAS when compared with these levels in the control
(Fig. 2C and D). In clinical ESCC
specimens, there was an inverse correlation between the
mRNA/protein levels of KRAS and the level of miR-27a expression
(Fig. 2E). These data suggest that
miR-27a acts as a tumor suppressor by targeting KRAS in ESCC.
miR-27a inhibits the ERK pathway
KRAS activation can trigger several important
signaling pathways, such as the Ras/Raf/MEK/ERK pathways, most of
which regulate cell proliferation, survival and invasion.
Therefore, we investigated the possibility that miR-27a regulates
those pathways by targeting KRAS. Upregulation of miR-27a through
transfection of miR-27a in TE-1 cells suppressed the levels of
phosphorylated ERK and its downstream effector c-Fos. We also
observed that knockdown of miR-27a, through transfection of
anti-miR-27a, in TE-1 cells increased the levels of ERK (Fig. 3A). Results of the western blot
analysis demonstrated that miR-27a is a negative regulator of the
ERK pathway.
Subsequently, rescue experiments were performed by
overexpressing the KRAS vector (without an endogenous 3′-UTR) in
miR-27a-treated cells. TE-1 cells were first transfected with
miR-27a and then with the KRAS-encoding vector 48 h later. The
miR-27a-induced downregulation of KRAS was rescued upon the
introduction of KRAS, and the phosphorylation level of ERK was
altered in a similar manner (Fig.
3B). These observations suggest that miR-27a inhibits the ERK
pathway by targeting KRAS.
miR-27a suppresses TE-1 cell
proliferation and invasion by targeting KRAS
Overexpression of miR-27a markedly attenuated cell
proliferation, and knockdown of miR-27a promoted cell proliferation
in TE-1 cells (Fig. 4A). Forced
expression of KRAS (without an endogenous 3′-UTR) rescued the cell
growth inhibition of miR-27a partially, suggesting that miR-27a
regulates cell growth through targeting KRAS (Fig. 4A). Scratch assays were used to
measure cell migration or to observe the healing of scratches in
cancer cell monolayers. Overexpression of miR-27a significantly
inhibited the ability of TE-1 cells to heal scratch assays.
However, knockdown of miR-27a significantly promoted this behavior
(Fig. 4B). In nude mouse xenograft
models, TE-1 cells with miR-27a overexpression developed smaller
tumors (both in tumor volume and weight) compared with TE-1 cells
with normal miR-27a (scramble). Accordingly, miR-27a knockdown in
TE-1 cells promoted tumor formation (both in tumor volume and
weight) in the nude mouse xenograft experiments (Fig. 4C).
Discussion
We first analyzed the miR-27a expression data
archived in GEO. GSE13937 recorded the miRNA array data to compare
miRNA expression between ESCC and paired adjacent normal tissues.
We found that miR-27a was significantly downregulated in ESCCs with
nodal involvement. However, miR-27a was not downregulated in ESCCs
without nodal involvement. The downregulation of miR-27a is present
only in ESCC specimens with nodal involvement, indicating it may
participate in the migratory capacity of ESCC. In order to confirm
the expression change of miR-27a, we further studied the miR-27a
expression in 25 ESCC specimens with nodal involvement using
qRT-PCR, and demonstrated miR-27a is downregulated in ESCCs. In six
esophageal carcinoma cell lines, the expression of miR-27a was also
decreased, particularly in TE-1 cells.
We next explored the possible targets of miR-27a in
ESCCs through different computational algorithms. In the present
study, we proved that miR-27a targets KRAS directly. KRAS is a
GTPase and an early player in many signal transduction pathways.
Active GTP-bound KRAS associates with a wide variety of effectors,
including Raf, PI3K, Ral-GDS, Rho GTPases and other molecules, to
transmit downstream signals that control distinct cellular events,
including cell proliferation, survival, differentiation and
invasion (21,22). KRAS promotes tumorigenesis and has
been proven to be downregulated by the let-7 family (23) and miR-96 (24). ESCC patients (16%) (5/30 cases) were
found to harbor KRAS gene mutations (25). In the present study, we demonstrated
that miR-27a may be a negative regulator of the ERK pathway,
through targeting KRAS and decreased expression of phosphorylated
ERK and its downstream effector c-Fos.
In order to investigate the role of miR-27a in
ESCCs, gain and loss of function experiments were conducted. Forced
expression of miR-27a significantly decreased the cell
proliferation and migration of TE-1 cells, while miR-27a inhibition
exerted an opposite effect. In the nude mouse assay, the result was
consistent with that in vitro. We hypothesized that miR-27a
is a tumor suppressor in ESCCs, although it is regarded as an
oncogene in several other tumor types (5,6). Our
results support the opinion that the same miRNA can have
antagonizing roles in two different cell types; i.e. in one cell
type the miRNA promotes proliferation whereas in another cell type
the same miRNA inhibits proliferation (7).
In conclusion, this is the first study to show that
miR-27a inhibits cell proliferation and invasion in ESCC cells. It
is also the first study to show that tumor promotor KRAS is
negatively regulated by miR-27a at the post-transcriptional level
via binding to 3′-UTR of KRAS mRNA in esophageal squamous cell
carcinoma cells.
Acknowledgements
The present study was supported by the Zhengzhou
University 211 Project-Phase II, The Basic and Clinical Research of
Stem Cells.
References
|
1
|
Gu J, Wang Y and Wu X: MicroRNA in the
pathogenesis and prognosis of esophageal cancer. Curr Pharm Des.
19:1292–1300. 2013.PubMed/NCBI
|
|
2
|
Yang M, Liu R, Sheng J, et al:
Differential expression profiles of microRNAs as potential
biomarkers for the early diagnosis of esophageal squamous cell
carcinoma. Oncol Rep. 29:169–176. 2013.PubMed/NCBI
|
|
3
|
Liu F, Zheng S, Liu T, et al: MicroRNA-21
promotes the proliferation and inhibits apoptosis in Eca109 via
activating ERK1/2/MAPK pathway. Mol Cell Biochem. 381:115–125.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kang M, Li Y, Liu W, et al: miR-129–2
suppresses proliferation and migration of esophageal carcinoma
cells through downregulation of SOX4 expression. Int J Mol Med.
32:51–58. 2013.
|
|
5
|
Liu T, Tang H, Lang Y, Liu M and Li X:
MicroRNA-27a functions as an oncogene in gastric adenocarcinoma by
targeting prohibitin. Cancer Lett. 273:233–242. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Z, Liu S, Shi R and Zhao G: miR-27
promotes human gastric cancer cell metastasis by inducing
epithelial-to-mesenchymal transition. Cancer Genet. 204:486–491.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chhabra R, Dubey R and Saini N:
Cooperative and individualistic functions of the microRNAs in the
miR-23a~27a~24-2 cluster and its implication in human diseases. Mol
Cancer. 9:2322010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Saumet A, Vetter G, Bouttier M, et al:
Transcriptional repression of microRNA genes by PML-RARA increases
expression of key cancer proteins in acute promyelocytic leukemia.
Blood. 113:412–421. 2009. View Article : Google Scholar
|
|
9
|
Xi Y, Shalgi R, Fodstad O, Pilpel Y and Ju
J: Differentially regulated micro-RNAs and actively translated
messenger RNA transcripts by tumor suppressor p53 in colon cancer.
Clin Cancer Res. 12:2014–2024. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Volinia S, Calin GA, Liu CG, et al: A
microRNA expression signature of human solid tumors defines cancer
gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dai Y, Sui W, Lan H, Yan Q, Huang H and
Huang Y: Comprehensive analysis of microRNA expression patterns in
renal biopsies of lupus nephritis patients. Rheumatol Int.
29:749–754. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kozaki K, Imoto I, Mogi S, Omura K and
Inazawa J: Exploration of tumor-suppressive microRNAs silenced by
DNA hypermethylation in oral cancer. Cancer Res. 68:2094–2105.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Prueitt RL, Yi M, Hudson RS, et al:
Expression of microRNAs and protein-coding genes associated with
perineural invasion in prostate cancer. Prostate. 68:1152–1164.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Acunzo M, Romano G, Palmieri D, et al:
Cross-talk between MET and EGFR in non-small cell lung cancer
involves miR-27a and Sprouty2. Proc Natl Acad Sci USA.
110:8573–8578. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang W, Cheng B, Miao L, Mei Y and Wu M:
Mutant p53-R273H gains new function in sustained activation of EGFR
signaling via suppressing miR-27a expression. Cell Death Dis.
4:e5742013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang H, Li M, Han Y, et al:
Down-regulation of miR-27a might reverse multidrug resistance of
esophageal squamous cell carcinoma. Dig Dis Sci. 55:2545–2551.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu C, Chen K, Zheng H, et al:
Overexpression of astrocyte elevated gene-1 (AEG-1) is associated
with esophageal squamous cell carcinoma (ESCC) progression and
pathogenesis. Carcinogenesis. 30:894–901. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reyes M, Lund T, Lenvik T, Aguiar D,
Koodie L and Verfaillie CM: Purification and ex vivo expansion of
postnatal human marrow mesodermal progenitor cells. Blood.
98:2615–2625. 2001. View Article : Google Scholar
|
|
19
|
Edgar R, Domrachev M and Lash AE: Gene
expression Omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mathe EA, Nguyen GH, Bowman ED, et al:
MicroRNA expression in squamous cell carcinoma and adenocarcinoma
of the esophagus: associations with survival. Clin Cancer Res.
15:6192–6200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Campbell PM, Groehler AL, Lee KM,
Ouellette MM, Khazak V and Der CJ: K-Ras promotes growth
transformation and invasion of immortalized human pancreatic cells
by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res.
67:2098–2106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Johnson SM, Grosshans H, Shingara J, et
al: RAS is regulated by the let-7 microRNA family. Cell.
120:635–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu S, Lu Z, Liu C, et al: miRNA-96
suppresses KRAS and functions as a tumor suppressor gene in
pancreatic cancer. Cancer Res. 70:6015–6025. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lyronis ID, Baritaki S, Bizakis I,
Krambovitis E and Spandidos DA: K-ras mutation, HPV infection and
smoking or alcohol abuse positively correlate with esophageal
squamous carcinoma. Pathol Oncol Res. 14:267–273. 2008. View Article : Google Scholar : PubMed/NCBI
|